



Starvation diabetes-like condition with low mTOR activity
If you read the Abstract, you might wonder whether rapamycin extends lifespan despite or because of “starvation-like diabetes”. As described by Lamming et al [1, 2] extending several previous observations [3-6], chronic administration of high doses of rapamycin causes insulin resistance in mice. Yet, at similar doses, rapamycin prolongs life span in mice [7, 8]. Moreover, in several studies, rapamycin prevented complications of diabetes such as nephropathy [9-14]. Also, theoretical considerations indicate rapamycin for retinopathy [15], which was recently confirmed in an animal model [16]. Rapamycin prevents atherosclerosis in rodents [17-20] and coronary re-stenosis in humans [21, 22]. In contrast, diabetes promotes nephropathy, retinopathy, atherosclerosis and coronary disease. How could this be reconciled? mTOR is a part of a nutrient-sensing pathway [23-27]. Nutrients and insulin activate mTOR. Rapamycin, which inhibits mTOR, is a “starvation-mimetic”, making the organism “think” that food is in a short supply. The most starvation-sensitive organ is the brain. The brain consumes only glucose and ketones. Therefore, to feed the brain during starvation, the liver produces glucose from amino acids (gluconeogenesis) and ketones from fatty acids (ketogenesis). Since insulin blocks both processes, the liver needs to become resistant to insulin. Also secretion of insulin by beta-cells is decreased. And adipocytes release fatty acids (lipolysis) to fuel ketogenesis by the liver. Thus, there are five noticeable metabolic alterations of starvation: gluconeogenesis, ketogenesis, insulin resistance, low insulin levels and increased lipolysis. This metabolic switch is known as starvation diabetes, a reversible condition, described 160 years ago (see for references [28]). Starvation diabetes could be explained by deactivation of mTOR, which otherwise is activated by nutrients. In theory, rapamycin can cause similar symptoms in the presence of nutrients.
Type II diabetes: insulin-resistance due to active mTOR
Starvation-diabetes is not a true type II diabetes. Type II diabetes is a consequence of insulin-resistance in part due to excessive nutrients and obesity. Even brief overfeeding may induce insulin resistance [29]. Nutrients and insulin activate mTOR. In turn, over-activated mTOR causes insulin resistance [30-42]. This feedback loop is shown in figure 1A. mTOR activates S6 kinase (S6K), which causes degradation of insulin-receptor substrates (IRS), thus impairing insulin signaling. Also, mTOR causes insulin resistance by an additional feedback mechanism [43, 44].
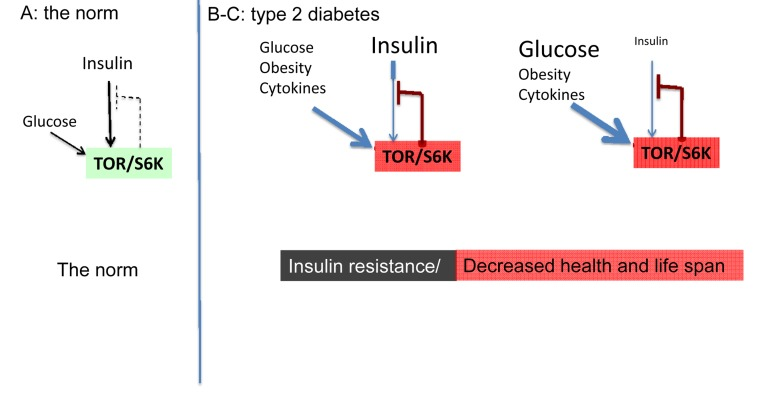
Figure 1. The norm and type 2 diabetes (simplified schema) (A) The norm. Insulin and nutrients such as glucose stimulate mTOR, which blocks insulin signaling (feedback loop).
(B-C) High mTOR/S6K activity: insulin resistance plus decreased lifespan. (B) Overactivated by nutrients, cytokins, insulin and other hormones, mTOR blocks insulin signaling causing insulin resistance. Nutrients overstimulate beta-cells and insulin is increased. (C) In type II diabetes, beta-cells eventually fail and levels of insulin may be decreased.
In high fat-fed obese rats, the mTOR pathway is activated in the liver and muscle, leading to insulin resistance [35]. In mice, sustained activation (by high fat feeding) of mTOR is associated with hepatic insulin resistance [45]. Chronic increase of insulin levels (hyperinsulinemia) causes insulin resistance, preventable by rapamycin [46]. In some animal models, removal of visceral fat prevents insulin resistance [47-49]. In humans, infusion of amino acids activate mTOR/S6K1, causing insulin resistance [38, 40]. In healthy men, rapamycin prevented activation of mTOR and insulin resistance caused by amino acid mixture [50]. Insulin stimulates glucose uptake and also activates mTOR. By a feedback loop, mTORC1 promotes insulin-resistance, decreasing glucose uptake by the cell. And most detrimentally, mTOR is involved in diabetic complications and age-related diseases [24-27, 51-54].
The two opposite conditions?
Type II diabetes and starvation diabetes seem to be the two opposite conditions: the first is associated with activation of nutrient-sensing pathways, whereas the second is associated with deactivation of nutrient sensing pathways such as mTOR. Type II diabetes is dangerous by its complications such as retinopathy, neuropathy and accelerated atherosclerosis and cancer. Long-term effects of prolonged “starvation diabetes” is not known of course: it could not last for a long time, otherwise an animal (or human) would die from starvation. Or would not? An outstanding study by Fontana et al provides some answers [55]. Among individuals who had been practicing sever CR for an average of 7 years, 40% of CR individuals exhibited “diabetic-like” glucose intolerance, despite low levels of fasting glucose, insulin and inflammatory cytokines as well as excellent other metabolic profiles. In comparison with the rest CR individuals, they had lower BMI, leptin, circulating IGF-I, testosterone, and high levels of adiponectin, which are key adoptations to CR in rodents, suggesting severe CR [55]. The authors speculated that the “insulin resistance” in this severe CR group might have the effect of slowing aging, also based on the finding that a number of insulin-resistant strains of mice are long-lived [55]. The same conclusion could be reached from the mTOR perspective (Appendix 1).
“The paradox of the insu-lin/IGF-1 signaling pathway in longevity” was first discussed by Nir Barzilai and co-workers, who precisely noticed that insulin-resistance, which is so detrimental in obese and aging mammals, can be associated with genetic manipulations that extend life span in model organisms [56]. Later Barzilai et al suggested that insulin-resistance might serve as an adaptive mechanism in some tissues by preventing excess uptake of nutrients by cells [57]. This very interesting idea implies that insulin resistance is partially beneficial and partially hazardous in the same condition such as type II diabetes. But still insulin resistance in type II diabetes is overall harmful (leading to retinopathy and other complications), whereas insulin resistance during severe CR is benevolent. These are clearly different conditions. In fact, they are the opposite conditions. So insulin resistance may be harmful or beneficial depending on the underlying condition.
The model of TOR-driven hyper-functional aging almost automatically solves paradoxes of aging, including the insulin paradox (see paradox 7 and figure 4 in “Paradoxes of aging” [58]). From the TOR perspective, insulin resistance is beneficial or harmful when it is associated with ether low or high TOR activity, respectively (Appendix, Fig. 1 and 3). And this should not be surprising. Consider insulin resistance as a symptom. The assessment of symptoms depends on the underlying cause. For example, weight loss due to calorie restriction is good, whereas weight loss in terminal cancer is bad. Positive Tuberculosis Skin (PPD) Test due to vaccination indicates protection from tuberculosis, whereas positive test due to tuberculosis is a symptom of tuberculosis. Similarly, hyperlipidemia in obesity is bad, whereas hyperlipidemia due to rapamycin-induced lipolysis is good (see figure 2 in reference [53]). The list of examples is endless. Similarly, insulin resistance, associated with TOR overactivation, is bad (Fig. 1 B-C). But either insulin sensitivity (Fig. 2) or insulin resistance (Fig. 3), associated with inactive TOR, is good.
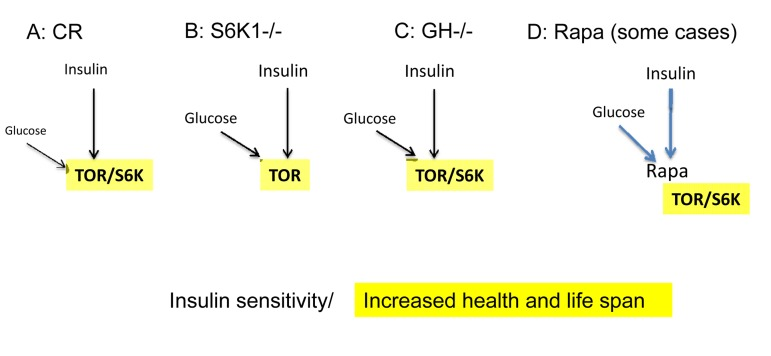
Figure 2. Low mTOR/S6K activity: insulin sensitivity plus longevity (A) Calorie restriction. Deactivation of the nutrient-sensing mTOR pathway results in insulin sensitivity.
(B) Knockout of S6K1 in mice abolishes feedback block of insulin signaling, resulting in insulin sensitivity [94].
(C) Decreased levels of growth hormone (GH). In mice, absence of GH or GH receptor leads to a remarkable extension of longevity [95]. GH receptor deficiency is associated with a reduction in pro-aging signaling, cancer, and diabetes in humans [96]. Growth hormone signaling accelerates aging in mammals [97]. Remarkably, growth stimulation promotes cellular aging, when cells cannot proliferate [98, 99]. Thus, the growth promoting pathways such as mTOR are involved in both organismal and cellular aging.
(D) Acute treatment with rapamycin. Deactivation of the nutrient-sensing mTOR pathway abolishes a feedback block of insulin signaling, resulting in insulin sensitivity [50].
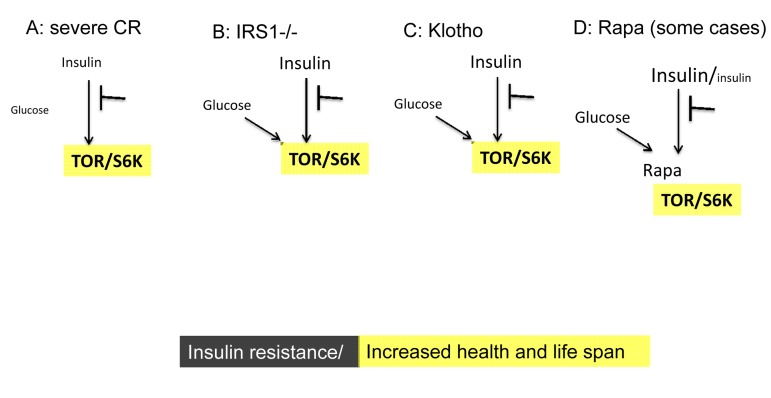
Figure 3. Low TOR/S6K activity: insulin resistance plus longevity (type 0 diabetes) (A) Severe CR and starvation. Insulin resistance and symptoms of diabetes are observed during starvation [28] and prolong severe CR [55]. Furthermore, CR may reduce rather than enhance insulin effects in the insulin-sensitive dwarf mice [100].
(B) IRS1 knockout. Insulin receptor substrate 1 null mice live longer despite insulin resistance [101].
(C) Klotho mice. Overexpression of Klotho in mice extends life span. Klotho protein represses intracellular signals of insulin and insulin-like growth factor 1 (IGF1), [102]. Also, Klotho interferes with insulin/IGF-like signaling to improve longevity in Caenorhabditis elegans [103].
(D) Chronic treatment with high doses of rapamycin causes insulin resistance and glucose intolerance. This condition can be associated with normal/increased and decreased levels of insulin. Noteworthy, rapamycin induces Klotho [64].
Type zero or benevolent diabetes
There are two types of diabetes, which at advanced stages may become similar. Insulin resistance may develop in type I diabetes (due to high glucose), whereas insulin insufficiency in type II diabetes (due to loss of beta-cells). Both types of diabetes lead to complications. In comparison, starvation diabetes [28] is only superficially resembles either type of diabetes. Also, diabetes-like symptoms may occur in rapamycin-treated mice and animals with genetically inhibited insulin/IGFI signaling (Fig. 3). To encompass all these cases, I suggest the term type 0 (zero) or benevolent diabetes. It is possible that some patients with diabetes have inactivating mutations in the insulin/IGFI pathway and thus “suffer” from benevolent diabetes. Furthermore, the condition can be imitated by chronic administration of rapamycin at least in some strains of mice. Both calorie restriction and rapamycin extend life span in mice. Rapamycin prevents retinopathy and nephropathy. Also CR prevents type II diabetes and other diseases [59], [60], [61], [62]. One can suggest that type 0 diabetes should prevent type 2 diabetes. Should type 0 diabetes be treated? Perhaps CR-associated type 0 diabetes should not. What about rapamycin-associated diabetes? Definitely, it should not be treated with insulin. It was discussed that in theory the most rational combinations with rapamycin are mild calorie and fat restriction, physical exercise and metformin [52]. Metformin may in theory counteract rapamycin-induced gluconeogenesis in the liver. And this rational drug combination may be also considered as treatment of type 0 diabetes.
Inconsistencies in the literature on rapamycin-induced insulin resistance
As demonstrated by Lamming et al, chronic administration of rapamycin caused insulin-resistance due to deactivation of mTORC2 and Akt [1]. This is consistent with previous data that IRS signaling and AKT activation was impaired in patients treated with rapamycin [63]. However, there are some inconsis-tencies. In another clinical study, rapamycin therapy in contrast caused activation of Akt [64]. Second, whereas Lamming et al found that rapamycin increased insulin levels after feeding [1], other studies reported that rapamycin in contrast inhibited insulin secretion [3, 4, 65]. Furthermore, inhibition of beta-cell adaptation and insulin production by rapamycin was considered as the main mechanism of rapamycin-induced diabetes in mice [6, 66-69]. On the other hand, selective inactivation of mTORC2 in the liver can cause hyperinsulinemia [70].
Finally, diabetic-like symptoms were not observed in numerous studies in mice. And rapamycin-induced diabetes is rare in human patients, even though most of them are prone to diabetes for other reasons.
Diabetes in patients receiving rapamycin
In renal transplant patients, who are prone to diabetes (due to several reasons), chronic administration of rapamycin modestly increases incidence of diabetes [71, 72]. Although the increase is statistically significant, it took many years to detect it. For many years it was thought that, unlike other agents used in these patients, rapamycin either do not increase the incidence of diabetes or increases it in combinations with tacrolimus [73-79]. In the study involving 20124 recipients of kidney transplant sirolimus (rapamycin) was independently associated with new onset diabetes [72]. And although it statistically significantly increases the incidence of diabetes in renal transplant patient, we do not know whether this is true diabetes, which is dangerous by its complications, or starvation-like diabetes, that prevents the complications of true diabetes . Will chronic high doses of rapamycin cause or prevent diabetes in humans without organ transplantation? More investigations are needed.
Intermittent administration of rapamycin
Is glucose intolerance a part of therapeutic effects of starvation-like drugs such as rapamycin? And may such condition be not only benign but also prevent true diabetes and its complications? Although these questions are very intriguing, the answers are not immediately crucial. Simply, the most rational anti-aging schedule is an intermittent (rather than chronic) administration of rapamycin [53, 80]. First, this will eliminate potential side effects. Second, intermittent administration of rapamycin may in theory rejuvenate stem and wound-healing cells and (in contrast to chronic treatment) improve wound healing [80]. And intermittent administration of rapamycin extended life span in mice [81-86]. Also, brief treatment with rapamycin does not affect mTORC2 [87].
Rapalogs (rapamycin and its analogs such evirolimus and temsirolimus) inhibit only one target (mTORC1). That was considered as a disadvantage of rapalogs for cancer therapy. Inhibitors of both mTORC1 and mTORC2 are under development [88, 89]. But if inhibition of mTORC2 is not needed for the longevity effect, then mTORC1 selectivity is an advantage for anti-aging therapy. Rapalogs (rapamycin and its analogs) are selective inhibitors of TORC1 and inhibitors of mTORC1 will have the same side effects as rapalogs. Yet, these (non-rapalog) inhibitors of the TOR kinase also have off-target effects and side effects. Therefore, rapamycin will remain the least toxic anti-aging drug in the near future [90].
Acknowledgments
I thank Nir Barzilai (Albert Einstein College of Medicine, Bronx NY) and Luigi Fontana (Washington University, St. Louis, MO) and helpful and expiring discussion and critical reading of the manuscript.
Conflicts of Interest
The author of this manuscript has no conflict of interest to declare.
References
- 1. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012; 335: 1638 -1643. [PubMed] .
- 2. Hughes KJ and Kennedy BK. Cell biology. Rapamycin paradox resolved. Science. 2012; 335: 1578 -1579. [PubMed] .
- 3. Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, Leibowitz G. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008; 57: 945 -957. [PubMed] .
- 4. Chang GR, Wu YY, Chiu YS, Chen WY, Liao JW, Hsu HM, Chao TH, Hung SW, Mao FC. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol. 2009; 105: 188 -198. [PubMed] .
- 5. Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010; 59: 1338 -1348. [PubMed] .
- 6. Yang SB, Lee HY, Young DM, Tien AC, Rowson-Baldwin A, Shu YY, Jan YN, Jan LY. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J Mol Med (Berl). 2011; .
- 7. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandezr E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Nature. 2009; 460: 392 -396. [PubMed] .
- 8. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, et al. Rapamycin, But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J Gerontol A Biol Sci Med Sci. 2011; 66: 191 -201. [PubMed] .
- 9. Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, Rama I, Vidal A, Grinyo JM. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol. 2006; 17: 1395 -1404. [PubMed] .
- 10. Sakaguchi M, Isono M, Isshiki K, Sugimoto T, Koya D, Kashiwagi A. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun. 2006; 340: 296 -301. [PubMed] .
- 11. Wittmann S, Daniel C, Stief A, Vogelbacher R, Amann K, Hugo C. Long-term treatment of sirolimus but not cyclosporine ameliorates diabetic nephropathy in the rat. Transplantation. 2009; 87: 1290 -1299. [PubMed] .
- 12. Flaquer M, Lloberas N, Franquesa M, Torras J, Vidal A, Rosa JL, Herrero-Fresneda I, Grinyo JM, Cruzado JM. The combination of sirolimus and rosiglitazone produces a renoprotective effect on diabetic kidney disease in rats. Life Sci. 2010; 87: 147 -153. [PubMed] .
- 13. Cruzado JM, Poveda R, Ibernon M, Diaz M, Fulladosa X, Carrera M, Torras J, Bestard O, Navarro I, Ballarin J, Romero R, Grinyo JM. Low-dose sirolimus combined with angiotensin-converting enzyme inhibitor and statin stabilizes renal function and reduces glomerular proliferation in poor prognosis IgA nephropathy. Nephrol Dial Transplant. 2011; 26: 3596 -3602. [PubMed] .
- 14. Lu MK, Gong XG, Guan KL. mTOR in podocyte function: is rapamycin good for diabetic nephropathy? Cell Cycle. 2011; 10: 3415 -3416. [PubMed] .
- 15. Demidenko ZN and Blagosklonny MV. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1alpha. Cell Cycle. 2011; 10: 1557 -1562. [PubMed] .
- 16. Kolosova NG, Muraleva NA, Zhdankina AA, Stefanova NA, Fursova AZ, Blagosklonny MV. Prevention of age-related macular degeneration (AMD)-like retinopathy by rapamycin in rats. Am J Pathol. 2012; in press .
- 17. Pakala R, Stabile E, Jang GJ, Clavijo L, Waksman R. Rapamycin attenuates atherosclerotic plaque progression in apolipoprotein E knockout mice: inhibitory effect on monocyte chemotaxis. J Cardiovasc Pharmacol. 2005; 46: 481 -486. [PubMed] .
- 18. Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR-/- mice despite severe hypercholesterolemia. Atherosclerosis. 2008; 198: 39 -48. [PubMed] .
- 19. Chen WQ, Zhong L, Zhang L, Ji XP, Zhang M, Zhao YX, Zhang C, Zhang Y. Oral rapamycin attenuates inflammation and enhances stability of atherosclerotic plaques in rabbits independent of serum lipid levels. Br J Pharmacol. 2009; 156: 941 -951. [PubMed] .
- 20. Zhao L, Ding T, Cyrus T, Cheng Y, Tian H, Ma M, Falotico R, Pratico D. Low-dose oral sirolimus reduces atherogenesis, vascular inflammation and modulates plaque composition in mice lacking the LDL receptor. Br J Pharmacol. 2009; 156: 774 -785. [PubMed] .
- 21. Keogh A, Richardson M, Ruygrok P, Spratt P, Galbraith A, O'Driscoll G, Macdonald P, Esmore D, Muller D, Faddy S. Sirolimus in de novo heart transplant recipients reduces acute rejection and prevents coronary artery disease at 2 years: a randomized clinical trial. Circulation. 2004; 110: 2694 -2700. [PubMed] .
- 22. Rodriguez AE, Granada JF, Rodriguez-Alemparte M, Vigo CF, Delgado J, Fernandez-Pereira C, Pocovi A, Rodriguez-Granillo AM, Schulz D, Raizner AE, Palacios I, O'neill W, Kaluza GL, Stone G, Investigators OI. Oral Rapamycin After Coronary Bare-Metal Stent Implantation to Prevent Restenosis The Prospective, Randomized Oral Rapamycin in Argentina (ORAR II) Study. J Am Coll Cardiol. 2006; 47: 1522 -1529. [PubMed] .
- 23. Sarbassov dos D, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005; 17: 596 -603. [PubMed] .
- 24. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471 -484. [PubMed] .
- 25. Blagosklonny MV and Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY). 2009; 1: 357 -362. [PubMed] .
- 26. Loewith R and Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011; 189: 1177 -1201. [PubMed] .
- 27. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21 -35. [PubMed] .
- 28. Blagosklonny MV. Rapamycin-induced glucose intolerance: Hunger or starvation diabetes. Cell Cycle. 2011; 10: 4217 -4224. [PubMed] .
- 29. Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001; 50: 2786 -2791. [PubMed] .
- 30. Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000; 14: 783 -794. [PubMed] .
- 31. Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, Donner DB. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci U S A. 2001; 98: 4640 -4645. [PubMed] .
- 32. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004; 431: 200 -205. [PubMed] .
- 33. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004; 14: 1650 -1656. [PubMed] .
- 34. Tzatsos A and Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006; 26: 63 -76. [PubMed] .
- 35. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005; 146: 1473 -1481. [PubMed] .
- 36. Manning BD, Logsdon MN, Lipovsky A, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005; 19: 1773 -1778. [PubMed] .
- 37. Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005; 30: 35 -42. [PubMed] .
- 38. Tremblay F, BržlŽ S, Hee, Um S, Li Y, Masuda K, Roden M, Sun X.J, Krebs M, Polakiewicz R.D, Thomas G, Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007; 104: 14056 -14061. [PubMed] .
- 39. Tremblay F and Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001; 276: 38052 -38060. .
- 40. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, WaldhŠusl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005; 54: 2674 -2684. [PubMed] .
- 41. Mordier S and Iynedjian PB. Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochem Biophys Res Commun. 2007; 362: 206 -211. [PubMed] .
- 42. Saha AK, Xu XJ, Balon TW, Brandon A, Kraegen EW, Ruderman NB. Insulin resistance due to nutrient excess: is it a consequence of AMPK downregulation? Cell Cycle. 2011; 10: 3447 -3451. [PubMed] .
- 43. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011; 332: 1317 -1322. [PubMed] .
- 44. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011; 332: 1322 -1326. [PubMed] .
- 45. Korsheninnikova E, van der Zon GC, Voshol PJ, Janssen GM, Havekes LM, Grefhorst A, Kuipers F, Reijngoud DJ, Romijn JA, Ouwens DM, Maassen JA. Sustained activation of the mammalian target of rapamycin nutrient sensing pathway is associated with hepatic insulin resistance, but not with steatosis, in mice. Diabetologia. 2006; 49: 3049 -3057. [PubMed] .
- 46. Ueno M, Carvalheira JB, Tambascia RC, Bezerra RM, Amaral ME, Carneiro EM, Folli F, Franchini KG, Saad MJ. Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia. 2005; 48: 506 -518. [PubMed] .
- 47. Barzilai N, She L, Liu BQ, Vuguin P, Cohen P, Wang J, Rossetti L. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes. 1999; 48: 94 -98. [PubMed] .
- 48. Gabriely I, Ma XH, Yang XM, Atzmon G, Rajala MW, Berg AH, Scherer P, Rossetti L, Barzilai N. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes. 2002; 51: 2951 -2958. [PubMed] .
- 49. Muzumdar R, Allison DB, Huffman DM, Ma X, Atzmon G, Einstein FH, Fishman S, Poduval AD, McVei T, Keith SW, Barzilai N. Visceral adipose tissue modulates mammalian longevity. Aging Cell. 2008; 7: 438 -440. [PubMed] .
- 50. Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, FŸrnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007; 56: 1600 -1607. [PubMed] .
- 51. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 52. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Disc Today. 2007; 12: 218 -224. .
- 53. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany NY). 2009; 1: 281 -288. [PubMed] .
- 54. Blagosklonny MV. mTOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 -4059. [PubMed] .
- 55. Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age (Dordr). 2010; 32: 97 -108. [PubMed] .
- 56. Rincon M, Muzumdar R, Atzmon G, Barzilai N. The paradox of the insulin/IGF-1 signaling pathway in longevity. Mech Ageing Dev. 2004; 125: 397 -403. [PubMed] .
- 57. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012; 61: 1315 -1322. [PubMed] .
- 58. Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007; 6: 2997 -3003. [PubMed] .
- 59. Weiss EP, Racette SB, Villareal DT, Fontana L, Steger-May K, Schechtman KB, Klein S, Holloszy JO. Group. WUSoMC. Improvements in glucose tolerance and insulin action induced by increasing energy expenditure or decreasing energy intake: a randomized controlled trial. Am J Clin Nutr. 2006; 84: 1033 -1042. [PubMed] .
- 60. Fontana L. The scientific basis of caloric restriction leading to longer life. Curr Opin Gastroenterol. 2009; 25: 144 -150. [PubMed] .
- 61. Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010; 328: 321 -326. [PubMed] .
- 62. Blagosklonny MV. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle. 2010; 9: 683 -688. [PubMed] .
- 63. Di Paolo S, Teutonico A, Leogrande D, Capobianco C, Schena PF. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: A crossroad between cancer and diabetes? J Am Soc Nephrol. 2006; 17: 2236 -2244. [PubMed] .
- 64. Tataranni T, Biondi G, Cariello M, Mangino M, Colucci G, Rutigliano M, Ditonno P, Schena FP, Gesualdo L, Grandaliano G. Rapamycin-Induced Hypophosphatemia and Insulin Resistance Are Associated With mTORC2 Activation and Klotho Expression. Am J Transplant. 2011; 11: 1656 -1664. [PubMed] .
- 65. Velazquez-Garcia S, Valle S, Rosa TC, Takane KK, Demirci C, Alvarez-Perez JC, Mellado-Gil JM, Ernst S, Scott DK, Vasavada RC, Alonso LC, Garcia-Ocana A. Activation of protein kinase C-zeta in pancreatic beta-cells in vivo improves glucose tolerance and induces beta-cell expansion via mTOR activation. Diabetes. 2011; 60: 2546 -2559. [PubMed] .
- 66. Rachdi L, Balcazar N, Osorio-Duque F, Elghazi L, Weiss A, Gould A, Chang-Chen KJ, Gambello MJ, Bernal-Mizrachi E. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A. 2008; 105: 9250 -9255. [PubMed] .
- 67. Leibowitz G, Cerasi E, Ketzinel-Gilad M. The role of mTOR in the adaptation and failure of beta-cells in type 2 diabetes. Diabetes Obes Metab. 2008; 10 Suppl 4 157 -169. [PubMed] .
- 68. Xie J and Herbert TP. The role of mammalian target of rapamycin (mTOR) in the regulation of pancreatic beta-cell mass: implications in the development of type-2 diabetes. Cell Mol Life Sci. 2011; .
- 69. Blandino-Rosano M, Chen AY, Scheys JO, Alejandro EU, Gould AP, Taranukha T, Elghazi L, Cras-Meneur C, Bernal-Mizrachi E. mTORC1 signaling and regulation of pancreatic beta-cell mass. Cell Cycle. 2012; 11 [PubMed] .
- 70. Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, Hall MN. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012; 15: 725 -738. [PubMed] .
- 71. Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. 2005; 16: 3128 -3135. [PubMed] .
- 72. Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol. 2008; 19: 1411 -1418. [PubMed] .
- 73. Schwarz C and Oberbauer R. The future role of target of rapamycin inhibitors in renal transplantation. Curr Opin Urol. 2002; 12: 109 -113. [PubMed] .
- 74. Legendre C, Campistol JM, Squifflet JP, Burke JT. Cardiovascular risk factors of sirolimus compared with cyclosporine: early experience from two randomized trials in renal transplantation. Transplant Proc. 2003; 35: 151S -153S. [PubMed] .
- 75. Gonwa T, Mendez R, Yang HC, Weinstein S, Jensik S, Steinberg S. Randomized trial of tacrolimus in combination with sirolimus or mycophenolate mofetil in kidney transplantation: results at 6 months. Transplantation. 2003; 75: 1213 -1220. [PubMed] .
- 76. Romagnoli J, Citterio F, Violi P, Nanni G, Castagneto M. Posttransplant diabetes mellitus after kidney transplantation with different immunosuppressive agents. Transplant Proc. 2004; 36: 690 -691. [PubMed] .
- 77. Sulanc E, Lane JT, Puumala SE, Groggel GC, Wrenshall LE, Stevens RB. New-onset diabetes after kidney transplantation: an application of 2003 International Guidelines. Transplantation. 2005; 80: 945 -952. [PubMed] .
- 78. Araki M, Flechner SM, Ismail HR, Flechner LM, Zhou L, Derweesh IH, Goldfarb D, Modlin C, Novick AC, Faiman C. Posttransplant diabetes mellitus in kidney transplant recipients receiving calcineurin or mTOR inhibitor drugs. Transplantation. 2006; 81: 335 -341. [PubMed] .
- 79. Pavlakis M and Goldfarb-Rumyantzev AS. Diabetes after transplantation and sirolimus: what's the connection? J Am Soc Nephrol. 2008; 19: 1255 -1256. [PubMed] .
- 80. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008; 11: 801 -808. [PubMed] .
- 81. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092 -2097. [PubMed] .
- 82. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10: 4230 -4236. [PubMed] .
- 83. Khanna A and Kapahi P. Rapamycin: killing two birds with one stone. Aging (Albany NY). 3: 1043 -1044. [PubMed] .
- 84. Spong A and Bartke A. Rapamycin slows aging in mice. Cell Cycle. 11 .
- 85. Longo VD and Fontana L. Intermittent supplementation with rapamycin as a dietary restriction mimetic. Aging (Albany NY). 2011; 3: 1039 -1040. [PubMed] .
- 86. Selman C and Partridge L. A double whammy for aging? Rapamycin extends lifespan and inhibits cancer in inbred female mice. Cell Cycle; 11: 17 -18. .
- 87. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004; 6: 1122 -1128. [PubMed] .
- 88. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010; 1: 530 -543. [PubMed] .
- 89. Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011; 10: 868 -880. [PubMed] .
- 90. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: Preliminary proposal. Cell Cycle. 2010; 9: 4788 -4794. [PubMed] .
- 91. Kirkwood TB. Understanding the odd science of aging. Cell. 2005; 120: 437 -447. [PubMed] .
- 92. Bartke A. Long-lived Klotho mice: new insights into the roles of IGF-1 and insulin in aging. Trends Endocrinol Metab. 2006; 17: 33 -35. [PubMed] .
- 93. Bartke A. Single-gene mutations and healthy ageing in mammals. Philos Trans R Soc Lond B Biol Sci. 2011; 366: 28 -34. [PubMed] .
- 94. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140 -144. [PubMed] .
- 95. Bartke A. Healthy Aging: Is Smaller Better? - A Mini-Review. Gerontology. 2012; .
- 96. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, Hwang D, Martin-Montalvo A, Saavedra J, Ingles S, de Cabo R, Cohen P, Longo VD. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011; 3: 70ra13 .
- 97. Bartke A. Pleiotropic effects of growth hormone signaling in aging. Trends Endocrinol Metab. 2011; 22: 437 -442. [PubMed] .
- 98. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 99. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888 -1895. [PubMed] .
- 100. Wang Z, Al-Regaiey KA, Masternak MM, Bartke A. Adipocytokines and lipid levels in Ames dwarf and calorie-restricted mice. J Gerontol A Biol Sci Med Sci. 2006; 61: 323 -331. [PubMed] .
- 101. Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, Piper MD, Al-Qassab H, Speakman JR, Carmignac D, Robinson IC, Thornton JM, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. Faseb J. 2008; 22: 807 -818. [PubMed] .
- 102. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005; 309: 1829 -1833. [PubMed] .
- 103. Chateau MT, Araiz C, Descamps S, Galas S. Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging (Albany NY). 2010; 2: 567 -581. [PubMed] .