



Oxidative stress is considered to be a major detrimental factor limiting longevity [1], as originally postulated in the free radical theory of aging [2]. The oxidative stress leads to accrual of damaged/misfolded proteins [3], increased mutagenesis rate [4] and inflammation [5,6]. Ironically, due to its ability to accumulate over time (as it was seen in many neurodegenerative disorders) [3], oxidative damage also emerged as a consequence of longevity per se. The human life-span exceeds that of most mammalian species at least 4 times (median life span records across 900 mammalian species is ~16 years) [7]. Importantly, anti-oxidative stress adaptations are not subjects of evolutionary pressure at post-reproductive age, which further contributes to the buildup of oxidative damage in aging individuals. Yet, paradoxically, in short-lived Caenorhabditis elegans, oxidative stress might have beneficial effect on longevity [8,9] by connecting to the nutrition signaling pathways [10,11]. It was suggested that aging is driven by over-activation of signal-transduction pathways such as the nutrient-sensing pathway, while oxidative stress may be both one of its activators and effectors [12,13].
Both blood cells and the circulatory system are permanently exposed to high oxygen tension; therefore, the signs of oxidative damage are expected to be present in blood substantially earlier than in other tissues. However, red blood cells are renewed before the damage achieves critical levels [14], and similar a mechanism might be in place for endothelial cells. It is likely that the decrease in renewal of endothelial cells is a significant contributing factor to cardiovascular disorders of the aging blood vessels [15]. Extra-cellular matrix (ECM) is a less dynamic component of the vessel wall, therefore, the signs of aging such as mineral deposits [16], amyloid-beta peptide [17] and viral particles [18] accumulate in this particular compartment. Likewise, ECM might serve as a reservoir for oxidative stress products.
Outside of the circulation, wounds and tumors (often referred to as “never healing wounds” [19]) are known to be associated with a high degree of oxidative stress [20,21]. In these pathological settings, the presence of an oxidative stress sensor able to activate stress-responsive protective mechanism is crucial. However, elevated oxygen or reactive oxygen species (ROS) have relatively short life times, which is a disadvantage in terms of adaptation to oxidative stress. In contrast, generation of oxidation products - oxidative signature - is able to accumulate and propagate “stress” signals over prolonged period of time.
One example of this molecular signature of oxidation is extracellular matrix modifications resulting from the oxidative stress [22]. Carboxy-alkyl-pyrroles (CAPs) protein adducts have all the anticipated properties of the oxidative stress signature: they are generated as end-products of lipid oxidation and are able to form stable conjugates with ECM proteins. CAPs were originally isolated from retinas of patients with advanced age-related macular degeneration (AMD) [23], and later were also found in other organs with substantial oxidative stress [22].
CEP (carboxy ethyl pyrrole), the most abundant CAP in AMD, has pronounced angiogenic effect both in vitro and in vivo [22,24]. CAP-induced angiogenesis may be considered as a part of coordinated tissue responses aimed to adapt to damaging conditions. CAP-induced neo-vascularization might represent an additional line of defense against tissue damage (e.g. wound) beneficial for tissue supply with oxygen and nutrients as well as augment inflammatory response.
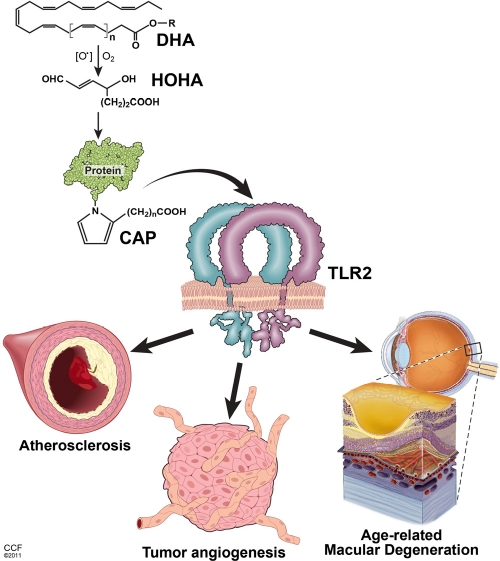
Figure 1. Key pathological effects of lipid oxidation products related to the aging Oxidation of poly-unsaturated fatty acids (exemplified by DHA - docosahexaenoic acid) leads to the generation of intermediate products (HOHA - 4-hydroxy-7-oxo hept-5-enoic acid) and, consequently, accumulation of carboxy-alkyl-pyrrole (CAP) protein adducts. CAPs stimulate TLR1/2 heterodimer on the surface of endothelial cells triggering angiogenesis. In turn, excessive CAP-induced angiogenesis contributes to the age-related macular degeneration and tumor progression. Accumulation of HOHA and CAPs in blood vessel walls may also constitute a substantial component of pathophysiological changes during atherosclerosis.
In our recent report [22] we identified Toll-like receptor 2 (TLR-2) as a main receptor and sensor for CAP protein adducts. The other likely candidates for this function - scavenger receptors, including CD36 [25,26], were not required for CAPs sensing. Originally described as innate immunity receptors [27], TLRs, in addition to the ligands of pathogen-associated molecular patterns (PAMP), have been shown to recognize a number of endogenous ligands associated with non-infectious (sterile) inflammation [28]. As a result, the concept of Damage Associated Molecular Patterns (DAMPs) which included PAMPs as a particular case, has evolved [28,29]. Thus, identification of CAPs as endogenous ligands for TLRs was not entirely surprising. However, this family of ligands was unique as it represented a “molecular signature” of the oxidative stress. Moreover, TLR2/CAPs axis exemplifies a novel oxidative-damage sensing mechanism aimed to promote vascularization. In an independent study, it was reported that TLR2/6 complex was involved in angiogenic response upon stimulation with bacterial lipoprotein [30].
CAPs-induced angiogenesis is a peculiar example of coordinated response by various tissues to the plethora of TLR ligands in vivo. Whereas PAMPs serve as potent modulators of immune system, CAPs promote angiogenesis via activation of endothelial cells. Whether CAPs are also able to affect immune system will be the subject of future studies. Noteworthy, robust CAPs-dependent angiogenesis was originally described in the eye [23] - an organ of immune privilege, which separates effects of TLR ligands on endothelial cells from that of invading leukocytes [31]. Likewise, angiogenesis was diminished in tumors grown in TLR2-null hosts, independently of TLR2 expression on bone marrow derived cells [22]. Experiments using isolated endothelial cells and in vivo wound closure assays on wild type and TLR2 knockout chimera animals confirmed the conclusion that effects of CEP was mediated by endothelial cells [22].
Another intriguing feature of CAP-induced angiogenesis is the independence from VEGF receptor tyrosine kinase activity [22]. Therefore, TLR2 signaling represents a unique pathway contributing to tumor vascularization, thereby providing an explanation for the unexpectedly low efficacy of VEGFR inhibitors in certain cancers [32]. However, under physiological conditions, it appears that both TLR2 and VEGFRs are aimed to promote formation of blood vessels in a well coordinated manner.
The pro-angiogenic function of TLR2 highlights the importance of both transcription-dependent and independent signaling pathways induced by Toll-like receptors. TLR2 has been shown to modulate levels of HIF-1alpha expression [33], which is also modulated by genotoxic streess [34] and aerobic glycolysis [35], providing additional node to the signaling network contributing to cancerogenesis [36]. Transcription-independent branch involves CEP-induced activation of Rac1 small G-protein, which modulates cell adhesion and migration, and known to be activated downstream of TLR2 [37]. Furthermore, Rac1 mediated ROS production could contribute both to genotoxic response and lipid oxidation, creating self-perpetuating cycle of oxidative damage [38].
Interestingly, different Toll-like receptors might have opposing effects on angiogenesis. Double stranded RNA, a known ligand for TLR3, has been reported to block angiogenesis in AMD [39]. The differential outcomes of TLR2 vs TLR3 stimulation might be result of interference between TLR2 and TLR3 -induced signaling pathways [40]. In the context of a whole tissue, the signaling induced by same TLR expressed on different cells combined with the interplay between different TLRs might allow coordination of tissue repair and immune response.
TLR2-dependent angiogenesis may be considered as a tissue adaptation to oxidative stress, most commonly found in wound [20]. Indeed, CEP is abundant in wounded tissue, and anti-CEP blocking antibodies decreased neo-vascularization and wound recovery, apparently without compromising immune response at the site of damage [22]. Both tissue remodeling and immune response induced by Toll like receptor 2 can be considered as manifestations of a general damage/stress response at the tissue level. In a far-fetched analogy, an intriguing example of systemic TLR dependent stress adaptation may be a radioprotective effect of TLR5 stimulation [41].
However, as it is often the case in complex systems, too much of a good thing is bad, and an aberrant TLR-dependent signaling is known to contribute to a number of pathologies ranging from allergies [42] to cancer [43,44]. While contributing to wound healing, CAPs seem to accumulate with age, most notably in blood vessels walls [22]. The accumulated CAPs might be involved in chronic inflammation linked to cardiovascular disorders. A significant example of this is the progression of atherosclerosis, which has been demonstrated to be a TLR2 dependent process [45]. Thus, elevated CAPs content could be an age-related risk factor associated with the disease.
In summary, oxidative stress creates a relatively stable molecular pattern in the form of carboxy-alkyl pyrroles-modified proteins. Accumulation of CAPs results in activation of TLR2 receptor complexes on endothelial cells, providing a stress-responsive mechanism for tissue remodeling through induction of angiogenesis. Thus, the concept of stress adaptation, put forth by Hans Selye more than 75 years ago [46], has received yet another mechanistic explanation.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001; 928: 22 -38. [PubMed] .
- 2. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11: 298 -300. [PubMed] .
- 3. Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005; 24: 1311 -1317. [PubMed] .
- 4. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. 2011; 711: 193 -201. [PubMed] .
- 5. Morgan MJ and Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011; 21: 103 -115. [PubMed] .
- 6. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2010; 469: 221 -225. [PubMed] .
- 7. Carey J and Judge D. Longevity Records - Life Spans of Mammals, Birds, Amphibians, Reptiles, and Fish. University Press of Southern Denmark 2000; .
- 8. Yang W and Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010; 8: 12 1 -14. .
- 9. Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010; 20: 2131 -2136. [PubMed] .
- 10. Haigis MC and Yankner BA. The aging stress response. Mol Cell. 2010; 40: 333 -344. [PubMed] .
- 11. Zuin A, Castellano-Esteve D, Ayte J, Hidalgo E. Living on the edge: stress and activation of stress responses promote lifespan extension. Aging (Albany NY). 2010; 2: 231 -237. [PubMed] .
- 12. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 13. Pani G. P66SHC and ageing: ROS and TOR? Aging (Albany NY). 2010; 2: 514 -518. .
- 14. Lang F, Lang KS, Lang PA, Huber SM, Wieder T. Mechanisms and significance of eryptosis. Antioxid Redox Signal. 2006; 8: 1183 -1192. [PubMed] .
- 15. Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009; 106: 326 -332. [PubMed] .
- 16. Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am. 2009; 93: 583 -604. Table of Contents. [PubMed] .
- 17. Nicolakakis N and Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab. 2011; 31: 1354 -1370. [PubMed] .
- 18. Neumann HA, Berretty PJ, Folmer SC, Cormane RH. Hepatitis B surface antigen deposition in the blood vessel walls of urticarial lesions in acute hepatitis B. Br J Dermatol. 1981; 104: 383 -388. [PubMed] .
- 19. Balkwill F and Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001; 357: 539 -545. .
- 20. Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009; 459: 996 -999. [PubMed] .
- 21. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004; 7: 97 -110. [PubMed] .
- 22. West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, Byzova TV. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010; 467: 972 -976. [PubMed] .
- 23. Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 14682 -14687. [PubMed] .
- 24. Ebrahem Q, Renganathan K, Sears J, Vasanji A, Gu X, Lu L, Salomon RG, Crabb JW, Anand-Apte B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc Natl Acad Sci U S A. 2006; 103: 13480 -13484. [PubMed] .
- 25. Courtois Y. The role of CD36 receptor in the phagocytosis of oxidized lipids and AMD. Aging (Albany NY). 2010; 2: 888 -889. [PubMed] .
- 26. Picard E, Houssier M, Bujold K, Sapieha P, Lubell W, Dorfman A, Racine J, Hardy P, Febbraio M, Lachapelle P, et al. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging (Albany NY). 2010; 2: 981 -989. [PubMed] .
- 27. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr.. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997; 388: 394 -397. [PubMed] .
- 28. Kono H and Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008; 8: 279 -289. [PubMed] .
- 29. Srikrishna G and Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009; 11: 615 -628. [PubMed] .
- 30. Grote K, Schuett H, Salguero G, Grothusen C, Jagielska J, Drexler H, Muhlradt PF, Schieffer B. Toll-like receptor 2/6 stimulation promotes angiogenesis via GM-CSF as a potential strategy for immune defense and tissue regeneration. Blood. 2010; 115: 2543 -2552. [PubMed] .
- 31. Hong S and Van Kaer L. Immune privilege: keeping an eye on natural killer T cells. J Exp Med. 1999; 190: 1197 -1200. [PubMed] .
- 32. Ebos JM and Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011; 8: 4 210 -221. [PubMed] .
- 33. Spirig R, Djafarzadeh S, Regueira T, Shaw SG, von Garnier C, Takala J, Jakob SM, Rieben R, Lepper PM. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS One. 2010; 5 -e0010983. .
- 34. Ousset M, Bouquet F, Fallone F, Biard D, Dray C, Valet P, Salles B, Muller C. Loss of ATM positively regulates the expression of hypoxia inducible factor 1 (HIF-1) through oxidative stress: Role in the physiopathology of the disease. Cell Cycle. 2010; 9: 2814 -2822. [PubMed] .
- 35. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010; 9: 2201 -2219. [PubMed] .
- 36. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010; 9: 3256 -3276. [PubMed] .
- 37. Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000; 1: 533 -540. [PubMed] .
- 38. Jun JI and Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY). 2010; 2: 627 -631. [PubMed] .
- 39. Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008; 452: 591 -597. [PubMed] .
- 40. Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010; 11: 70 -75. [PubMed] .
- 41. Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D, Kurnasov OV, Fort FL, Osterman AL, Didonato JA, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science. 2008; 320: 226 -230. [PubMed] .
- 42. Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009; 457: 585 -588. [PubMed] .
- 43. Rakoff-Nahoum S and Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007; 317: 124 -127. [PubMed] .
- 44. Coste I, Le Corf K, Kfoury A, Hmitou I, Druillennec S, Hainaut P, Eychene A, Lebecque S, Renno T. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest. 2010; 120: 3663 -3667. [PubMed] .
- 45. Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005; 115: 3149 -3156. [PubMed] .
- 46. Selye H. A Syndrome produced by Diverse Nocuous Agents Nature. 1936; 138 -32. .