



Paraphrasing the famous quote “Nothing in Biology Makes Sense Except in the Light of Evolution”, one can say that nothing in aging makes sense except in the light of TOR-driven quasi-programmed aging, a continuation of developmental growth driven by growth-promoting pathways. And life span extension by mild damage makes no sense, if aging is a decline caused by accumulation of damage.
Conventional view on aging
It is believed that aging is a decline, deterioration due to accumulation of random molecular and cellular damage caused by free radicals, radiation, stresses, pathogens, toxins, carcinogens, mistakes in replication/translation, protein misfolding and even mechanical forces. If aging is caused by damage, then damaging stresses would accelerate aging (Figure 1A). However, mild stresses (including oxidative stress) can extend life span in different species [1-30].
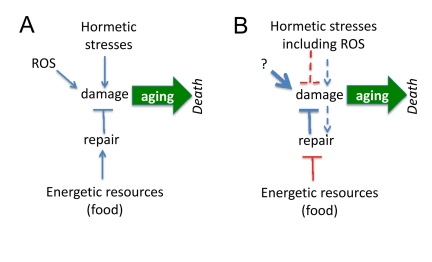
Figure 1. Paradoxical links between damage and aging (A) If agings is caused by damage, then hormetic damage should accelerate aging. Also food by providing resources should becelerate aging. Both prediction contradict observations, making the model incorrect. (B) Paradoxical model assumes that (a) damage decrease damage and (b) the less resources (food), the more resources can be used for anti-aging repair. These assumptions are paradoxical but nevertheless are needed to fit predictions and observations. Paradoxical links are shown in red.
How this can be reconciled with the conventional theory of aging. There are 3 options:
First, hormesis is an artefact. Certainly there are many artefacts in this field. Yet, there are also solid data especially on the life-extension by calorie restriction, ROS, heat shock and phytochemicals.
Second, the phenomenon of hormesis rules out the conventional theory of aging. Furthermore, as it was already reviewed, “damage-induced aging” theory was ruled out by other evidence too [31-38]. It was discussed that aging is not the life-long accumulation of molecular damage, is not decline and is not caused by reactive oxygen species (ROS) [35].
Third, instead of rejecting the damage-induced theory, paradoxical assumptions were suggested to reconcile it with hormesis (Figure 1B). To explain extension of lifespan by mild and repeated stresses, it was suggested that (a) mild stresses stimulate maintenance and repair pathways and (b) cause adaptation of cells and the ability to tolerate stronger stresses. Let us briefly review the attempt to reconcile hormesis and molecular damage-driven aging.
Conventional explanations of hormesis
Heat shock
Repeated exposure to mild heat shock increases life span in Drosophila [11]. It was suggested that mild heat shock reduces damage and protein aggregation by activating internal antioxidant, repair and degradation processes [2, 3]. In other words, chronic cellular stress may prolong life span by either activating repair mechanisms or by causing cell adaptation or both [1-3].
Calorie restriction
It was suggested that calorie restriction (CR) is a low-intensity stressor, which enhances the ability of animals to cope with intense stressors [4]. For example, in young rodents, CR causes an increase in the afternoon peak concentration of plasma corticosterone, a stress hormone [5, 39]. Another explanation is completely paradoxical. If damage is not completely repaired because resources for repair are limited by food supply, as suggested by Kirkwood [40], this predicts that CR (less resources) must accelerate aging (Figure 1A). It was also suggested by Kirkwood that, although the repair is limited by insufficient resources, the more resources (food), the less repair (Figure 1B). The reason of self-contradiction is that aging, according to the same theory, is purposefully regulated and the organism may choose to age slower [40]. The paradoxes of this point of view were recently discussed [34, 38, 41] and will not be discussed here further.
ROS
In some studies, an increased production of reactive oxygen species (ROS) correlated with extended life span in different species. To explain such paradoxical results it was suggested that an increased ROS in turn increases resistance to ROS, thus extending life span [1, 12, 14]. It was suggested that ROS leads to a condition of mild stress, which in turn enhances vitality [12]. In C. elegans reduced glucose availability promoted formation of ROS, induces catalase activity, and increased oxidative stress resistance, cumulating in extension of life span [15].
Two noticeable problems
First, it is paradoxical to decrease damage by causing damage (Figure 1B). There is no similar example in medicine. If one wishes to prevent stroke due to high blood pressure, one needs to decrease blood pressure not to increase it. Examples are endless including weight control to prevent diabetes and quitting smoking to prevent lung cancer. No one will advocate “mild and repeated” smoking to prevent lung cancer even though it might activate the defenses. The simple reason is that DNA damage is actually involved in cancer initiation. But even cancer-promoting damage is not random: mutations activate growth-promoting pathways including PI3K/mTOR, the most universal alteration in cancer [42-45]. And cancer-initiating damage does not cause cellular decline (in contrast, cancer cells are very robust and hyper-functional), is not sufficient to cause cancer, requiring rounds of cell replication, selection [46, 47] and organismal aging [48]. By slowing down aging, CR and rapamycin delay cancer (without affecting mutations). The notion that aging promotes cancer is beyond the topic of this article and cannot be discussed here. The point here is that since DNA damage contributes to cancer, no one will suggest hormetic smoking or radioactivity (at any doses) to delay cancer. In analogy, if damaging hormesis may delay aging, aging cannot be possibly be caused by damage.
Second problem is the suggestion that mild hormetic stresses protect against severe stresses. What are these severe stresses? Even according to the conventional view, aging is not caused by accidental injures that are stronger than hormetic damage. It is caused by ‘everyday’ ROS and other background stresses. Let's ask a straightforward question. Are hormetic stresses stronger or weaker than those that cause aging? And there is no answer. If damage that drives aging does not sufficiently induce protective response, then hormesis is stronger than aging-causing stresses. Then why is the purpose of hormesis to protect from stronger stresses? This question will be easily answered in the light of TOR-driven aging.
Solution: a new view on aging
If aging is driven by damage, then damage must accelerate aging. If hormesis induces damage and slows down aging, then aging is not driven by damage. So a straightforward explanation is that aging is not caused by accumulation of molecular damage [36]. It was predicted “that five years from now, current opponents will take the TOR-centric model for granted, which then will become new dogma (ironically)” [35].
It is becoming evident that ROS do not cause aging and furthermore often is associated with longevity [26, 27, 30, 49, 50-66].
While rejecting ROS-driven aging, scientists still do not dare to reject the view that aging is a decline due to accumulation of random damage. Yet data exclude not only ROS but also damage as a cause of aging. For example, in C elegans, CR did not decrease the accumulation of spontaneous mutations with age but nevertheless extended life span [54].
Yes, perhaps, molecular damage accumulates but organisms do not live long enough to age from this accumulation. Even humans, long-living organisms, do not die from a decline due to such an accumulation. (And of course worm that lives just 5 days [67] cannot possibly accumulate deadly levels of molecular damage). Instead any human being has died from age-related diseases, which are caused by active cellular processes initiated by hyper-activation of signaling pathways including mTOR [36, 68]. The pathogenesis of diseases is well known. In contrast, a mysterious cellular decline due to accumulation of molecular damage is the fiction of gerontology, unknown in medical science.
Instead of trying to adopt the phenomenon of hormesis to the view on aging as accumulation of random molecular damage, we will reconsider the view on aging.
Aging: TOR-driven process
The nutrient-sensing TOR pathway is activated by insulin, growth factors and nutrients (Figure 2). In turn, it increases protein synthesis, stimulates ribosomal biogenesis and cell mass growth (causing cell hypertrophy), inhibits autophagy, induces accumulation of aggregation-prone proteins, increases growth factors (GF) secretion and causes resistance to GF and insulin [69-79].
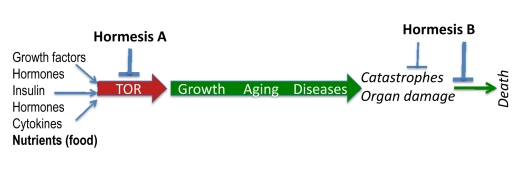
Figure 2. TOR-centric model of aging Nutrients (food), growth factors, cytokines, insulin and hormones activate the nutrient-sensing TOR (Target of Rapamycin) pathway, which promotes growth and then aging, causing age-related diseases. In turn, diseases cause non-random organ damage and death. Hormesis type A inhibits TOR thus slowing down aging. Hormesis type B increases aging-tolerance and tolerance to complications of age-related diseases.
The TOR pathway drives cellular mass growth. In proliferating cells, cellular growth is balanced by cell division [80]. In quiescent (resting) cells, growth-promoting pathways drive senescence [80, 81]. When the cell cycle is blocked but mTOR is still active, it causes hypertrophic, hyperactive, hyper-functional (for example, hyper-secretory) phenotype, with compensatory resistance to signals such as insulin and growth factors [82, 83] and compensatory lysosomal activation. In other words, mTOR converted quiescence into senescence, the process that was named gerogenic conversion or geroconversion or gerogenesis [83].
In brief, TOR causes cellular hyper-functions, specific to each tissue such as bone resorption by osteoclasts and arterial wall tonus by smooth muscle cells, manifested as osteoporosis and hypertension, for instance. This eventually damages organs (aging-induced catastrophes). As discussed, secretory phenotype [84] or (more generally) hyper-functional phenotype [31] links cell senescence to organismal aging and specifically to age-related diseases. The TOR pathway is involved in diseases such as cancers, type II diabetes and its complications (retinopathy and renal hypertrophy), age-related macular degeneration, obesity, atherosclerosis, cardiac hypertrophy, organ fibrosis (liver, renal and cardiac fibrosis), osteoporosis, Alzheimer's and Parkinson's diseases and arthritis [31, 32, 38, 68, 74, 85-89]. Organisms die from age-related diseases. TOR is involved in all of them [31, 68]. In other words, TOR limits life span by accelerating age-related diseases. In humans (and other mammals), age-related diseases are manifestations of aging that actually limit life span.
Age-related diseases culminate in sudden catastrophes (Figure 2). For example, death of cardiocytes, during myocardial infarction, is often caused ischemia due to the arterial occlusion. Such occlusions result from increased coagulation and platelet hyper-function, atherosclerosis, inflammatory state and high blood pressure. Age-related osteoporosis culminates in the broken hip, diabetes – in renal failure, hypertension – in stroke, just to name a few. Inhibition of the TOR pathway prolongs life span in yeast, worm, flies and mice [77, 90-107]. Genes for aging (named gerogenes [37]) constitute the TOR pathway [37]. Genes for longevity (named gerosuppressors) antagonize the TOR pathway [37]. Furthermore, some “anti-aging” hormetic agents antagonize this pathway too.
Longevity: (a) slow aging and (b) aging-tolerance
Life span can be extended by either (a) slowing down aging and (b) by increasing aging tolerance, defined as the ability to survive complications (catastrophes) of aging [36].
a. Obviously, inhibition of aging should extend life span and delay age-related diseases. For example, calorie restriction (CR) slows aging. CR delays age-related diseases such as cancer and atherosclerosis, thus extending life span. In other words, inhibition of mTOR-driven aging delays catastrophic complications of aging: namely, complications of age-related diseases such as stroke, myocardial infarction (Figure 2). These non-random catastrophes actually cause death.
b. But inhibition of aging (and delaying diseases) is not the only way to extend life span. The second way is to increase aging tolerance, which allows an organism to survive catastrophes caused by age-related diseases.
Why organisms age
The existence of aging is well understood from the evolutionary perspective and was discussed in detail. Roughly speaking, in the wild, organisms do not live long enough to experience aging. Therefore forces of natural selection against aging are weak. Only in protected environment (humans, domestic and laboratory animals) die from aging.
Why organisms have low aging-tolerance
From the evolutionary perspective, organisms do not tolerate aging for the same reason why they age in the first place. In the wild, organisms do not live long enough to experience aging and therefore organisms are not naturally shaped to experience complications of aging. Organisms are not selected by nature for aging-tolerance. For example, parts of myocardium depend on a single coronary artery. The occlusion of a coronary artery causes life-threatening ischemia. Collateral arteries would prevent ischemia. Natural selection would favor such anatomical re-design, if it will extend reproductive life span. If humans were routinely reproducing after the age of 70, then variations with additional branches of coronary arteries would be selected.
Thus myocardial ischemia, due to artery occlusion is one of the most common causes of death. The occlusion may result from thrombosis of atherosclerotic coronary arteries. But if the ischemic zone would receive blood supply from a collaterally artery, the organism would survive the catastrophe. Thus, anatomical modifications of a myocardial blood supply would increase aging-tolerance without affecting aging itself. Noteworthy, this is how coronary stents extend lifespan without affecting aging. Most medical treatments increase aging tolerance, thus extending an average life span despite chronic age-related diseases. In contrast, pharmacological suppression of aging would increase healthy lifespan by postponing diseases [108].
Two types of hormesis: (a) slowing down aging and (b) increasing aging-tolerance
Hormetic stresses include two groups of agents that (a) slow down aging by inhibiting the TOR pathway and (b) increase aging tolerance, without affecting the aging process (Figure 2). We will call them hormesis A and hormesis B. Examples of hormesis A are calorie restricttion, rapamycin, resveratrol and p53-inducing agents. Examples of hormesis B are adaptive preconditioning to ischemia and coronary bypass. Heat shock, hypoxia and physical exercise belong to both groups.
Hormesis A
Calorie restriction
Caloric restriction (CR) markedly extends life span in diverse species from yeast to mammals and delays the occurrence and/or slows progression of age-associated diseases [109-118]. It was suggested that CR slows down aging via the TOR pathway in yeast, C. elegans and Drosophila and mammals [34, 93, 94, 119-[122]. In humans, it has been shown that nutrients activate TOR in the muscle tissue, causing insulin-resistance, preventable by rapamycin [123]. Starvation or CR deactivates the TOR pathway [71, 96, 97, 124]. Thus, by inhibiting TOR, CR may slow down aging and extend lifespan.
Chemical hormesis
Plants, microorganisms and sea animals produce toxic agents that inhibit or damage microtubules, DNA and many other vital targets. Due to their toxicity, some of them are used as anti-cancer drugs, although nature did not created them for that purpose. Nature of course created these poisons to hurt predators and competitors [125, 126]. Similarly, rapamycin is an antifungal antibiotic produced by bacteria. TOR stimulates growth in response to nutrients. Therefore, soil bacteria produce rapamycin to inhibit yeast growth. While inhibiting TOR-dependent growth, rapamycin slows down TOR-dependent aging in older yeast [93, 94]. Given that cancer (like aging) is “a form of growth”, the mTOR pathway is activated in cancer. And, although not created for that purpose by nature, inhibitors of mTOR are used as anticancer agents [42, 43, 127, 128]. I wish to emphasize again that bacteria produce rapamycin neither as a medicine for longevity nor as an anticancer drug, but as an antifungal antibiotic. Simply the same signaling pathways that are involved in growth also are involved in cancer and aging [88]. Growth suppressants may suppress aging because aging is a continuation of growth, driven by the same TOR/S6K pathway [129]. To extend lifespan, they either should inhibit the TOR pathway or increase aging tolerance (Figure 2).
Rapamycin
Rapamycin extends life span in yeast, drosophila and mice [94, 98, 99, 130, 101, 103, 130]. It is indicated for almost all age-related diseases [31, 48, 68, 131]. Rapamycin is not toxic for normal cells at concentrations that exceed therapeutic levels 1000 fold [132], [133]. There are no side effects of high dose rapamycin in healthy volunteers [123, 134]. Rapamycin has been used in children [135] and in pregnant women [136]. Despite common misconception that rapamycin is an immunosuppressant, rapamycin improves immunity in mice when used appropriately [137-139]. As an anti-aging modality, rapamycin could be used in doses and schedules that do not cause side effects [132].
Resveratrol
Resveratrol, a natural agent found in grape skins, prevents age-related diseases and extends lifespan in several species [10, 140-145], including mice on high-fat diet [144]. Resveratrol activates sirtuins [10, 146-147], which inhibit the TOR pathway (see for references [148]). Resveratrol indirectly antagonizes the mTOR/S6K pathway upstream and downstream [129, 149-153], in part via activation of AMPK and sirtuins [154-160]. Plants produce resveratrol to protect grapes from parasites. But, coincidentally, inhibition of the TOR pathway slows down aging. Thus, the anti-aging effect of resveratrol may be just a side effect of targeting mTOR. Yet, at concentrations that inhibit mTOR the gerosuppressive effect of resveratrol is limited by its toxicity [149]. This is not surprising, given that resveratrol inhibits mTOR at micro-molar concentrations at which it also inhibits multiple unrelated targets. This may explain why anti-aging effects in mice may be limited by resveratrol toxicity [161]. As a potential solution, resveratrol at sub-therapeutic doses could be combined with rapamycin.
Metformin
The anti-diabetic drug metformin activates AMPK, which in turn antagonizes the mTOR pathway [24, 162-165]. Metformin decreases insulin resistance, prevents diabetes and its complications, decrease incidence of heart diseases and cancer [166-168]. In rodents, metformin prolongs life span or prevents cancer or both [169-174].
Physical exercise
As an example of hormesis A, chronic increase in physical activity inhibits mTOR/S6K1 in rat skeletal muscle [175]. Physical activity can also increase aging-tolerance, acting as hormesis B.
Heat shock
The TOR pathway stimulates Cap-dependent protein synthesis. Elevated temperature inhibits cap-dependent protein synthesis. Thus, heat shock blocks TOR-stimulated protein synthesis. For example, heat shock protein Hsp27 inhibits translation during heat shock by binding eIF4G [176]. Therefore, heat shock acts as “hormesis A” by imitating TOR inhibition. The small heat-shock proteins also delay the onset of polyglutamine-expansion protein aggregation, suggesting that these proteins couple the normal aging process to this type of age-related disease [177]. Also, HSPs and chaperones can increase resistance or tolerance to catastrophic complications of aging, defining them also as hormesis B.
Hypoxia
Depending on conditions, HIF-1 and hypoxia have different effects on longevity [30, 65, 178]. mTOR via phosphorylation of S6K/S6 and 4EBP1 induce cap-dependent translation. In contrast, hypoxia decreases cap-dependent translation. Hypoxia inhibits protein synthesis by deactivation of the mTOR pathway as well as by inactivation of eIF2α and eEF2 factors [179-186].
p53-inducing stresses
DNA damage induces p53, which is known to inhibit mTOR pathway both upstream and downstream of mTOR [187-197]. Induction of p53 by nutlin-3a can suppress senescent phenotype or suppress conversion of quiescence into senescence [197-200]. The gero-suppressive effect is evident only when p53 is capable to inhibit mTOR [198, 201]. In certain conditions, p53 may act as an anti-aging agent [202-207].
Hormesis B
Hormesis B extends life span by increasing aging tolerance. Mild stresses prepare organism to catastrophes caused by diseases of aging. Examples of catastrophes include stroke and myocardial ischemia. The occlusion of a cerebral artery for 60 min (injurious ischemia) damages the brain. The occlusion of the same cerebral artery for 15 min (preconditioning) protects from the damage caused by injurious ischemia [208]. Similarly, severe myocardial ischemia causes irreversible injury. Mild ischemia protects the heart from severe ischemia. Similarly, by inducing HSPs, heat shock may protect the myocardium from severe ischemia. Repeated, transient ischemic episodes or heat shock augment the ischemic tolerance of affected myocardium. Upregulation of immediate early genes and heat shock genes plays an important role in myocardial adaptation to acute ischemic stress [209]. Also, hormetic stresses can cause growth of collateral arteries. This coronary collateral function can preserve ischemic myocardium [210].
The cardioprotection against myocardial injury by regular exercise may include the development of collateral coronary arteries and induction of myocardial heat shock proteins [211].
Similarly, coronary bypass protected heart from ischemia. Although we do not call such medical procedures hormesis, there is no strict borderline between them and hormesis B. For example, reconditioning, hypoxia, and stresses may “train” cardiomyocutes to survive acute episode of coronary thrombosis. Also, it develops small blood vessels that could compensate for the occlusion of main artery.
Now we can solve the second problem of hormesis (see “two noticeable problems”). The answer is: hormetic stresses protect from stronger stresses. But these stronger stresses are not those that cause aging (aging is not caused by any stresses). These are complications of aging or age-related diseases. We call them catastrophes. Hormesis B protects from lethal catastrophes.
Conclusion
The hypothesis that aging is NOT driven by accumulation of random damage allows us to explain hormesis. Type A hormesis antagonizes the TOR pathway (Figure 2). Hormesis B causes stresses including damaging stresses. Since aging is not caused by damage, this does not contribute to aging but instead may cause aging-tolerance, thus protecting organisms from lethal consequences of aging-induced catastrophes.
Conflicts of Interest
The author of this manuscript has no conflict of interest to declare.
References
- 1. Radak Z, Chung HY, Goto S. Exercise and hormesis: oxidative stress-related adaptation for successful aging. Biogerontology. 2005; 6: 71 -75. [PubMed] .
- 2. Rattan SIS. Anti-ageing strategies: prevention or therapy. EMBO Rep. 2005; 6: S25 -29. [PubMed] .
- 3. Rattan SI. Hormetic modulation of aging and longevity by mild heat stress. Dose Response. 2006; 3: 533 -546. [PubMed] .
- 4. Masoro EJ. Role of hormesis in life extension by caloric restriction. Dose Response. 2006; 5: 16 -173. .
- 5. Sabatino F, Masoro EJ, McMahan CA, Kuhn RW. Assessment of the role of the glucocorticoid system in aging processes and in the action of food restriction. J Gerontol. 1991; 46: B171 -179. [PubMed] .
- 6. Shama S, Lai CY, Antoniazzi JM, Jiang JC, Jazwinski SM. Heat stress-induced life span extension in yeast. Exp Cell Res. 1998; 245: 379 -388. [PubMed] .
- 7. Caratero A, Courtade M, Bonnet L, Planel H, Caratero C. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology. 1998; 44: 272 -276. [PubMed] .
- 8. Le Bourg E, Valenti P, Lucchetta P, Payre F. Effects of mild heat shocks at young age on aging and longevity in Drosophila melanogaster. Biogerontology. 2001; 2: 155 -164. [PubMed] .
- 9. Yashin AI, Cypser JR, Johnson TE, Michalski AI, Boyko SI, Novoseltsev VN. Ageing and survival after different doses of heat shock: the results of analysis of data from stress experiments with the nematode worm Caenorhabditis elegans. Mech Ageing Dev. 2001; 122: 1477 -1495. [PubMed] .
- 10. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003; 425: 191 -196. [PubMed] .
- 11. Hercus MJ, Loeschcke V, Rattan SI. Lifespan extension of Drosophila melanogaster through hormesis by repeated mild heat stress. Biogerontology. 2003; 4: 149 -156. [PubMed] .
- 12. Kharade SV, Mittal N, Das SP, Sinha P, Roy N. Mrg19 depletion increases S. cerevisiae lifespan by augmenting ROS defence. FEBS Lett. 2005; 579: 6809 -6813. [PubMed] .
- 13. Olsen A, Vantipalli MC, Lithgow GJ. Lifespan extension of Caenorhabditis elegans following repeated mild hormetic heat treatments. Biogerontology. 2006; 7: 221 -230. [PubMed] .
- 14. Brys K, Vanfleteren JR, Braeckman BP. Testing the rate-of-living/oxidative damage theory of aging in the nematode model Caenorhabditis elegans. Exp Gerontol. 2007; 42: 845 -851. [PubMed] .
- 15. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6: 280 -293. [PubMed] .
- 16. Scannapieco AC, Sorensen JG, Loeschcke V, Norry FM. Heat-induced hormesis in longevity of two sibling Drosophila species. Biogerontology. 2007; 8: 315 -325. [PubMed] .
- 17. Moskalev A. Radiation-induced life span alteration of Drosophila lines with genotype differences. Biogerontology. 2007; 8: 499 -504. [PubMed] .
- 18. Wu D, Cypser JR, Yashin AI, Johnson TE. The U-shaped response of initial mortality in Caenorhabditis elegans to mild heat shock: does it explain recent trends in human mortality? J Gerontol A Biol Sci Med Sci. 2008; 63: 660 -668. [PubMed] .
- 19. Wiegant FA, Surinova S, Ytsma E, Langelaar-Makkinje M, Wikman G, Post JA. Plant adaptogens increase lifespan and stress resistance in C. elegans. Biogerontology. 2009; 10: 27 -42. [PubMed] .
- 20. Richardson RB. Ionizing radiation and aging: rejuvenating an old idea. Aging. 2009; 1: 887 -902. [PubMed] .
- 21. Wu D, Cypser JR, Yashin AI, Johnson TE. Multiple mild heat-shocks decrease the Gompertz component of mortality in Caenorhabditis elegans. Exp Gerontol. 2009; 44: 607 -612. [PubMed] .
- 22. Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leao C, Costa V, Rodrigues F, Burhans WC, Ludovico P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci U S A. 2010; 107: 15123 -15128. [PubMed] .
- 23. Ristow M and Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010; 45: 410 -418. [PubMed] .
- 24. Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010; 9: 1057 -1064. [PubMed] .
- 25. Rattan SI. Targeting the age-related occurrence, removal, and accumulation of molecular damage by hormesis. Ann N Y Acad Sci. 2010; 1197: 28 -32. [PubMed] .
- 26. Hunt PR, Son TG, Wilson MA, Yu QS, Wood WH, Zhang Y, Becker KG, Greig NH, Mattson MP, Camandola S, Wolkow CA. Extension of lifespan in C. elegans by naphthoquinones that act through stress hormesis mechanisms. PLoS One. 2011; 6: e21922 [PubMed] .
- 27. Ristow M and Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011; 51: 327 -336. [PubMed] .
- 28. Pietsch K, Saul N, Chakrabarti S, Sturzenbaum SR, Menzel R, Steinberg CE. Hormetins, antioxidants and prooxidants: defining quercetin-, caffeic acid- and rosmarinic acid-mediated life extension in C. elegans. Biogerontology. 2011; 12: 329 -347. [PubMed] .
- 29. Martins I, Galluzzi L, Kroemer G. Hormesis, cell death and aging. Aging. 2011; 3: 821 -828. [PubMed] .
- 30. Hwang AB and Lee SJ. Regulation of life span by mitochondrial respiration: the HIF-1 and ROS connection. Aging. 2011; 3: 304 -310. [PubMed] .
- 31. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 32. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Disc Today. 2007; 12: 218 -224. .
- 33. Blagosklonny MV. Program-like aging and mitochondria: instead of random damage by free radicals. J Cell Biochem. 2007; 102: 1389 -1399. [PubMed] .
- 34. Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007; 6: 2997 -3003. [PubMed] .
- 35. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 36. Blagosklonny MV. mTOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 -4059. [PubMed] .
- 37. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 -3156. [PubMed] .
- 38. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging. 2010; 2: 265 -273. [PubMed] .
- 39. Klebanov S, Diais S, Stavinoha WB, Suh Y, Nelson JF. Hyperadrenocorticism, attenuated inflammation, and the life-prolonging action of food restriction in mice. J Gerontol A Biol Sci Med Sci. 1995; 50: B79 -882. .
- 40. Kirkwood TB and Shanley DP. Food restriction, evolution and ageing. Mech Ageing Dev. 2005; 126: 1011 -1016. [PubMed] .
- 41. Blagosklonny MV. Why the disposable soma theory cannot explain why women live longer and why we age. Aging. 2010; 2: 884 -887. [PubMed] .
- 42. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010; 1: 530 -543. [PubMed] .
- 43. Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010; 1: 89 -103. [PubMed] .
- 44. Schmidt-Kittler O, Zhu J, Yang J, Liu G, Hendricks W, Lengauer C, Gabelli SB, Kinzler KW, Vogelstein B, Huso DL, Zhou S. PI3Kalpha inhibitors that inhibit metastasis. Oncotarget. 2010; 1: 339 -348. [PubMed] .
- 45. Zhao L and Vogt PK. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle. 2010; 9: 596 -600. [PubMed] .
- 46. Blagosklonny MV. Oncogenic resistance to growth-limiting conditions. Nat Rev Cancer. 2002; 2: 221 -225. [PubMed] .
- 47. Blagosklonny MV. Carcinogenesis, cancer therapy and chemoprevention. Cell Death Differ. 2005; 12: 592 -602. [PubMed] .
- 48. Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008; 7: 1520 -1524. [PubMed] .
- 49. Keaney M, Matthijssens F, Sharpe M, Vanfleteren J, Gems D. Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic Biol Med. 2004; 37: 239 -250. [PubMed] .
- 50. Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008; 22: 3236 -3241. [PubMed] .
- 51. Van Raamsdonk JM and Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009; 5: e1000361 [PubMed] .
- 52. Gems D and Doonan R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle. 2009; 8: 1681 -1687. [PubMed] .
- 53. Lapointe J and Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2010; 67: 1 -8. [PubMed] .
- 54. Edman U, Garcia AM, Busuttil RA, Sorensen D, Lundell M, Kapahi P, Vijg J. Lifespan extension by dietary restriction is not linked to protection against somatic DNA damage in Drosophila melanogaster. Aging Cell. 2009; 8: 331 -338. [PubMed] .
- 55. Van Raamsdonk JM, Meng Y, Camp D, Yang W, Jia X, Benard C, Hekimi S. Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics. 2010; 185: 559 -571. [PubMed] .
- 56. Van Raamsdonk JM and Hekimi S. Reactive Oxygen Species and Aging in Caenorhabditis elegans: Causal or Casual Relationship? Antioxid Redox Signal. 2010; 13: 1911 -1953. [PubMed] .
- 57. Yang W, Li J, Hekimi S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics. 2007; 177: 2063 -2074. [PubMed] .
- 58. Guachalla LM and Rudolph KL. ROS induced DNA damage and checkpoint responses: influences on aging? Cell Cycle. 2010; 9: 4058 -4060. [PubMed] .
- 59. Yang W and Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010; 8: e1000556 [PubMed] .
- 60. Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011; 21: 569 -576. [PubMed] .
- 61. Sanz A, Fernandez-Ayala DJ, Stefanatos RK, Jacobs HT. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging (Albany NY). 2010; 2: 200 -223. [PubMed] .
- 62. Pani G. P66SHC and ageing: ROS and TOR? Aging. 2010; 2: 514 -518. [PubMed] .
- 63. Cabreiro F, Ackerman D, Doonan R, Araiz C, Back P, Papp D, Braeckman BP, Gems D. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med. 2011; 51: 1575 -1582. [PubMed] .
- 64. Speakman JR and Selman C. The free-radical damage theory: Accumulating evidence against a simple link of oxidative stress to ageing and lifespan. Bioessays. 2011; 33: 255 -259. [PubMed] .
- 65. Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010; 20: 2131 -2136. [PubMed] .
- 66. Rodriguez KA, Wywial E, Perez VI, Lambert AJ, Edrey YH, Lewis KN, Grimes K, Lindsey ML, Brand MD, Buffenstein R. Walking the Oxidative Stress Tightrope: A Perspective from the Naked Mole-Rat, the Longest Living Rodent. Curr Pharm Des. .
- 67. Gardner MP, Gems D, Viney ME. Extraordinary plasticity in aging in Strongyloides ratti implies a gene-regulatory mechanism of lifespan evolution. Aging Cell. 2006; 5: 315 -323. [PubMed] .
- 68. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging. 2009; 1: 281 -288. [PubMed] .
- 69. Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, Yonezawa K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006; 25: 6361 -6372. [PubMed] .
- 70. Hands SL, Proud CG, Wyttenbach A. mTOR's role in ageing: protein synthesis or autophagy? Aging. 2009; 586 -597. [PubMed] .
- 71. Tremblay F and Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001; 276: 38052 -38060. [PubMed] .
- 72. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004; 14: 1650 -1656. [PubMed] .
- 73. Sarbassov dos D, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005; 17: 596 -603. [PubMed] .
- 74. Tee AR and Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol. 2005; 16: 29 -37. [PubMed] .
- 75. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471 -484. [PubMed] .
- 76. Glazer HP, Osipov RM, Clements RT, Sellke FW, Bianchi C. Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle. 2009; 8: 1738 -1746. [PubMed] .
- 77. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010; 11: 453 -465. [PubMed] .
- 78. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005; 146: 1473 -1481. [PubMed] .
- 79. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, WaldhŠusl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005; 54: 2674 -2684. [PubMed] .
- 80. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358 -362. [PubMed] .
- 81. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 82. Blagosklonny MV. Aging-suppressants: cellular senescence (hyperactivation) and its pharmacologic deceleration. Cell Cycle. 2009; 8: 1883 -1887. [PubMed] .
- 83. Blagosklonny MV. Cell cycle arrest is not senescence. Aging. 2011; 3: 94 -101. [PubMed] .
- 84. Patil CK, Mian IS, Campisi J. The thorny path linking cellular senescence to organismal aging. Mech Ageing Dev. 2005; 126: 1040 -1045. [PubMed] .
- 85. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005; 37: 19 -24. [PubMed] .
- 86. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008; 11: 801 -808. [PubMed] .
- 87. Tsang CK, Qi H, Liu LF, Zheng XFS. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Disc Today. 2007; 12: 112 -124. .
- 88. Blagosklonny MV and Hall MN. Growth and aging: a common molecular mechanism. Aging. 2009; 1: 357 -362. [PubMed] .
- 89. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21 -35. [PubMed] .
- 90. Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003; 426: 620 [PubMed] .
- 91. Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004; 131: 3897 -3906. [PubMed] .
- 92. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004; 14: 885 -890. [PubMed] .
- 93. Kaeberlein M, Powers RWr, KK S, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310: 1193 -1196. [PubMed] .
- 94. Powers RWr, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20: 174 -184. [PubMed] .
- 95. Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2009; 457: 726 -730. [PubMed] .
- 96. Masternak MM, Panici JA, Bonkowski MS, Hughes LF, Bartke A. Insulin sensitivity as a key mediator of growth hormone actions on longevity. J Gerontol A Biol Sci Med Sci. 2009; 64: 516 -521. [PubMed] .
- 97. Estep PWr, Warner JB, Bulyk ML. Short-term calorie restriction in male mice feminizes gene expression and alters key regulators of conserved aging regulatory pathways. PLoS One. 2009; 4: e5242 [PubMed] .
- 98. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010; 11: 35 -46. [PubMed] .
- 99. Moskalev AA and Shaposhnikov MV. Pharmacological Inhibition of Phosphoinositide 3 and TOR Kinases Improves Survival of Drosophila melanogaster. Rejuvenation Res. 2010; 13: 246 -247. [PubMed] .
- 100. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140 -144. [PubMed] .
- 101. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandezr E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Nature. 2009; 460: 392 -396. [PubMed] .
- 102. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, et al. Rapamycin, But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J Gerontol A Biol Sci Med Sci. 2011; 66: 191 -201. [PubMed] .
- 103. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092 -2097. [PubMed] .
- 104. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10: 4230 -4236. .
- 105. Bjedov I and Partridge L. A longer and healthier life with TOR down-regulation: genetics and drugs. Biochem Soc Trans. 2011; 39: 460 -465. [PubMed] .
- 106. Blagosklonny MV. Rapamycin and quasi-programmed aging: Four years later. Cell Cycle. 2010; 9: 1859 -1862. [PubMed] .
- 107. Katewa SD and Kapahi P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Exp Gerontol. 2011; 46: 382 -390. [PubMed] .
- 108. Blagosklonny MV. Why human lifespan is rapidly increasing: solving “longevity riddle” with “revealed-slow-aging” hypothesis. Aging. 2010; 2: 177 -182. [PubMed] .
- 109. Bordone L and Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005; 6: 298 -305. [PubMed] .
- 110. Longo VD and Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003; 299: 1342 -1346. [PubMed] .
- 111. Kennedy BK, Steffen KK, Kaeberlein M. Ruminations on dietary restriction and aging. Cell Mol Life Sci. 2007; 64: 1323 -1328. [PubMed] .
- 112. Holloszy JO and Fontana L. Caloric restriction in humans. Exp Gerontol. 2007; 42: 709 -712. [PubMed] .
- 113. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009; 325: 201 -204. [PubMed] .
- 114. Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003; 54: 131 -152. [PubMed] .
- 115. Heilbronn LK and Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr. 2003; 78: 361 -369. [PubMed] .
- 116. Ingram DK, Roth GS, Lane MA, Ottinger MA, Zou S, de Cabo R, Mattison JA. The potential for dietary restriction to increase longevity in humans: extrapolation from monkey studies. Biogerontology. 2006; 7: 143 -148. [PubMed] .
- 117. Ingram DK, Roth GS, Lane MA, Ottinger MA, Zou S, de Cabo R, Mattison JA. The potential for dietary restriction to increase longevity in humans: extrapolation from monkey studies. Biogerontology. 2006; 7: 143 -148. [PubMed] .
- 118. Yu BP. Why calorie restriction would work for human longevity. Biogerontology. 2006; 7: 179 -182. [PubMed] .
- 119. Walker G, Houthoofd K, Vanfleteren JR, Gems D. Dietary restriction in C. elegans: from rate-of-living effects to nutrient sensing pathways. Mech Ageing Dev. 2005; 126: 929 -937. [PubMed] .
- 120. Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 Link Calorie Restriction and TOR to Sirtuin-Mediated Lifespan Extension in Saccharomyces cerevisiae. PLoS Biol. 2007; 5: e261 [PubMed] .
- 121. Houthoofd K, Gems D, Johnson TE, Vanfleteren JR. Dietary restriction in the nematode Caenorhabditis elegans. Interdiscip Top Gerontol. 2007; 35: 98 -114. [PubMed] .
- 122. Blagosklonny MV. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle. 2010; 9: 683 -688. [PubMed] .
- 123. Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, FŸrnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007; 56: 1600 -1607. [PubMed] .
- 124. Jiang W, Zhu Z, Thompson HJ. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res. 2008; 68: 5492 -5499. [PubMed] .
- 125. Blagosklonny MV. Teratogens as anti-cancer drugs. Cell Cycle. 2005; 4: 1518 -1521. [PubMed] .
- 126. Blagosklonny MV. Overcoming limitations of natural anticancer drugs by combining with artificial agents. Trends Pharmacol Sci. 2005; 26: 77 -81. [PubMed] .
- 127. Choo AY and Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009; 8: 567 -572. [PubMed] .
- 128. Janes MR and Fruman DA. Targeting TOR dependence in cancer. Oncotarget. 2010; 1: 69 -76. [PubMed] .
- 129. Blagosklonny MV. Inhibition of S6K by resveratrol: in search of the purpose. Aging. 2009; 1: 511 -514. [PubMed] .
- 130. Pan Y and Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging. 2009; 1: 131 -135. [PubMed] .
- 131. Dazert E and Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011; .
- 132. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: Preliminary proposal. Cell Cycle. 2010; 9: 4788 -4794. [PubMed] .
- 133. Foster DA. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex1 and suppressing phosphorylation of 4E-BP1. Cell Cycle. 2011; 10: 3948 -3956. [PubMed] .
- 134. Leelahavanichkul A, Areepium N, Vadcharavivad S, Praditpornsilpa K, Avihingsanon Y, Karnjanabuchmd T, Eiam-Ong S, Tungsanga K. Pharmacokinetics of sirolimus in Thai healthy volunteers. J Med Assoc Thai. 2005; 88: S157 -162. [PubMed] .
- 135. Major P. Potential of mTOR inhibitors for the treatment of subependymal giant cell astrocytomas in tuberous sclerosis complex. Aging. 2011; 3: 189 -191. [PubMed] .
- 136. Sifontis NM, Coscia LA, Constantinescu S, Lavelanet AF, Moritz MJ, Armenti VT. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006; 82: 1698 -1702. [PubMed] .
- 137. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009; 460: 108 -112. [PubMed] .
- 138. Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2: ra75 [PubMed] .
- 139. Wang Y, Wang XY, Subjeck JR, Shrikant PA, Kim HL. Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br J Cancer. 2011; 104: 643 -652. [PubMed] .
- 140. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000; 403: 795 -800. [PubMed] .
- 141. Berdichevsky A and Guarente L. A Stress Response Pathway Involving Sirtuins, Forkhead and 14-3-3 Proteins. Cell Cycle. 2006; 5 .
- 142. Baur JA and Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006; 5: 493 -506. [PubMed] .
- 143. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004; 430: 686 -689. [PubMed] .
- 144. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006; 444: 337 -342. [PubMed] .
- 145. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006; 127: 1109 -1122. [PubMed] .
- 146. Baur JA, Chen D, Chini EN, Chua K, Cohen HY, de Cabo R, Deng C, Dimmeler S, Gius D, Guarente LP, Helfand SL, Imai S, Itoh H, Kadowaki T, Koya D, Leeuwenburgh C, et al. Dietary restriction: standing up for sirtuins. Science. 2010; 329: 1012 -1013. author reply 1013-1014. [PubMed] .
- 147. Haigis MC and Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010; 5: 253 -295. [PubMed] .
- 148. Blagosklonny MV. Linking calorie restriction to longevity through sirtuins and autophagy: any role for TOR. Cell Death Dis. 2010; 1: e12 doi:101038/cddis200917 [PubMed] .
- 149. Demidenko ZN and Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009; 8: 1901 -1904. [PubMed] .
- 150. Armour SM, Baur Joseph A., Sherry N., Hsieh SN, Land-Bracha A, Thomas SM, Sinclair DA. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging. 2009; 1: 515 -528. [PubMed] .
- 151. Brito PM, Devillard R, Negre-Salvayre A, Almeida LM, Dinis TC, Salvayre R, Auge N. Resveratrol inhibits the mTOR mitogenic signaling evoked by oxidized LDL in smooth muscle cells. Atherosclerosis. 2009; 205: 126 -134. [PubMed] .
- 152. Rajapakse AG, Yepuri G, Carvas JM, Stein S, Matter CM, Scerri I, Ruffieux J, Montani JP, Ming XF, Yang Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS One. 2011; 6: e19237 [PubMed] .
- 153. Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem. 2010; 285: 36387 -36394. [PubMed] .
- 154. Dasgupta B and Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007; .
- 155. Chan AY, Dolinsky VW, Soltys CL, Viollet B, Baksh S, Light PE, Dyck JR. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem. 2008; 283: 24194 -24201. [PubMed] .
- 156. Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010; 285: 9100 -9113. [PubMed] .
- 157. Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008; 8: 347 -358. [PubMed] .
- 158. Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007; 450: 712 -716. [PubMed] .
- 159. Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010; 5: e9199 [PubMed] .
- 160. Guo W, Qian L, Zhang J, Zhang W, Morrison A, Hayes P, Wilson S, Chen T, Zhao J. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res. 2011; 89: 1723 -1736. [PubMed] .
- 161. Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Woollett LA, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008; 8: 157 -168. [PubMed] .
- 162. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005; 310: 1642 -1646. [PubMed] .
- 163. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009; 8: 88 -96. [PubMed] .
- 164. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010; 9: 4461 -4468. [PubMed] .
- 165. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. If mammalian target of metformin indirectly is mammalian target of rapamycin, then the insulin-like growth factor-1 receptor axis will audit the efficacy of metformin in cancer clinical trials. J Clin Oncol. 2009; 27: e207 -209. author reply e210 [PubMed] .
- 166. DeFronzo RA and Abdul-Ghani M. Type 2 diabetes can be prevented with early pharmacological intervention. Diabetes Care. 2011; 34: Suppl 2 S202 -209. [PubMed] .
- 167. Muti P, Berrino F, Krogh V, Villarini A, Barba M, Strano S, Blandino G. Metformin, diet and breast cancer: an avenue for chemoprevention. Cell Cycle. 2009; 8: 2661 [PubMed] .
- 168. Anisimov VN. Metformin for aging and cancer prevention. Aging. 2010; 2: 760 -74. [PubMed] .
- 169. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005; 40: 685 -693. [PubMed] .
- 170. Anisimov VN, Egormin PA, Bershtein LM, Zabezhinskii MA, Piskunova TS, Popovich IG, Semenchenko AV. Metformin decelerates aging and development of mammary tumors in HER-2/neu transgenic mice. Bull Exp Biol Med. 2005; 139: 721 -723. [PubMed] .
- 171. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, Semenchenko AV. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008; 7: 2769 -2773. [PubMed] .
- 172. Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, Zabezhinski MA, Anikin IV, Karkach AS, Romanyukha AA. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010; 9: 188 -197. [PubMed] .
- 173. Anisimov VN, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Egormin PA, Yurova MV, Rosenfeld SV, Semenchenko AV, Kovalenko IG, Poroshina TE, Berstein LM. Gender differences in metformin effect on aging, life span and spontaneous tumorigenesis in 129/Sv mice. Aging. 2010; 2: 945 -958. [PubMed] .
- 174. Blagosklonny MV. Metformin and sex: Why suppression of aging may be harmful to young male mice. Aging. 2010; 2: 897 -899. [PubMed] .
- 175. Glynn EL, Lujan HL, Kramer VJ, Drummond MJ, DiCarlo SE, Rasmussen BB. A chronic increase in physical activity inhibits fed-state mTOR/S6K1 signaling and reduces IRS-1 serine phosphorylation in rat skeletal muscle. Appl Physiol Nutr Metab. 2008; 33: 93 -101. [PubMed] .
- 176. Cuesta R, Laroia G, Schneider RJ. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000; 14: 1460 -1470. [PubMed] .
- 177. Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003; 300: 1142 -1145. [PubMed] .
- 178. Kaeberlein M and Kapahi P. The hypoxic response and aging. Cell Cycle. 2009; 8: 2324 [PubMed] .
- 179. Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003; 278: 29655 -29660. [PubMed] .
- 180. Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006; 26: 3955 -3965. [PubMed] .
- 181. Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006; 21: 521 -531. [PubMed] .
- 182. DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008; 22: 239 -251. [PubMed] .
- 183. Young RM, Wang SJ, Gordan JD, Ji X, Liebhaber SA, Simon MC. Hypoxia-mediated selective mRNA translation by an internal ribosome entry site-independent mechanism. J Biol Chem. 2008; 283: 16309 -16319. [PubMed] .
- 184. Schneider A, Younis RH, Gutkind JS. Hypoxia-induced energy stress inhibits the mTOR pathway by activating an AMPK/REDD1 signaling axis in head and neck squamous cell carcinoma. Neoplasia. 2008; 10: 1295 -1302. [PubMed] .
- 185. Magagnin MG, van den Beucken T, Sergeant K, Lambin P, Koritzinsky M, Devreese B, Wouters BG. The mTOR target 4E-BP1 contributes to differential protein expression during normoxia and hypoxia through changes in mRNA translation efficiency. Proteomics. 2008; 8: 1019 -1028. [PubMed] .
- 186. Knaup KX, Jozefowski K, Schmidt R, Bernhardt WM, Weidemann A, Juergensen JS, Warnecke C, Eckardt KU, Wiesener MS. Mutual regulation of hypoxia-inducible factor and mammalian target of rapamycin as a function of oxygen availability. Mol Cancer Res. 2009; 7: 88 -98. [PubMed] .
- 187. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005; 102: 8204 -8209. [PubMed] .
- 188. Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A. 2007; 104: 16633 -16638. [PubMed] .
- 189. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007; 67: 3043 -3053. [PubMed] .
- 190. Hu W, Feng Z, Levine AJ. The regulation of human reproduction by p53 and its pathway. Cell Cycle. 2009; 8: 3621 -3622. [PubMed] .
- 191. Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006; 20: 267 -275. [PubMed] .
- 192. Budanov AV and Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134: 451 -460. [PubMed] .
- 193. Hay N. p53 strikes mTORC1 by employing sestrins. Cell Metab. 2008; 8: 184 -185. [PubMed] .
- 194. Matthew EM, Hart LS, Astrinidis A, Navaraj A, Dolloff NG, Dicker DT, Henske EP, El-Deiry WS. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle. 2009; 8 .
- 195. Constantinou C, Elia A, Clemens MJ. Activation of p53 stimulates proteasome-dependent truncation of eIF4E-binding protein 1 (4E-BP1). Biol Cell. 2008; 100: 279 -289. [PubMed] .
- 196. Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol. 2009; 29: 5645 -5656. [PubMed] .
- 197. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -9664. [PubMed] .
- 198. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging. 2010; 2: 344 -352. [PubMed] .
- 199. Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323 -4327. [PubMed] .
- 200. Leontieva OV and Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging. 2010; 2: 924 -935. [PubMed] .
- 201. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging. 2010; 2: 748 [PubMed] .
- 202. Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007; 448: 375 -379. [PubMed] .
- 203. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9: 4256 -4257. [PubMed] .
- 204. Huang B and Vassilev LT. Reduced transcriptional activity in the p53 pathway of senescent cells revealed by the MDM2 antagonist nutlin-3. Aging. 2009; 1: 845 -854. [PubMed] .
- 205. Tower J. The genetic architecture of aging: sexual antagonistic pleiotropy of p53 and foxo. Cell Cycle. 2010; 9: 3840 -3841. [PubMed] .
- 206. Biteau B and Jasper H. It's all about balance: p53 and aging. Aging. 2009; 1: 884 -886. [PubMed] .
- 207. de Keizer PL, Laberge RM, Campisi J. p53: Pro-aging or pro-longevity? Aging. 2010; .
- 208. Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003; 362: 1028 -1037. [PubMed] .
- 209. Kingma JGJ. Cardiac adaptation to ischemia-reperfusion injury. Ann N Y Acad Sci. 1999; 874: 83 -99. [PubMed] .
- 210. Heilmann C, Beyersdorf F, Lutter G. Collateral growth: cells arrive at the construction site. Cardiovasc Surg. 2002; 10: 570 -578. [PubMed] .
- 211. Powers SK, Lennon SL, Quindry J, Mehta JL. Exercise and cardioprotection. Curr Opin Cardiol. 2002; 17: 495 -502. [PubMed] .