



Introduction
The excellent study by Miller et al “Big mice die young: early life body weight predicts longevity in genetically heterogeneous mice” [1] has set in stone the paradigm that large mice live shorter [2-9]. Weight in 2-month-old mice is a significant predictor of life span [1]. The rule is common in mammals, within (but not between) species, and even including humans in some special studies [10-16] (note: we will also discuss opposite tendency favoring taller people to live longer).
Yet, it has been known for millennia and is absolutely obvious that larger animals of different species live longer. Elephants live longer than mice and 75 ton bowhead whales may live up to 150-200 years, which is at least 3000 times longer than the lifespan of small C. elegans. So how do two rules co-exist? Based on a new view on aging, named for brevity “hyperfunction theory” [17-19], we can explain why and how the two phenomena coexist. Various applications and aspects of hyperfunction theory have been already extensively reviewed [19-35] (with references on other articles within), so here only a brief summary is needed in order to answer a particular question: “Why big mice die young but large animals live longer”.
Mechanistic (formally, mammalian) Target of Rapamycin (MTOR) is activated by nutrients (glucose, amino acids, fatty acids), oxygen, hormones (such as insulin), growth factors and cytokines and, in turn, stimulates growth and metabolism and is involved in pathological conditions such as diseases of aging [20, 38-53].
MTOR mechanistically links cellular mass growth and senescence, whereas aging is a continuation of growth [26, 54]. When actual cellular growth becomes impossible (post-mitotic and arrested cells), MTOR drives cellular aging/senescence [55-61], a process named geroconversion [62]. Importantly, MTOR stimulates cellular functions such as secretion and lipogenesis [63-65]. Senescent cells are hyperfunctional, leading to age-related diseases and conditions, an increasing the probability of death (organism aging) [34, 62]. This topic is beyond the scope of this article, it was discussed before [34] and cannot be discuss here in detail.
When the developmental program of an organism is completed, full MTOR activity is not needed. Then, instead of actual growth, MTOR drives aging and age-related diseases [19, 20]. Thus, aging is an aimless continuation of developmental programs (Figure 1), driven by the same “MoTOR” in the same direction (at first) and may be at almost the same speed [27]. A quasi-program for aging is not a program (it has no purpose) but a blindly-running program of developmental growth that has been already completed but not switched off [31]. At least initially, the MTOR-driven quasi-program causes no visible harm to the organism. In humans, adulthood may seem very healthy indeed, despite subclinical changes of homeostasis [19]. Overt diseases and organ damages arise much later in life. But natural selection is not at play at such ages, because rare animals survive until deep aging in the wild. Exceptions will be discussed here.
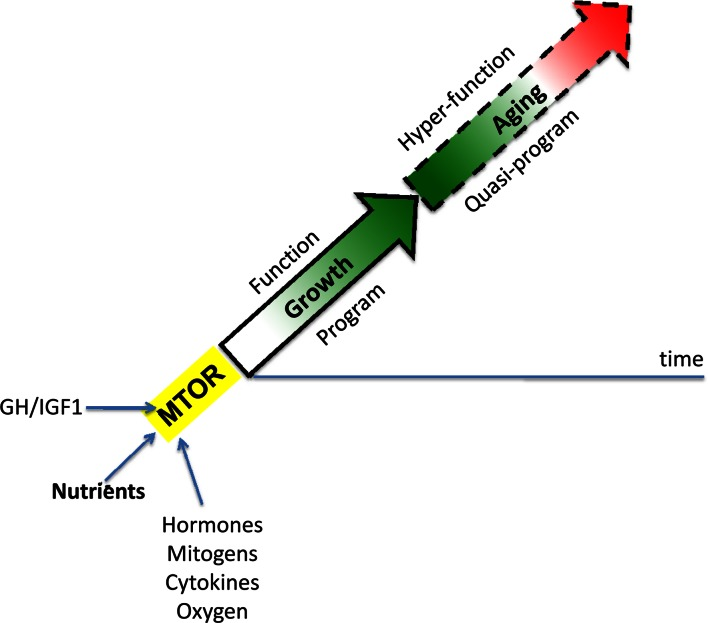
Figure 1. MTOR-driven quasi-programmed aging (hyperfunction model) Aging is a continuation of developmental growth driven by growth-promoting pathways such as MTOR. Green: robustness. Red: hyperfunction-driven diseases leading to death. See Ref. 19.
Why do large animals live longer?
Larger animals live in more protected environments with less accidental death from extrinsic causes, so natural selection favors slow aging. For example, elephants or whales (unless hunted by men) may die from “old age”: so natural selection has been at work to further increase their life span. Large size by itself is protective from predators. Large, complex organisms with sophisticated behaviors require prolonged periods of development, so large animals develop slower. Then aging, a continuation of developmental growth, is also slow (Figure 2). Second, due to low extrinsic death rate, natural selection may favor slow aging in large animal types, and the most natural way to do so is to repress growth-promoting/gerogenic pathways such as MTOR. This will automatically decelerate developmental growth: the most certain way to slow aging is to slow the developmental growth. In other words, slow development turns into the slow quasi-program of aging and age-related diseases. Taking these two reasons together, there is a positive feedback loop between slow development and low incidental death rate, ensuring that large animals are long-lived. In brief, species with large body size and low accidental death rate have undergone (or even undergo now) selection for longevity. Because aging is a quasi-progammed hyperfuntcion and continuation of growth, slow aging can be a result of slow development in large animals.
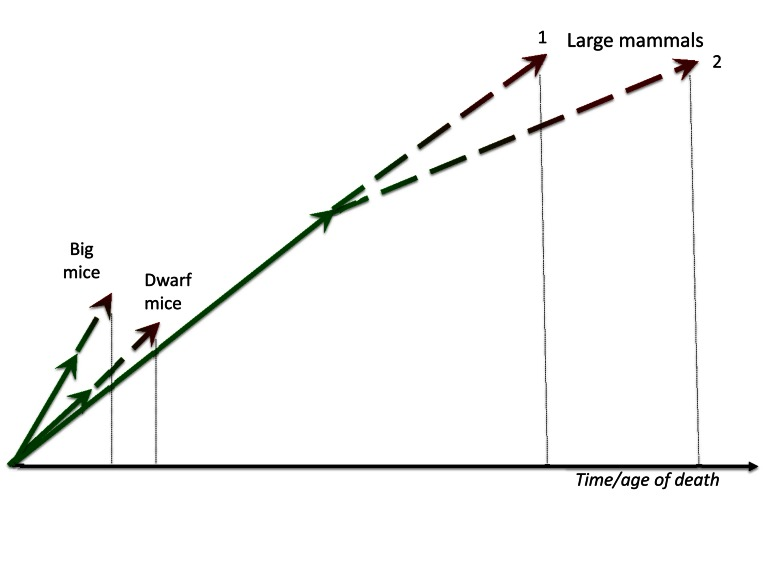
Figure 2. Big mice and large mammals Big mice grow faster than normal and especially dwarf mice. Fast growth is translated in fast aging. In contrast, large mammals develop for a prolonged period of time and aging (a continuation of developmental growth) is slow too. Given that very large animals may have very low extrinsic death rate and therefore may die from aging, natural selection may, in theory, favor deceleration of aging (curve 2).
Why the correlation is not perfect
Yet low accidental death rate can be dissociated from body size. Then, even smaller animals, which live in protected environments in the wild, live longer than equally-sized animals. Examples include bats and naked mole-rats, compared with rats. In such animals, natural selection favors slow aging and aging-tolerance (the term discussed in [27, 33]). Hypothetically, in durable safe environments with low accidental death rates, natural selection may favor a “decelerator” of MTOR, slowing down aging after development. Another way is to increase aging-tolerance, despite the same rate of aging. For example, changes in the developmental program that increases the number of collateral arteries in vital organs such as the heart (aging-tolerance) can extend lifespan despite the same rate of atherosclerosis (aging).
Why do big mice age faster?
Big mice grow faster than slow-growing mice. They are bigger than slow-growing counterpart at the same age [1]. This is an advantage earlier in life in any environment. (Note: one may argue that this may be disadvantage in the wild because large mice need to eat more to stay alive. However, a cause-effect relationship may be opposite: it is food, which activates MTOR and growth, that makes mice bigger at the time of plenty). But if they are larger because nutrient- and growth factor-sensing pathways like MTOR are over-active, then such fast-growing mice should age faster too (Figure 2).
Calorie restriction decreases size, prolongs development, delays reproduction and, on the other hand, delays aging, age-related diseases, loss of reproduction [28, 66]. In contrast, by stimulating MTOR, overeating causes the opposite effects. While nutrients, insulin, growth factors all activate the MTOR pathway, they accelerate gerogenic conversion and hyperfunctional aging.
Similarly, mice overproducing growth hormone and IGF are bigger and live shorter lives [6-70]. In contrast, small (slow-growing) mice which have defects at different points of the growth pathway (such as RasGrf1−/−, growth hormone-deficient, GH receptor-deficient, IRS1−/−, S6K1−/−) live longer [5, 8, 71-78]. For all these examples, short life span is associated with increased MTOR activity, while long-lived strains have a decreased MTOR activity [36]. This was discussed in detail [36]. Whether long-lived strains are insulin sensitive (see Figure 2 in Ref. [36] or insulin-resistant (Figure 3 in Ref. [36]) is co-incidental. Thus, insulin resistance may result from MTOR overactivation (bad condition). Other types of insulin/IGF1 resistance may result in deactivation of MTOR (benevolent condition) (see figures in [36]).
The activity of MTOR, in theory, determines both body size and lifespan, as discussed in detail [19-35] (with references on other articles within).
Noteworthy, fibroblasts from long-lived mutant mice exhibit lower MTOR activity [79]. In contrast, in the muscle of long-lived Ames dwarf mice, the PI3K/Akt/MTOR pathway is deactivated compared with their normal size siblings at the same age of 2 months [80].
In sum, to achieve prematurely big body weight, a mouse needs hyperactive growth-promoting pathways (genetic mutation, overfeeding), which later drives accelerated aging. This happens because two processes are continuation of one another and because growth-promoting pathways such as MTOR are involved in both growth and aging (Figure 2)
Importantly, slow-aging mice were developed in the laboratory. Small size and late reproduction and infertility are disadvantages in the wild with high risk of accidental death. Yet slow-growing, slow-aging GH- and GH receptor-deficient mice are an excellent model to illustrate that aging is a continuation of developmental growth and is driven by the same growth-promoting signals. What if this gerogenic force could be diminished to decelerate aging after the completion of development. In an outstanding study by the Miller group [81], long-lived Snell dwarf mice received 11 weeks of GH that increased their weight, although they remained much smaller than controls. The treatment also restored fertility to male dwarf mice and did not diminish life span [81]. I suggest that GH induces IGF-I and thus activates the MTOR pathway [82], which drives growth and then aging [26].
IGF levels correlates with body size and short longevity in mice [83-85]. In the Miller study, GH had been discontinued before mice reached full size and therefore only developmental growth was affected and promoted. Aging remained slow. The quasi-program of aging was not affected. (Another study, however, demonstrated that seemingly similar treatment with GH affected the speed of aging [86]. The difference may depend on dosage, duration and age of treatment [86]. As noted by Panici et al [86], the dosages were effectively declining in the Miller study [81]. Also, the age of nutritional intervention may switch the mouse to a slow aging trajectory [87].
And vice versa, deceleration of a quasi-program can be achieved in normal mice by administration of rapamycin, even when developmental growth program has been completed [88-90].
Males age faster than females
In many species, males are bigger than females. Could that be a special case of the rule that “big mice age fast.” Males must be strong and robust because of the competition for mates, fights and dangerous behavior. In the wild, an accidental death rate is very high for young males, so there is no need to age slower, especially given that fast aging is “a continuation” of the program of robustness and growth. Natural selection does not need to turn off the “MoTOR of aging” (MTOR). Especially because, as discussed previously [91, 92], MTOR brings about robustness earlier in life. Besides increasing strength and muscle size, testosterone stimulates MTOR. And rapamycin decreases both spermatogenesis and testosterone, although completely reversibly [93]. So low MTOR is a disadvantage for young males, where high MTOR activity is an advantage, providing hypertrophy and increased of functions. But hypertrophic functions become later hyperfunctions, causing loss of homeostasis, diseases and organ damage. In sum, males age faster and develop age-related diseases earlier than females. It was recently shown that young male mice have increased MTOR activity in the heart and the liver, and this activity correlates with body weight [94]. This supports the hypotheses explaining why males live shorter [94].
Emergence of slow-aging individuals in extremely protected environment
Several studies demonstrated that people with low body weight live longer. This could be explained by the low activity of the GH/IGF1/MTOR pathway, consistent with the “within species” rule. On the other hand, human life span is constantly increasing but people become taller. One hypothetical explanation is that (in the past) long-developing individuals died young from infections, starvation and accidents [95]. Now slow-developing and therefore slow-aging individuals survive until aging: the average lifespan is increasing. In principle, as speculated, their phenotype can be associated with “weak” MTOR and prolonged development [95]. In analogy, C. elegans lacking PI3K, an upstream component of the MTOR pathway, have prolonged developmental times and (under very protective environments) they mature into extremely long-lived adults (10-fold extension of both median and maximum adult lifespan) [96]. In humans, delayed puberty (slow development) is associated with exceptional longevity [97].
Instead of conclusion
Fast versus slow aging may depend on whether the organism “grows fast” or “develops longer”: first case should be associated with high MTOR. Exceptions may be numerous. Small size is not always related to the GH/IGF/MTOR pathway but instead may be caused by defects that shorten life span. But understanding of each exception will further illuminate the rules [98, 99]. On a wider scale (from worm to whale), large animals live longer because aging is quasi-programmed. In contrast, “big” mice live shorter because they grow faster than dwarf mice and growth is driven by the same pathways that drive aging. Fast-growing mice are expected to have over-activation of growth-promoting pathways (either by excessive calorie consumption or due to genetic mutations), which drive aging and age-related diseases later. Cellular hyperfunction is the key feature of aging cell, leading to organismal death [17-19] Yet, there are also two other crucial aspects of hyperfunction theory: (a) aging as a quasi-program of developmental growth and (b) both processes are driven by the same growth-promoting-signaling pathways including MTOR.
Acknowledgments
I thank Andrzej Bartke, Vera Gorbunova, Richard Miller and David Stipp for their insightful comments and valuable suggestions.
Conflicts of Interest
The author of this manuscript has no conflicts of interest to declare.
References
- 1. Miller RA, Harper JM, Galecki A, Burke DT. Big mice die young: early life body weight predicts longevity in genetically heterogeneous mice. Aging Cell. 2002; 1: 22 -29. [PubMed] .
- 2. Miller RA, Chrisp C, Atchley W. Differential longevity in mouse stocks selected for early life growth trajectory. J Gerontol A Biol Sci Med Sci. 2000; 55: B455 -461. [PubMed] .
- 3. Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001; 98: 6736 -6741. [PubMed] .
- 4. Bartke A, Coschigano K, Kopchick J, Chandrashekar V, Mattison J, Kinney B, Hauck S. Genes that prolong life: relationships of growth hormone and growth to aging and life span. J Gerontol A Biol Sci Med Sci. 2001; 56: B340 -349. [PubMed] .
- 5. Bartke A and Brown-Borg H. Life extension in the dwarf mouse. Curr Top Dev Biol. 2004; 63: 189 -225. [PubMed] .
- 6. Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003; 299: 1346 -1351. [PubMed] .
- 7. Rocha JS, Bonkowski MS, de França LR, Bartke A. Effects of mild calorie restriction on reproduction, plasma parameters and hepatic gene expression in mice with altered GH/IGF-I axis. Mech Ageing Dev. 2007; 128: 317 -331. [PubMed] .
- 8. Bartke A. Healthy aging: is smaller better? - a mini-review. Gerontology. 2012; 58: 337 -343. [PubMed] .
- 9. Huffman DM and Barzilai N. Contribution of adipose tissue to health span and longevity. Interdiscip Top Gerontol. 2011; 37: 1 -19. [PubMed] .
- 10. Urfer SR, Greer K, Wolf NS. Age-related cataract in dogs: a biomarker for life span and its relation to body size. Age (Dordr). 2011; 33: 451 -460. [PubMed] .
- 11. Li Y, Deeb B, Pendergrass W, Wolf N. Cellular proliferative capacity and life span in small and large dogs. J Gerontol A Biol Sci Med Sci. 1996; 51: B403 -408. [PubMed] .
- 12. Brosnahan MM and Paradis MR. Demographic and clinical characteristics of geriatric horses: 467 cases (1989-1999). J Am Vet Med Assoc. 2003; 223: 93 -98. [PubMed] .
- 13. Samaras TT, Storms LH, Elrick H. Longevity, mortality and body weight. Ageing Res Rev. 2002; 1: 673 -691. [PubMed] .
- 14. McCarron P, Okasha M, McEwen J, Smith GD. Height in young adulthood and risk of death from cardiorespiratory disease: a prospective study of male former students of Glasgow University, Scotland. Am J Epidemiol. 2002; 155: 683 -687. [PubMed] .
- 15. Salaris L, Poulain M, Samaras TT. Height and survival at older ages among men born in an inland village in Sardinia (Italy), 1866-2006. Biodemography Soc Biol. 2012; 58: 1 -13. [PubMed] .
- 16. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, Hwang D, Martin-Montalvo A, Saavedra J, Ingles S, de Cabo R, Cohen P, Longo VD. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011; 3: 70 -ra13. .
- 17. Gems D and Partridge L. Genetics of Longevity in Model Organisms: Debates and Paradigm Shifts. Annu Rev Physiol. 2013; 75: 621 -644. [PubMed] .
- 18. Gems DH and de la Guardia YI. Alternative Perspectives on Aging in C. elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2012; .
- 19. Blagosklonny M. Answering the ultimate question “What is the Proximal Cause of Aging?”. Aging (Albany NY). 2012; 4: 861 -877. [PubMed] .
- 20. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 21. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Disc Today. 2007; 12: 218 -224. .
- 22. Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007; 6: 2997 -3003. [PubMed] .
- 23. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008; 11: 801 -808. [PubMed] .
- 24. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 25. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging. 2009; 1: 281 -288. [PubMed] .
- 26. Blagosklonny MV and Hall MN. Growth and aging: a common molecular mechanism. Aging. 2009; 1: 357 -362. [PubMed] .
- 27. Blagosklonny MV. mTOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 -4059. [PubMed] .
- 28. Blagosklonny MV. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle. 2010; 9: 683 -688. [PubMed] .
- 29. Blagosklonny MV. Rapamycin and quasi-programmed aging: Four years later. Cell Cycle. 2010; 9: 1859 -1862. [PubMed] .
- 30. Blagosklonny MV. Linking calorie restriction to longevity through sirtuins and autophagy: any role for TOR. Cell Death Dis 1. 2010; e12 doi:101038/cddis200917 .
- 31. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 -3156. [PubMed] .
- 32. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY). 2011; 3: 1130 -1141. [PubMed] .
- 33. Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging (Albany NY). 2011; 3: 1051 -1062. [PubMed] .
- 34. Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012; 181: 1142 -1146. [PubMed] .
- 35. Blagosklonny MV. Rapalogs in cancer prevention: Anti-aging or anticancer? Cancer Biol Ther. 2012; 13: 1349 -1354. [PubMed] .
- 36. Blagosklonny MV. Once again on rapamycin-induced insulin resistance and longevity: despite of or owing to. Aging (Albany NY). 2012; 4: 350 -358. [PubMed] .
- 37. Blagosklonny MV. How to save Medicare: the anti-aging remedy. Aging (Albany NY). 4: 547 -552. [PubMed] .
- 38. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471 -484. [PubMed] .
- 39. Inoki K and Guan KL. Complexity of the TOR signaling network. Trends Cell Biol. 2006; 16: 206 -212. [PubMed] .
- 40. Tsang CK, Qi H, Liu LF, Zheng XFS. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Disc Today. 2007; 12: 112 -124. .
- 41. Kaeberlein M and Kennedy BK. Ageing: A midlife longevity drug? Nature. 2009; 460: 331 -332. [PubMed] .
- 42. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010; 40: 310 -322. [PubMed] .
- 43. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21 -35. [PubMed] .
- 44. Duran RV and Hall MN. Glutaminolysis feeds mTORC1. Cell Cycle. 2012; 11: 4107 -4108. [PubMed] .
- 45. Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013; 23: 53 -62. [PubMed] .
- 46. Passtoors WM, Beekman M, Deelen J, van der Breggen R, Maier AB, Guigas B, Derhovanessian E, van Heemst D, de Craen AJ, Gunn DA, Pawelec G, Slagboom PE. Gene expression analysis of mTOR pathway: association with human longevity. Aging Cell. 2013; 12: 24 -31. [PubMed] .
- 47. Sharp ZD. Aging and TOR: interwoven in the fabric of life. Cell Mol Life Sci. 2011; 68: 587 -597. [PubMed] .
- 48. Khanna A and Kapahi P. Rapamycin: killing two birds with one stone. Aging (Albany NY). 2011; 3: 1043 -1044. [PubMed] .
- 49. Stipp D. A new path to longevity. Sci Am. 2012; 306: 32 -39. [PubMed] .
- 50. Spong A and Bartke A. Rapamycin slows aging in mice. Cell Cycle. 2012; 11: .
- 51. Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012; 11: 675 -682. [PubMed] .
- 52. Katewa SD and Kapahi P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Exp Gerontol. 2011; 46: 382 -390. [PubMed] .
- 53. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013; 493: 338 -345. [PubMed] .
- 54. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358 -362. [PubMed] .
- 55. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 56. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -9664. [PubMed] .
- 57. Leontieva OV and Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2: 924 -935. [PubMed] .
- 58. Leontieva OV, Demidenko ZN, Gudkov AV, Blagosklonny MV. Elimination of proliferating cells unmasks the shift from senescence to quiescence caused by rapamycin. PLoS One. 2011; 6: e26126 [PubMed] .
- 59. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012; 109: 13314 -13318. [PubMed] .
- 60. Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012; 11: 4642 -4649. [PubMed] .
- 61. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, Blagosklonny MV, Lisanti MP. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181: 278 -293. [PubMed] .
- 62. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4: 159 -165. [PubMed] .
- 63. Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavare S, Inoki K, Shimizu S. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011; 332: 966 -970. [PubMed] .
- 64. Pani G. From growing to secreting: new roles for mTOR in aging cells. Cell Cycle. 2011; 10: 2450 -2453. [PubMed] .
- 65. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2011; 107: 3441 -3446. [PubMed] .
- 66. Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010; 328: 321 -326. [PubMed] .
- 67. Bartke A. Can growth hormone (GH) accelerate aging? Evidence from GH-transgenic mice. Neuroendocrinology. 2003; 78: 210 -216. [PubMed] .
- 68. Bartke A, Cecim M, Tang K, Steger RW, Chandrashekar V, Turyn D. Neuroendocrine and reproductive consequences of overexpression of growth hormone in transgenic mice. Proc Soc Exp Biol Med. 1994; 206: 345 -359. [PubMed] .
- 69. Diaz ME, Gonzalez L, Miquet JG, Martinez CS, Sotelo AI, Bartke A, Turyn D. Growth hormone modulation of EGF-induced PI3K-Akt pathway in mice liver. Cell Signal. 2011; 24: 514 -523. [PubMed] .
- 70. Kucia M, Shin DM, Liu R, Ratajczak J, Bryndza E, Masternak MM, Bartke A, Ratajczak MZ. Reduced number of VSELs in the bone marrow of growth hormone transgenic mice indicates that chronically elevated Igf1 level accelerates age-dependent exhaustion of pluripotent stem cell pool: a novel view on aging. Leukemia. 2011; 25: 1370 -1374. [PubMed] .
- 71. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140 -144. [PubMed] .
- 72. Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, Piper MD, Al-Qassab H, Speakman JR, Carmignac D, Robinson IC, Thornton JM, Gems D, Partridge L, Withers DJ. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008; 22: 807 -818. [PubMed] .
- 73. Selman C, Partridge L, Withers DJ. Replication of extended lifespan phenotype in mice with deletion of insulin receptor substrate 1. PLoS One. 2011; 6: e16144 [PubMed] .
- 74. Bonkowski MS, Dominici FP, Arum O, Rocha JS, Al Regaiey KA, Westbrook R, Spong A, Panici J, Masternak MM, Kopchick JJ, Bartke A. Disruption of growth hormone receptor prevents calorie restriction from improving insulin action and longevity. PLoS One. 2009; 4: e4567 [PubMed] .
- 75. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012; 61: 1315 -1322. [PubMed] .
- 76. Borras C, Monleon D, Lopez-Grueso R, Gambini J, Orlando L, Pallardo FV, Santos E, Vina J, Font de Mora J. RasGrf1 deficiency delays aging in mice. Aging (Albany NY). 3: 262 -276. [PubMed] .
- 77. Davies KJ and Forman HJ. RasGrf1 and aging. Aging (Albany NY). 3: 455 [PubMed] .
- 78. Ratajczak MZ, Kucia M, Liu R, Shin DM, Bryndza E, Masternak MM, Tarnowski M, Ratajczak J, Bartke A. RasGrf1: genomic imprinting, VSELs, and aging. Aging (Albany NY). 2011; 3: 692 -697. [PubMed] .
- 79. Wang M and Miller RA. Fibroblasts from long-lived mutant mice exhibit increased autophagy and lower TOR activity after nutrient deprivation or oxidative stress. Aging Cell. 2012; 11: 668 -674. [PubMed] .
- 80. Sharp ZD and Bartke A. Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J Gerontol A Biol Sci Med Sci. 2005; 60: 293 -300. [PubMed] .
- 81. Vergara M, Smith-Wheelock M, Harper JM, Sigler R, Miller RA. Hormone-treated snell dwarf mice regain fertility but remain long lived and disease resistant. J Gerontol A Biol Sci Med Sci. 2004; 59: 1244 -1250. [PubMed] .
- 82. Hayashi AA and Proud CG. The rapid activation of protein synthesis by growth hormone requires signaling through mTOR. Am J Physiol Endocrinol Metab. 2007; 292: E1647 -1655. [PubMed] .
- 83. Yuan R, Meng Q, Nautiyal J, Flurkey K, Tsaih SW, Krier R, Parker MG, Harrison DE, Paigen B. Genetic coregulation of age of female sexual maturation and lifespan through circulating IGF1 among inbred mouse strains. Proc Natl Acad Sci U S A. 109: 8224 -8229. [PubMed] .
- 84. Yuan R, Tsaih SW, Petkova SB, Marin de Evsikova C, Xing S, Marion MA, Bogue MA, Mills KD, Peters LL, Bult CJ, Rosen CJ, Sundberg JP, Harrison DE, Churchill GA, Paigen B. Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell. 2009; 8: 277 -287. [PubMed] .
- 85. Flurkey K and Yuan R. How the evolutionary theory of aging can guide us in the search for aging genes. Aging (Albany NY). 2012; 4: 318 -319. [PubMed] .
- 86. Panici JA, Harper JM, Miller RA, Bartke A, Spong A, Masternak MM. Early life growth hormone treatment shortens longevity and decreases cellular stress resistance in long-lived mutant mice. Faseb J. 2010; 24: 5073 -5079. [PubMed] .
- 87. Sun L, Sadighi Akha AA, Miller RA, Harper JM. Life-span extension in mice by preweaning food restriction and by methionine restriction in middle age. J Gerontol A Biol Sci Med Sci. 2009; 64: 711 -722. [PubMed] .
- 88. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandezr E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Nature. 2009; 460: 392 -396. [PubMed] .
- 89. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66: 191 -201. [PubMed] .
- 90. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10: 4230 -4236. [PubMed] .
- 91. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging (Albany NY). 2010; 2: 265 -273. [PubMed] .
- 92. Blagosklonny MV. Why the disposable soma theory cannot explain why women live longer and why we age. Aging (Albany NY). 2010; 2: 884 -887. [PubMed] .
- 93. Skrzypek J and Krause W. Azoospermia in a renal transplant recipient during sirolimus (rapamycin) treatment. Andrologia. 2007; 39: 198 -199. [PubMed] .
- 94. Leontieva OV, Geraldine M, Paszkiewicz GM, Blagosklonny MV. Mechanistic or mammalian target of rapamycin (mTOR) may determine robustness in young male mice at the cost of accelerated aging. Aging (Albany ny). 2012; 4: 899 -916. [PubMed] .
- 95. Blagosklonny MV. Why human lifespan is rapidly increasing: solving “longevity riddle” with “revealed-slow-aging” hypothesis. Aging (Albany NY). 2010; 2: 177 -182. [PubMed] .
- 96. Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ. Remarkable longevity and stress resistance of nematode PI3K-null mutants. Aging Cell. 2008; 7: 13 -22. [PubMed] .
- 97. Tabatabaie V, Atzmon G, Rajpathak SN, Freeman R, Barzilai N, Crandall J. Exceptional longevity is associated with decreased reproduction. Aging (Albany NY). 2011; 3: 1202 -1205. [PubMed] .
- 98. Gorbunova V, Bozzella MJ, Seluanov A. Rodents for comparative aging studies: from mice to beavers. Age (Dordr). 2008; 30: 111 -119. [PubMed] .
- 99. Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell. 2008; 7: 813 -823. [PubMed] .