



Introduction
Life expectancy is currently increasing in both developed and developing countries. In turn, age-related health problems represent a growing socioeconomic challenge for society. To date, the interplay of life expectancy and healthy aging of different organs is not well understood. In the past, genetic model systems like the fruit fly Drosophila have allowed the identification of evolutionarily conserved genes that control longevity [1, 2], such as insulin-like peptides [3] and TOR signaling [4, 5]. These studies have generally used lifespan of population cohorts as a primary readout. However, we still know little about the control of organ aging or organ “healthspan” in individual aging animals. Healthspan is defined as the period of time in an organism's life during which its physiological good health is maintained, and is currently viewed by many as a more important readout of aging than lifespan [6]. Importantly, many tissues in a multicellular organism age differently at the cellular level. For example, C. elegans muscles show early signs of deterioration, while the nervous system remains remarkably intact [7]. This likely reflects the fact that individual tissues make different contributions to lifespan, suggesting that their healthspans may be regulated differently. Hence, an open question about the relationship between lifespan and healthspan is how longevity genes differentially control individual tissue-specific aging programs. To examine this issue, simple and early visual tissue biomarkers of “aging in progress” are urgently needed. Recently, physiological biomarkers of aging have been identified in the Drosophila heart [8], muscles [9], nervous system [10] and sleep patterns [11]. However, most of these organs and behaviors either have a complex morphology and are not easily visualized from the exterior, or are not easily measured and only show changes that arise after a week or two into adulthood. A more easily accessible and earlier tissue aging readout would thus provide a valuable model for further studies on organ aging.
The skin shows the first obvious changes with age of any organ. Altered skin morphology is externally visible and is influenced by both extrinsic environmental factors as well as the intrinsic aging program. In humans, some of these changes include a gradual atrophy and thinning of the epidermal layer by up to 50% from the age of 30 to 80 years [12]. In addition to externally visible changes, both membrane protein levels [13] and global gene expression profiles [14] are known to change in aging skin. Because the skin sits at the interface between the organism and its environment it is a promising tissue to search for biomarkers of aging. A genetically tractable Drosophila model of skin aging would provide numerous advantages, including short lifespan, a simple monolayer epidermis, and a powerful genetic toolbox for manipulation and visualization.
Recently, research has suggested that one of the most important cellular processes to impact aging is autophagy, the process by which an intracellular membrane engulfs organelles and cytoplasmic material to form an autophagosome which is then digested after fusion with a lysosome [15]. Autophagy maintains tissue homeostasis by clearance of aggregates of damaged proteins and other molecules. This function is particularly important in the nervous system [16], where the intricate cell morphology renders the cells especially sensitive to the accumulation of aggregates. Autophagy can also lead to cell death [15] and is important for cellular remodeling processes [17-19]. In the context of aging, autophagy is often seen as protective, since it reduces damaged organelles that accumulate with age [20]. Moreover, senescent keratinocytes in culture die by autophagy, and blocking autophagy delays this process [21]. Whether autophagy can also drive tissue remodeling or deterioration in aging skin remains unclear.
Here we introduce the adult epidermis of the fly as an early onset tissue readout for the aging program. Age-related changes in this model include a loss of epidermal membrane labeling followed by a decrease in the number of nuclei. We also observed a strong thinning of the epidermal layer with age. Importantly, our study reveals that epidermal aging is a plastic process that is decelerated in a long-lived fly mutant and accelerated in a Progeria-like mutant with shortened lifespan. Deceleration of epidermal aging in an autophagy mutant suggests that autophagy is a driving force behind age-related alterations in the fly epidermis, in contrast to its protective role in other tissues.
Results
Age-related morphology changes in the adult epidermis
We first tested if Drosophila adult epidermal morphology changes with age. The membranes of the ventro-lateral abdominal epidermis (see Figure 1A) were visualized with a Fasciclin (Fas) III antibody (Figure 1C-F). In 1d old flies the epidermis was a uniform monolayer of mononuclear cells (Figure 1C). Surprisingly, although laboratory-reared flies can live for several months, loss of epidermal membrane labeling was apparent within a few days of eclosion (Figure 1D) and progressed steadily so that very little epidermal membrane labeling remained by six weeks (Figure 1F). A comparable deterioration was also observed using a FasIII-GFP fusion construct or anti-Coracle labeling (data not shown). To quantify the morphological changes in control flies we defined four classes reflecting an increasing loss of epidermal membrane labeling (see Figure 1C-F). All pair wise comparisons of class distributions at 1, 3, 7, 14, and 42 days were significant (Figure 1B). Co-labeling of epidermal nuclei revealed that these initially persisted despite the loss of epidermal membranes (Figure 1D, E). In contrast, by six weeks of age, epidermal nuclei were also strongly decreased in number (Figure 1F and G).
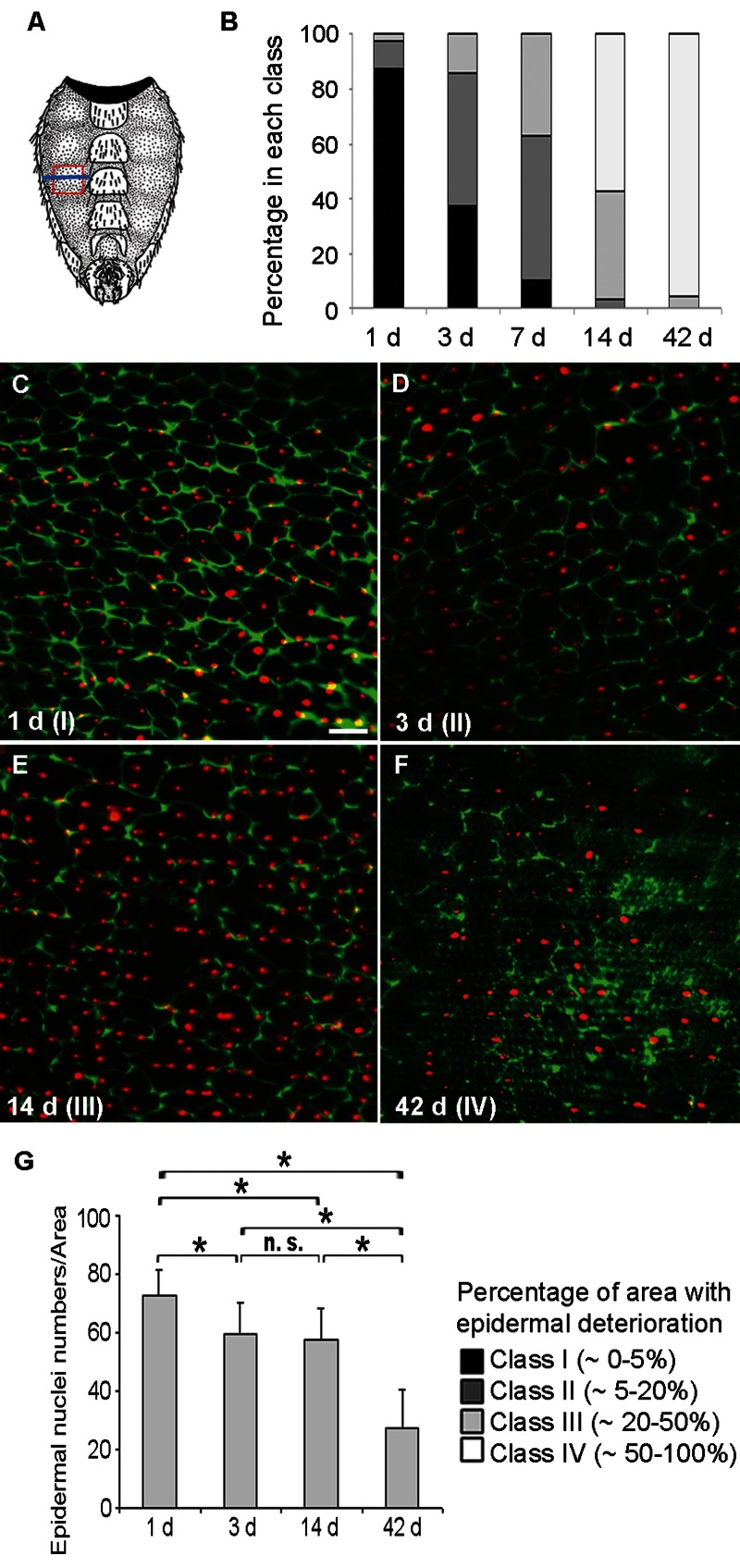
Figure 1. Loss of Membranes and Nuclei in the Aging Adult Epidermis (A) Schematic of the ventral adult abdominal epidermis (reprinted with kind permission by Cold Spring Harbor Laboratory Press from [56]). Red frame, section of the pleura analyzed by immunofluorescence; Blue line, approximate location of TEM cross-sections. (B) Quantification of epidermal deterioration in control w1118 flies with age (n ≥ 10). See C-F for a representative example of each morphology class. (C-F) Control (w;UAS-DsRed2-Nuc2/CyO; NP2108-GAL4/TM6C) epidermal whole mounts of different ages expressing nuclear DsRed2-Nuc (red) and labeled with anti-Fasciclin III (green). Bar, 20 μm. C, 1 d. D, 3 d. E, 14 d. F, 42 d. Green channel intensity elevated in F for visualization of weakly-labeled membranes. All comparisons between different time points were significantly different using the Chi square test (p < 0.05). (G) Quantification of nuclear numbers in epidermal whole mounts of flies of different ages bearing NP2108-GAL4 and UAS-DsRed2-Nuc (n = 7 for 1 d and 14 d; n = 8 for 3 d and n = 3 for 42 d). Asterisks, significant comparisons by Single-Factor Anova (p < 0.05); n. s., not significant.
We next examined epidermal changes at the structural level by transmission electron microscopy (TEM) in control flies (Figure 2). On day one, a continuous epidermal monolayer (average thickness 1.34 μm, n = 4) (Figure 2A) was observed distinct from the underlying muscles. A strong loss of cytoplasmic volume was detected as early as 3 d (average thickness 1.03 μm, n = 5), followed by a further gradual reduction at later time points (Figure 2B-D). From 3 to 14 days epidermal nuclei were condensed, while at 42 days clearly defined nuclei were often absent. Despite these other markers of epidermal deterioration, a thin but continuous epidermal layer and its basal lamina were maintained even at 42 d. Together, these fluorescence and TEM data suggest that progressive changes in the epidermis occur as the fly ages, and may represent one of the earliest reporters of insect tissue aging.

Figure 2. Epidermal Thickness Decreases in Aging Flies (A-D) TEM analysis of ventral pleura in w1118 controls. Bar, 2 μm. A, 1 d. B, 3 d. C, 14 d. D, 42 d. Red, epidermal boundaries; Blue, epidermal nuclei. c, cuticle; m, muscles. The presence of muscles underlying the epidermis varies with the precise plane of section and was not dependent on age.
A gene that prolongs lifespan decelerates epidermal aging
We next asked if epidermal aging would be altered in a mutant affecting Drosophila lifespan. Decreasing insulin signaling prolongs lifespan in diverse organisms [3, 22-26].
In Drosophila, flies that are homozygous mutant for the insulin-like receptor substrate (chico) have increased lifespan [27-29]. Therefore, we assessed markers of epidermal aging in chico1 flies. On day one, epidermal morphology in chico1 mutants was indistinguishable from controls (Figure 3A, E). However, epidermal membrane labeling over time was strongly preserved (Figure 3A-C) in comparison to controls (compare Figure 3B and C to Figure 1E and F). Morphological class distributions at 14 and 42 days were significantly different from age-matched controls (Figure 3E). Indeed, the 14 d old chico1 mutant epidermis most closely resembled 3 d old controls and the 42 d old chico1 mutant epidermis still looked younger than that of 14 d old controls. By TEM, the decrease in cytoplasmic volume and epidermal thickness also appeared to be decelerated (Figure S1A-C). These results are consistent with the idea that chico1 mutants, which have an extended lifespan [27] also retain the characteristics of a younger epidermis.
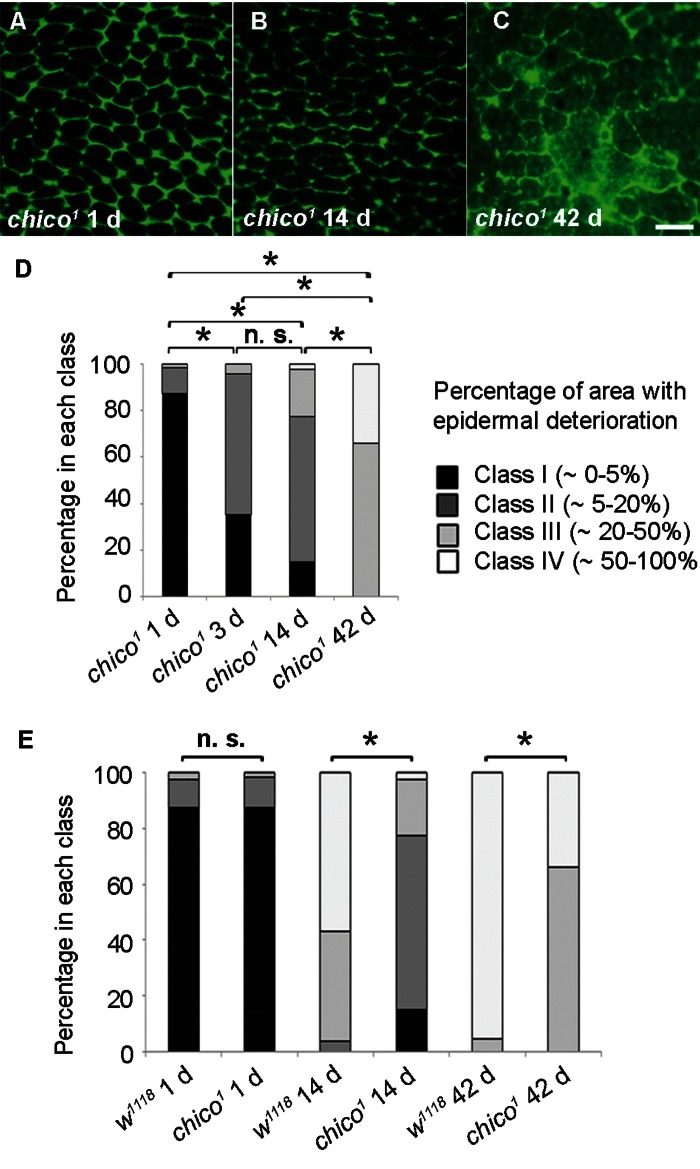
Figure 3. Decelerated Epidermal Aging in Long-lived chico Mutants (A-C) Anti-Fasciclin III immunofluorescence (green) of chico1 epidermal whole mounts (n ≥ 10). Bar, 20 μm. A, 1 d. B, 14 d. C, 42 d. (D) Quantification of chico1 epidermal deterioration with age. Asterisks, significant comparisons by Chi square test (p < 0.05); n. s., not significant. (E) Quantitative comparison of epidermal deterioration in chico1 mutants and w1118 controls. Significance indicators as in Figure 3D.
A mutant with shortened lifespan exhibits accelerated epidermal aging
Conversely, we investigated if a mutation that shortens lifespan would affect epidermal aging. In humans, disorders that resemble premature aging, including atypical Werner syndrome and Hutchinson-Gilford progeria syndrome, have been linked to mutations in genes encoding lamin proteins [30-32]. The premature aging phenotype in these disorders is accompanied by a reduced lifespan. Whether laminopathy disorders truly represent accelerated aging is vigorously debated [33] but it is clear that many of these diseases are characterized by an early onset of gene expression changes and morphological skin changes resembling those of normal aging [34]. We thus investigated if a mutation in a Drosophila lamin gene would alter epidermal morphology. Although many lamin mutant alleles do not survive to the adult stage, adult escapers of a B-type lamin allele, lamG262[35], have a shortened lifespan [36]. In 1 d old lamG262 flies epidermal morphology resembled controls (Figure 4A), suggesting that development of the adult epidermis during pupariation proceeded normally. By 3 days and 7 days, the lamG262 mutant epidermis showed more morphological deterioration than control epidermis of the same age (Figure 4B and C) and the classification of lamG262 mutant epidermal pictures (Figure 4D) supported an accelerated epidermal deterioration at these time points compared to controls (Figure 4E). In 14 d old lamG262 flies epidermal morphology did not look significantly different. Finally, TEM revealed strong nuclear condensation at 3 days compared to control nuclei at this time point (Figure S2B). Although there was no obvious difference in epidermal thickness in 3 d old lamG262 mutants compared to age-matched controls, TEM samples of lamG262 mutants exhibited altered cellular morphology. In summary, these results suggest that epidermal aging is accelerated at early time points in lamG262 mutants compared to control flies.
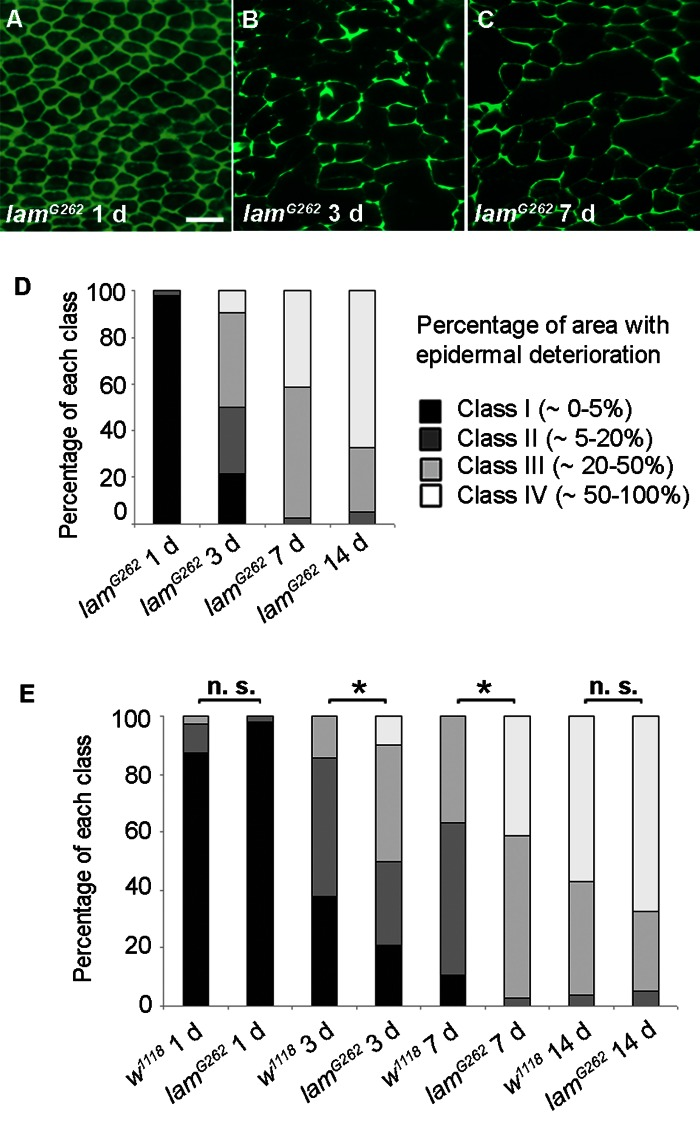
Figure 4. Epidermal Aging is Accelerated in Short-lived lamin Mutants (A-C) Anti-Fasciclin III immunofluorescence (green) of lamG262 epidermal whole mounts (n ≥ 10). Bar, 20 μm. A, 1 d. B, 3 d. C, 7 d. (D) Quantitative comparison of epidermal deterioration in lamG262 mutants and w1118 controls. All comparisons between different time points were siginificantly different using the Chi square test (p < 0.05). E, Quantitative comparison of epidermal deterioration in lamG262 mutants and w1118 controls. Significance indicators as in Figure 3D.
Autophagy correlates with and drives epidermal deterioration
The dramatic loss of membrane and cytoplasm in aging epidermal cells suggested that autophagy might drive the observed changes in morphology. We thus examined the aging epidermis for markers of autophagy. Consistent with the hypothesis that autophagy was responsible for the loss of membrane and cytoplasm, we found large autophagosomes in 14 d old control flies (Figure 5A). We next quantified epidermal autophagosomes at different ages and in different genotypes using a transgene, UAS-LC3-GFP, that labels autophagosomal membranes. We compared 3 d and older flies to avoid high autofluorescence levels in the 1 d old epidermis. Autophagosome numbers increased significantly from 3 d to 42 d in controls (Figure 5B and C). As expected, autophagosome numbers remained low in Atg7d77 flies with disrupted autophagy [37] and in chico1 mutants regardless of age. By contrast, autophagic activity on day 3 (but not day 7) was higher in lamG262 flies than in controls. In conclusion, an early rise in autophagy levels (lamin mutant) compared to controls correlates with accelerated epidermal aging. Conversely, a decrease in autophagy levels (chico or Atg7 mutants) correlates with decelerated epidermal aging.
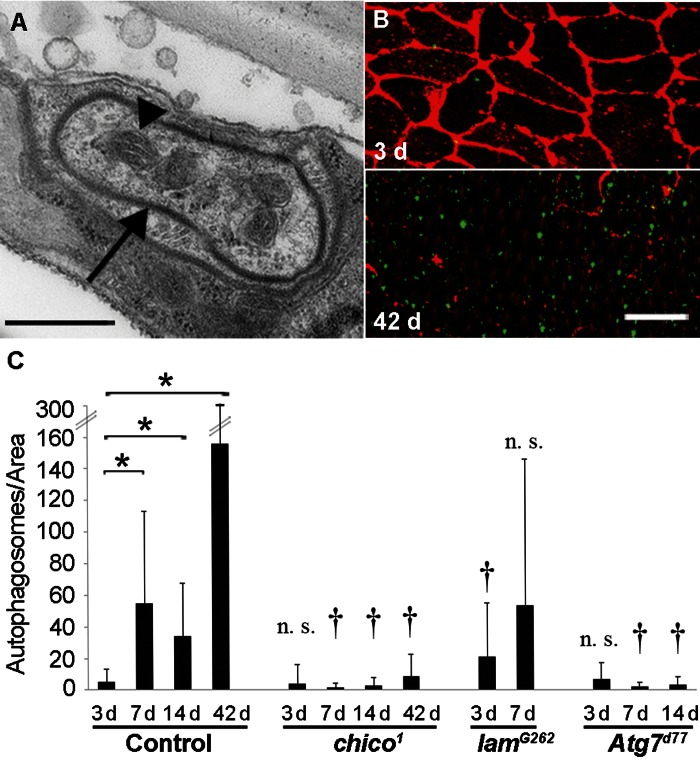
Figure 5. Autophagy Levels Correlate with Epidermal Deterioration (A) TEM of autophagosome (arrow) in 14 d old w1118 epidermis. Bar, 500 nm. Arrow, autophagosome membrane. Arrowhead, Mitochondrion. (B) 3 d (top) and 42 d (bottom) old control (NP2108-GAL4 and UAS-LC3-GFP) epidermal whole mounts labeled with anti-Fasciclin III (red) and LC3-GFP signals amplified with anti-GFP (green). Bar, 20 μm. (C) Quantification of autophagosome numbers (using NP2108-GAL4 and UAS-LC3-GFP) in epidermal whole mounts of the indicated genetic backgrounds. (n ≥ 20 for each time point). Asterisks, statistically significant comparisons (Single-factor Anova test (p < 0.05)) of control time points; Daggers, statistically significant comparisons of mutants versus controls of the corresponding time points; n. s., not significant.
These findings prompted us to test if autophagy drives age-related epidermal morphology changes since autophagy can drive developmental [38] and physiological remodeling of tissues [39]. Like chico1 and lamG262 mutants, the initial epidermal morphology of Atg7d77 flies resembled controls (Figure 6A). Interestingly, at 14 d loss of epidermal membrane labeling was strongly decelerated in comparison to controls (compare Figure 6B to Figure 1E and Figure 6C to Figure 1B). Additionally, TEM analysis revealed a thicker epidermal layer in 14 d old Atg7d77 flies compared to control epidermis of the same age (Figure S4).
To test if Atg7 controls epidermal aging in a tissue-autonomous manner, we used NP2108-GAL4 to express an RNAi targeting Atg7. NP2108-GAL4 is expressed in the adult epidermis. It is also expressed transiently in the larval fat body at eclosion, but this expression persists less than 24 hours and is therefore unlikely to affect epidermal aging at later stages (Figure S5). No expression in the adult fat body was observed. Epidermal knockdown of Atg7 resulted in decelerated epidermal aging similar to the mutant, suggesting a tissue-autonomous role for autophagy (Fig. 6D). Together, these results suggest that changes in epidermal morphology with age are driven by autophagy.
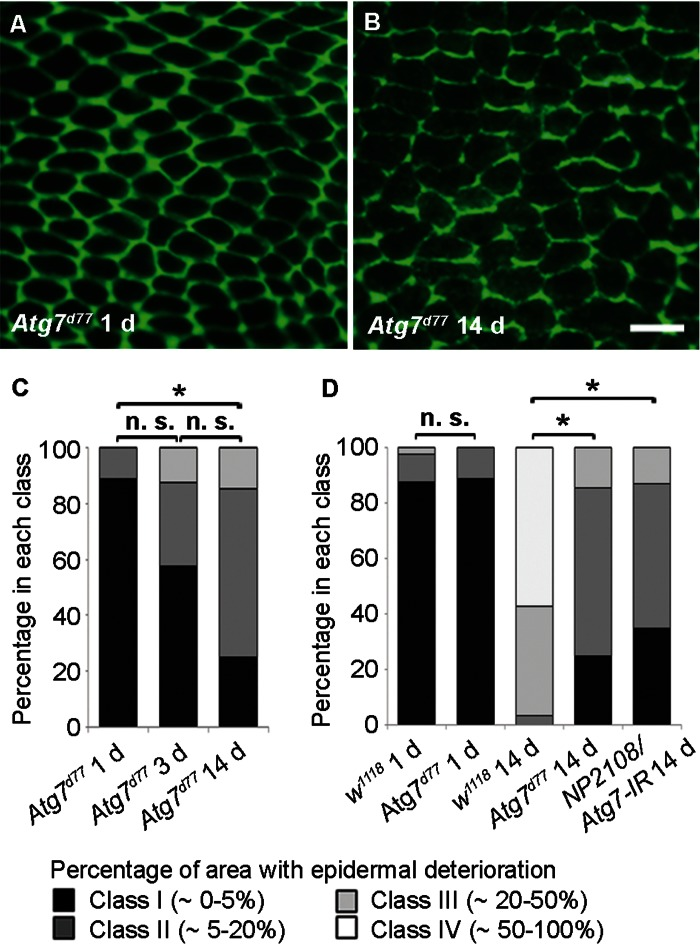
Figure 6. Autophagy is Required for Epidermal Aging (A-B) Anti-Fasciclin III immunofluorescence (green) of Atg7d77 epidermal whole mounts (n ≥ 10). Bar, 20 μm. A, 1 d. B, 14 d. (C) Quantification of Atg7d77 epidermal deterioration with age. Asterisks, significant comparisons by Chi square test (p < 0.05). n. s., not significant. (D) Quantitative comparison of epidermal deterioration in Atg7d77 mutants, NP2108-GAL4/Atg7-IR flies and w1118 controls. Significance indicators as in C.
Discussion
Here we establish an adult Drosophila model of epidermal aging in a search for early healthspan biomarkers and the genes that regulate them. By 3 d post-eclosion, earlier than other established biomarkers of tissue aging, both loss of epidermal membrane staining and cytoplasmic volume were apparent. Later, some epidermal nuclei were lost as well. Because blockade of insulin signaling decelerated epidermal aging and a Drosophila lamin mutant with shortened lifespan accelerated epidermal aging, age-related changes in this tissue are analogously regulated by these two established aging pathways. Surprisingly, we find that in contrast to its protective role in many aging models autophagy is driving morphological deterioration of the Drosophila adult epidermis (comparisons of epidermal morphology quantifications from different time points and genotypes are compiled in Figure S3). The autophagy mutant that exhibited decelerated epidermal aging is itself short-lived [40], suggesting that in this tissue healthspan can be uncoupled from lifespan, at least in the case of the cellular process that drives tissue aging. We discuss this and other curious features of our model in more detail below.
Complex Roles for Autophagy in Tissue Aging
In the context of aging of the organism as a whole, it is generally believed that autophagy is primarily protective [20]. Increased autophagy correlates with increased lifespan in worms [41] and normal levels of systemic autophagy are required for the lifespan extension observed upon reduction of insulin signaling [42]. In Drosophila, maintenance of a sufficiently high level of autophagy in the nervous system and muscles seems to be particularly important for lifespan extension since neuronal-specific induction of autophagy leads to healthier neurons and lifespan extension [16, 43]. The role of autophagy in other aging tissues such as the epidermis has not been investigated until now. Unexpectedly, we observe that autophagy both correlates with and drives morphological deterioration of an aging adult tissue. Our results highlight the complex relationship between autophagy and the aging of individual tissues. Given the contrasting roles of autophagy in neuronal and epidermal tissue aging we hypothesize that the relationship between tissue-aging and lifespan may be unique for each organ. In other words, the net effect of autophagy on lifespan is likely determined by its particular role in those tissues most critical for survival. Specifically, we propose that in tissues whose healthspan is essential for setting lifespan, such as the nervous system, autophagy is protective, while some other tissues whose healthspan is not critical, such as the epidermis, may be degraded by autophagy.
While it is expected that all organs will deteriorate with age, it is surprising that this seems to be an actively regulated process in the case of the adult fly epidermis. Why? One possibility is that once the cuticle barrier structure is formed shortly after eclosion the adult epidermis becomes dispensable for survival. Our TEM data suggests that cuticle secretion stops after two to three days into adulthood (data not shown) and the expression of known adult cuticle proteins ceases around this same time [44]. The majority of epidermal degradation we observed occurred after cuticle secretion was complete, suggesting that aging, rather than development, drives the degradation process. We hypothesize that the adult fly normally degrades its epidermis as a nutrition source and that autophagy orchestrates this process. This controlled destruction is reminiscent of the autophagic elimination of certain organs that become superfluous at the onset of metamorphosis [38, 45] as well as the elimination of wing epidermal cells in the first hours of adult life [46]. A requirement for autophagy in the epidermis is consistent with previously demonstrated roles of autophagy in cellular remodeling [17, 18]. This proposed role is not inconsistent with a potential protective role of autophagy in the epidermis since a residual epidermis is maintained even in old flies.
Finally, our results imply that in the particular case of autophagy epidermal aging can be uncoupled from lifespan. The short-lived Atg7d77 mutant [37] maintains a young skin. Conversely, the Progeria-like lamG262 mutant, which has a short lifespan and higher epidermal autophagy, rapidly develops a very old skin. Interestingly, Progeria mice also exhibit an elevated autophagy level in the heart and other tissues [47]. In both flies and mice it remains unclear if the elevated autophagy levels are a cytoprotective reaction to Progeria or if autophagy is part of the pathology driving organ deterioration.
Parallels between Drosophila and vertebrate epidermal aging
In contrast to the epidermal monolayer in flies, vertebrate skin consists of a multilayered epidermis replenished by basal stem cells and separated from an underlying dermis. The vertebrate epidermis is continuously needed to maintain a functional permeability barrier, and thus is likely to have a higher contribution to overall lifespan than the epidermis of the fly. Given these differences, how then do the changes observed in the aging Drosophila adult epidermis relate to vertebrate skin? A decreased integrin expression in the basal epidermis has been reported for aging human skin [13] and epidermal thinning is one of the hallmarks of aged human skin [12]. Relative to average lifespan, epidermal morphology changes in the fly have an even earlier onset than age-related changes in human skin. This difference may be explained by the fact that fly skin cells are not replenished by stem cell-mediated replacement.
Despite the morphological differences between fly and vertebrate skin, are there parallels on the molecular level that control the skin aging program? Intriguingly, mice bearing an insulin receptor substrate null mutation exhibit delayed skin aging [48] and progeroid mice with a mutant lamin exhibit hallmarks of accelerated skin aging [49]. Whether autophagy correlates with and plays a causal role in the cellular or molecular control of vertebrate skin aging in vivo remains to be tested. However, senescent keratinocytes in cell culture undergo autophagic cell death [21], suggesting that autophagy may play a conserved role in controlling the morphological progression of vertebrate skin aging.
A major need in the aging field is the identification of readouts of “healthspan”, which has been defined as the length of time that an individual maintains good health [6]. Drosophila has proven itself an important model for the identification of genes that regulate lifespan [50]. However, by contrast Drosophila tissue aging has only been examined in a few organ systems and behaviours including the heart, gut, muscles, nervous system, and sleep patterns [8-10]. The changes in the epidermis observed here arise earlier, can be detected using a simple visual assay, and represent a chronometer for the healthspan of an individual organism. Our model thus complements more commonly used population-based longevity assays. In addition, the epidermal aging model provides a simple platform for testing the impact of extrinsic factors such as UV radiation or caloric restriction. We expect that further molecular dissection of the epidermal aging program will reveal other regulators of tissue healthspan and how these genes are connected to organismal lifespan.
Methods
Fly stocks and husbandry
Aging Drosophila were reared at 25 °C on standard cornmeal medium under a 12 h light-dark cycle. For consistency, only virgin females were analyzed. Aging flies were switched to new food every two to three days to avoid starvation effects.
Except where noted, w1118 was used as a control strain for the rate of epidermal aging. In some cases, a Fasciclin III (FasIII)-GFP fusion transgene (YD0853; [51]) was used to visualize epidermal cell membranes. Epidermal morphology was also analyzed in long-lived chico1 mutants [27], short-lived lamG262 mutants [35], and Atg7d77 mutants, which have reduced autophagic activity [37]. The GAL4/UAS system [52] was used for tissue-specific expression of transgenes under UAS control. The NP2108-GAL4 driver (strain 112783; DGRC Japan) expresses in the larval and adult epidermis, as well as the persistent larval fat body. UAS-Atg7-IR (CG5489-IR #45558 from VDRC) was used for knockdown of Atg7. UAS-DsRed2-Nuc and UAS-LC3-GFP allowed labeling of epidermal nuclei and autophagosomes, respectively. chico1/CyO;NP2108 -GAL4/TM6B and chico1/CyO;UAS-LC3-GFP were crossed to each other to visualize autophagosomes in the chico1 mutant background. Similarly, w;lamG262/ CyO;NP2108-GAL4/TM6B crossed to w;lamG262/CyO;UAS-LC3-GFP and w;Atg7d77/CyO;NP2108-GAL4/ TM6B crossed to w;Atg7d77/CyO;UAS-LC3-GFP were used to examine epidermal autophagosomes in the lamG262 and Atg7d77 mutant backgrounds, respectively.
Dissection of the adult abdominal epidermis
Flies were anaesthetized and the head and appendices removed. The thorax and abdomen were pinned dorsal side up on a Sylgard (Dow Corning, Midland, Michigan, United States) dissection plate with 0.1 mm diameter dissection needles (Fine Science Tools). We covered the specimen with 1x phosphate-buffered saline (PBS), flushed out attached air bubbles, and used dissecting scissors (Fine Science Tools #15000-02, Foster City, California, United States) to make an incision along the abdominal dorsal midline. The two dorsal halves were pinned to the sides, the thorax and inner organs removed to expose the ventral abdominal epidermis. Finally, the epidermis was flattened by careful repositioning of the dissection needles. Samples were fixed for 1 to 3 h in 3.7% formaldehyde in PBS. After washing with PBS, the needles were detached and the samples transferred to 1.5 ml microtubes for subsequent staining. A more detailed protocol is available on request.
Analysis of NP2108-GAL4 driver expression
1118/+; NP2108-GAL4 or UAS-2x eYFP/+; NP2108-GAL4/+ animals were collected immediately after eclosion and aged for 1, 7, or 14 days. Fat bodies were dissected and fixed in 3.7% formaldehyde in PBS for 30 minutes at room temperature. Fat bodies were then washed in PBS, mounted in Vectashield, and imaged with a Leica MZ16FA microscope with a Planapo 1.6x objective.
Immunofluorescence
Immunostaining of adult epidermal whole-mounts was performed as described earlier for larval samples [53]. Primary antibodies were mouse anti-Fasciclin III [54] diluted 1:50, mouse anti-Coracle monoclonal antibody 9C [55] diluted 1:500 and rabbit anti-GFP antibody (A11122, Invitrogen) diluted 1:500. Secondary antibodies were Fluorescein (FITC)-conjugated goat anti-mouse (1: 250), Cy3-conjugated goat anti-mouse (1:1000) and FITC-labeled goat anti-rabbit antibody (1:250), all obtained from Jackson ImmunoResearch Laboratories Inc. All primary and secondary antibodies were diluted in PHT buffer (phosphate-buffered saline containing heat-inactivated normal goat serum and 3% Triton X100). Immuno-labeled epidermal whole mounts were imaged using a Leica MZ16FA microscope and a Planapo 1.6x objective. Photos were taken with a color Leica DFC350FX digital camera.
Quantification of epidermal morphology
To quantify epidermal deterioration pictures of epidermal samples of different genotypes and time points, we defined four classes I to IV reflecting distinct stages of epidermal membrane labeling loss. Class I was characterized as a sample with 0 to 5% epidermal deterioration in respect to the total epidermal area, class II corresponded to 5-20%, class III to 20-50% and class IV to over 50% epidermal deterioration. Four independent individuals were asked to assign ten individual pictures for each genotype-time point combination into these pre-defined classes. The individual groupings of the four judges were then averaged for each genotype-time point combination. Subsequently the classification numbers for different genotypes and time points were compared using an exact one-sided analysis contingency table (Chi square test). Nuclear numbers and LC3-GFP signals were compared using a single-factor Anova test in Microsoft Excel.
Autophagosome quantification
LC3-GFP-labeled epi-dermal whole mounts were visualized using an Olympus Fluoview 500 confocal microscope. Green channel settings were kept constant to allow for quantitative comparison. For each sample a Z series of optical sections in 0.7 μm increments spanning the entire depth of the epidermis and covering an area of 210 by 210 μm in the XY plane was collected and merged into a Z-stack. The resulting images were further analyzed as follows using ImagePro Analyzer 6.2. LC3-GFP signals, apparent as small punctae, were first converted to grayscale and then counted. To exclude background signals from cuticular autofluorescence, only bright punctae were counted (those >30 pixels on a 0-255 range of selected areas). Since the lamG262 allele contains a GFP insertion that labels epidermal nuclei, in lamG262 samples GFP-labeled nuclei were substracted from overall GFP counts in one of two ways: 1. By setting a size threshold that excluded large (area threshold here) labeled objects. 2. By then manually subtracting the remaining labeled objects that by morphological criteria and comparison to the original image seemed to result from nuclear labeling. Because some autophagosomes appear to be tightly associated with nuclei, the autophagosome counts for lamG262 samples likely represent an undercount of the actual autophagosome numbers. The autophagosome numbers from all individual pictures were averaged for each genotype and time point and compared using the Student's t-test in Microsoft Excel.
Transmission Electron Microscopy (TEM)
For transmission electron microscopy whole fly abdomens were individually dissected in 0.2 M sodium phosphate buffer (pH 7.2) and then immediately fixed for 4 h at room temperature in 3% glutaraldehyde, 2% paraformaldehyde and 2.5% dimethylsulfoxide in this same buffer. Samples were post-fixed in 1% Osmium tetroxide (Electron Microscopy Services, EMS, Hatfield, PA), briefly rinsed with water, stained with 2% aqueous uranyl acetate (EMS), dehydrated in graded ethanols, and embedded in Poly-Bed 812 resin (Polysciences Inc., Warrington, PA). Ultrathin sections were cut on a Leica EM UC6RT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with lead citrate, and viewed in a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA) at an accelerating voltage of 80 kV and 10,000x magnification. Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA). TEM pictures shown in Figure 2 and Figure S1, S3 and S4 consist of several overlapping pictures merged in Adobe Photoshop CS4. The cuticle-epidermis boundaries could be clearly distinguished by color, while the muscle-epidermis boundaries were defined by the intervening basal laminae.
Supplementary Materials
Acknowledgments
We thank Scott Pletcher, Mitch Dushay, Andreas Bergmann, Thomas Neufeld, the Kyoto Drosophila Stock Center and the Vienna Drosophila RNAi Center for fly stocks and the Developmental Studies Hybridoma Bank for the FasIII antibody. TEM was performed with support of Kenneth Dunner, Jr. at the High Resolution Electron Microscopy Facility, UTMDACC. We thank Galko lab members, Nicholas Buchon and Ernesto Perez for helpful comments on the manuscript. This study was supported by UTMDACC startup funds to MJG, NIH R01 GM083031 and AARA supplement to this same parent award to MJG, and NIH T32 CA009299 to AEA.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010; 328: 321 -326. [PubMed] .
- 2. Vijg J and Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008; 454: 1065 -1071. [PubMed] .
- 3. Grönke S, Clarke DF, Broughton S, Andrews TD, Partridge L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010; 6: e1000857 [PubMed] .
- 4. Katewa SD and Kapahi P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Exp Gerontol. 2011; 46: 382 -390. [PubMed] .
- 5. Katewa SD and Kapahi P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Experimental gerontology. 2011; 46: 382 -390. [PubMed] .
- 6. Tatar M. Can we develop genetically tractable models to assess healthspan (rather than life span) in animal models? J Gerontol A Biol Sci Med Sci. 2009; 64: 161 -163. [PubMed] .
- 7. Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002; 419: 808 -814. [PubMed] .
- 8. Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004; 36: 1275 -1281. [PubMed] .
- 9. Demontis F and Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010; 143: 813 -825. [PubMed] .
- 10. Beramendi A, Peron S, Casanova G, Reggiani C, Cantera R. Neuromuscular junction in abdominal muscles of Drosophila melanogaster during adulthood and aging. J Comp Neurol. 2007; 501: 498 -508. [PubMed] .
- 11. Koudounas S, Green EW, Clancy D. Reliability and variability of sleep and activity as biomarkers of ageing in Drosophila. Biogerontology. 2012; 13: 489 -499. [PubMed] .
- 12. Makrantonaki E and Zouboulis CC. Molecular mechanisms of skin aging: state of the art. Ann NY Acad Sci. 2007; 1119: 40 -50. [PubMed] .
- 13. Giangreco A, Goldie SJ, Failla V, Saintigny G, Watt FM. Human skin aging is associated with reduced expression of the stem cell markers beta1 integrin and MCSP. J Invest Dermatol. 2010; 130: 604 -608. [PubMed] .
- 14. Adler AS, Kawahara TL, Segal E, Chang HY. Reversal of aging by NFkappaB blockade. Cell Cycle. 2008; 7: 556 -559. [PubMed] .
- 15. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010; 221: 3 -12. [PubMed] .
- 16. Marino G, Madeo F, Kroemer G. Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol. 2010; .
- 17. Kadandale P, Stender JD, Glass CK, Kiger AA. Conserved role for autophagy in Rho1-mediated cortical remodeling and blood cell recruitment. Proc Natl Acad Sci U S A. 2010; 107: 10502 -10507. [PubMed] .
- 18. Shen W and Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009; 187: 71 -79. [PubMed] .
- 19. Malagoli D, Abdalla FC, Cao Y, Feng Q, Fujisaki K, Gregorc A, Matsuo T, Nezis IP, Papassideri IS, Sass M, Silva-Zacarin EC, Tettamanti G, Umemiya-Shirafuji R. Autophagy and its physiological relevance in arthropods: Current knowledge and perspectives. Autophagy. 2010; 6: 575 -588. [PubMed] .
- 20. Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008; 24: 604 -612. [PubMed] .
- 21. Gosselin K, Deruy E, Martien S, Vercamer C, Bouali F, Dujardin T, Slomianny C, Houel-Renault L, Chelli F, De Launoit Y, Abbadie C. Senescent keratinocytes die by autophagic programmed cell death. Am J Pathol. 2009; 174: 423 -435. [PubMed] .
- 22. Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993; 366: 461 -464. [PubMed] .
- 23. Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003; 299: 572 -574. [PubMed] .
- 24. Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003; 421: 182 -187. [PubMed] .
- 25. Broughton SJ, Piper MD, Ikeya T, Bass TM, Jacobson J, Driege Y, Martinez P, Hafen E, Withers DJ, Leevers SJ, Partridge L. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 3105 -3110. [PubMed] .
- 26. Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, Piper MD, Al-Qassab H, Speakman JR, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008; 22: 807 -818. [PubMed] .
- 27. Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001; 292: 104 -106. [PubMed] .
- 28. Tu MP, Epstein D, Tatar M. The demography of slow aging in male and female Drosophila mutant for the insulin-receptor substrate homologue chico. Aging cell. 2002; 1: 75 -80. [PubMed] .
- 29. Tu MP and Tatar M. Juvenile diet restriction and the aging and reproduction of adult Drosophila melanogaster. Aging cell. 2003; 2: 327 -333. [PubMed] .
- 30. Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiological reviews. 2006; 86: 967 -1008. [PubMed] .
- 31. Capell BC and Collins FS. Human laminopathies: nuclei gone genetically awry. Nature reviews Genetics. 2006; 7: 940 -952. .
- 32. Mattout A, Dechat T, Adam SA, Goldman RD, Gruenbaum Y. Nuclear lamins, diseases and aging. Current opinion in cell biology. 2006; 18: 335 -341. [PubMed] .
- 33. Burtner CR and Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010; 11: 567 -578. [PubMed] .
- 34. Kyng KJ, May A, Kolvraa S, Bohr VA. Gene expression profiling in Werner syndrome closely resembles that of normal aging. Proc Natl Acad Sci USA. 2003; 100: 12259 -12264. [PubMed] .
- 35. Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci U S A. 2001; 98: 15050 -15055. [PubMed] .
- 36. Munoz-Alarcon A, Pavlovic M, Wismar J, Schmitt B, Eriksson M, Kylsten P, Dushay MS. Characterization of lamin mutation phenotypes in Drosophila and comparison to human laminopathies. PloS one. 2007; 2: e532 [PubMed] .
- 37. Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007; 21: 3061 -3066. [PubMed] .
- 38. Berry DL and Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007; 131: 1137 -1148. [PubMed] .
- 39. Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Developmental cell. 2004; 7: 167 -178. [PubMed] .
- 40. Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes & development. 2007; 21: 3061 -3066. [PubMed] .
- 41. Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K, Orosz L, Kovacs AL, Csikos G, Sass M, Vellai T. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008; 4: 330 -338. [PubMed] .
- 42. Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003; 301: 1387 -1391. [PubMed] .
- 43. Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008; 4: 176 -184. [PubMed] .
- 44. Qiu J and Hardin PE. Temporal and spatial expression of an adult cuticle protein gene from Drosophila suggests that its protein product may impart some specialized cuticle function. Dev Biol. 1995; 167: 416 -425. [PubMed] .
- 45. Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009; 19: 1741 -1746. [PubMed] .
- 46. Kimura K, Kodama A, Hayasaka Y, Ohta T. Activation of the cAMP/PKA signaling pathway is required for post-ecdysial cell death in wing epidermal cells of Drosophila melanogaster. Development. 2004; 131: 1597 -1606. [PubMed] .
- 47. Marino G, Ugalde AP, Salvador-Montoliu N, Varela I, Quiros PM, Cadinanos J, van der Pluijm I, Freije JM, Lopez-Otin C. Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum Mol Genet. 2008; 17: 2196 -2211. [PubMed] .
- 48. Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, Piper MD, Al-Qassab H, Speakman JR, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008; 22: 807 -818. [PubMed] .
- 49. Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003; 423298 -301. .
- 50. Libert S, Zwiener J, Chu X, Vanvoorhies W, Roman G, Pletcher SD. Regulation of Drosophila life span by olfaction and food-derived odors. Science. 2007; 315: 1133 -1137. [PubMed] .
- 51. Kelso RJ, Buszczak M, Quinones AT, Castiblanco C, Mazzalupo S, Cooley L. Flytrap, a database documenting a GFP protein-trap insertion screen in Drosophila melanogaster. Nucleic Acids Res. 2004; 32: (Database issue) D418 -D420. [PubMed] .
- 52. Brand AH and Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993; 118: 401 -415. [PubMed] .
- 53. Galko MJ and Krasnow MA. Cellular and Genetic Analysis of Wound Healing in Drosophila Larvae. PLoS Biol. 2004; 2: E239 .
- 54. Patel NH, Snow PM, Goodman CS. Characterization and cloning of fasciclin III: a glycoprotein expressed on a subset of neurons and axon pathways in Drosophila. Cell. 1987; 48: 975 -988. [PubMed] .
- 55. Fehon RG, Dawson IA, Artavanis-Tsakonas S. A Drosophila homologue of membrane-skeleton protein 4.1 is associated with septate junctions and is encoded by the coracle gene. Development. 1994; 120: 545 -557. [PubMed] .
- 56. Demerec M. Biology of Drosophila. Woodbury, NY Cold Spring Harbor Laboratory Press 1950; .