



Introduction
Cellular senescence can be categorized in two groups. The replicative senescence, seen after certain rounds of cell division in cultures (“Hayflick's limit”) [1], is a consequence of a progressive erosion of telomeres at each division which leads to a telomere dysfunction and irreversible cell cycle arrest [2]. The second category defined as premature cellular senescence is unrelated to telomere shortening [review, 3]. Persistent cellular stress including replicative stress caused by oxidative DNA damage [4,5], activation of oncogenes [6] and loss of tumor suppressor genes [7] are among mechanisms inducing premature senescence. While in certain malignancies, particularly in acute leukemia, chemo- or radio- therapy induces apoptosis, the mechanism of elimination of cancer cells in some solid tumors often relies on irreversible impairment of cell reproductive capability defined as a drug- or radiation-induced senescence, that also belongs to the category of premature senescence [8,9]. Likewise, premature senescence of induced pluripotent stem cells (iPSCs) is a barrier in tumor development [10]. The stress-induced premature senescence of normal cells in vivo is considered to be a critical mechanism affecting organismal aging and longevity [11-14].
Extensive attempts have been made to develop gero-suppressive modalities that can slow down processes of senescence and aging extending longevity. Assessment of their effectiveness by analysis of animals' life span, especially when it involves vertebrates [15,16], is cumbersome and time consuming. It is therefore desirable to have relatively rapid in vitro approach that can be used for this purpose. Cumulative DNA damage caused by reactive oxygen species (ROS) produced during oxidative phosphorylation for long time was thought to be the major factor promoting aging (ROS mechanism) [17-19]. More recently, however, the persistent stimulation of the mitogen- and nutrient-sensing pathways including mammalian target of rapamycin (mTOR) signaling mechanism has been advanced as an alternative to ROS mechanism [20-28]. Activation of these pathways enhances translation and leads to cell growth in size/mass resulting in cell hypertrophy and senescence. Activation of mTOR/S6K pathway when combined with oxidative DNA damage that leads to replication stress appears to be particularly effective factor promoting aging and senescence [29].
The background level of constitutive activation of ATM and expression of γH2AX seen in untreated normal or cancer cells reports the ongoing DNA oxidative damage and replication stress induced by endogenous ROS [30-32]. Using flow- and laser scanning- cytometry as major methodologies we have recently shown that several reported gero-suppressive agents, namely, rapamycin, metformin, berberine (BRB), 1,25-dihydroxyvitamin D3, the calorie-restriction mimetic 2-deoxyglucose, and acetylsalicylic acid (ASA; aspirin), all depressed the level of constitutive DNA damage signaling [33-35]. Specifically, these substances reduced expression of γH2AX and activation of ATM in a variety of cell types, including tumor A549 and TK6 cells, as well as normal WI-38 cells or mitogenically stimulated human lymphocytes [33]. These agents also decreased the level of intracellular ROS and mitochondrial trans-membrane potential ΔΨm, the marker of mitochondrial energizing [33-35]. The above observations would be consistent with the ROS mechanism of aging. However, all these agents also distinctly reduced the constitutive level of phosphorylation of Ser235/236 of ribosomal S6 protein (rpS6), Ser2448 of mTOR and Ser65 of 4EBP1 [33], the major elements of the mTOR signaling [27,36-38]. Collectively, these data indicated that the reduction of mTOR/S6K signaling, that in turn reduces the translation rate, was coupled with a decrease in oxidative phosphorylation (revealed by ΔΨm) that led to reduction of ROS and attenuated oxidative DNA damage [33]. Thus, while the decreased rate of translation induced by these agents may slow down cells hypertrophy and alleviate other features of cell aging/senescence reduction of oxidative DNA damage may lower predisposition to neoplastic transformation. The latter may result from damage to DNA sites coding for oncogenes or tumor suppressor genes. Our data suggested that combined assessment of constitutive γH2AX expression, mitochondrial activity (ROS, ΔΨm) and mTOR signaling by cytometry can provide an adequate gamut of cell responses to evaluate effectiveness of potential gero-suppressive agents [33].
In continuation of these studies we attempted to explore whether gero-suppressive agents can also attenuate the level of premature, stress-induced cellular senescence. Toward this end we initiated experiments designed to reveal possible effects of these agents on induction of cellular senescence upon exposure of A549 cells to very low concentration of the DNA damaging drugs, shown by us before to trigger DNA replication stress manifesting by ATM activation and induction of γH2AX, that leads to senescence [39,40]. In preliminary experiments we observed that one of the gero-suppressive agents, the isoquinoline alkaloid berberine (BRB), was the most effective, suppressing the induction of cellular senescence at its low, clinically relevant, concentration. The present study, therefore, was designed to explore this effect of BRB in more detail and at the same time to demonstrate utility of flow- and laser scanning- cytometry in multiparametric analysis [41] of the depth of cellular senescence and its modulation by this alkaloid.
Results
One of the most characteristic features of cells undergoing senescence is change in their morphology revealed by an increase in cellular and nuclear size. In the case of cells growing attached this manifests by their dramatic “flattening” appearance combined with markedly reduced cell density at confluence [42,43]. The morphometric analysis of cell nucleus by laser scanning cytometry (LSC) is a sensitive detector of such a change [39,40]. Premature senescence of A549 cells was induced by their exposure to 2 nM DNA topoisomerase inhibitor mitoxantrone (Mxt), the drug that interacts with DNA by intercalation and is a type II DNA topoisomerase inhibitor [44]. Fig. 1 illustrates the features of these cells undergoing senescence, including their morphological changes that were measured by LSC. The morphometric analysis of the nucleus stained with the DNA fluorochrome DAPI shows an increase in nuclear area concomitant with the decrease in intensity of DAPI maximal pixel fluorescence [39]. The integral of intensity of DAPI fluorescence over the nucleus reports DNA content and thus the cell cycle phase, which as seen Fig. 1 (insets), indicates cell arrest in G1 and G2M with paucity of S-phase cells in the Mxt-treated cultures. Since nuclear area in senescent cells is increased and DAPI maximal pixel decreased the ratio of maximal pixel to the area (Mp:area) provides an even more sensitive marker reporting this change in cell morphology than either of these measurements alone (Fig. 1B). The cells arrested in G1 and G2M in the Mxt-treated cultures show 10 to 19-fold increase in expression of the CDK inhibitor p21WAF1, the another marker of cellular senescence [42,43]. Activation of the senescense-associated β-galactosidase A-β-gal), another hallmark of cellular senescence [42,43] is also markedly elevated in cells growing in the presence of Mxt. The rise in SA-β-gal activity measured by LSC is expressed either as percent of the enzyme-positive cells, which is augmented from 1.4 to 23% and 44%, or the mean value of the light absorption of the SA-β-gal product, increased 8.4- and 17.7-fold, in cells from cultures growing with Mxt for 48 and 72 h respectively (Fig. 1; D,E).
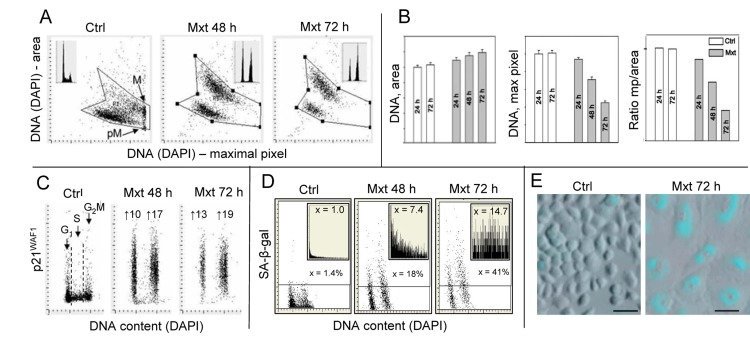
Figure 1. Induction of premature cellular senescence of A549 cells measured by laser scanning cytometry Human pulmonary non-small cell lung carcinoma A549 were untreated (Ctrl) or treated with 2 nM DNA topoisomerase II inhibitor mitoxantrone (Mxt) for 48 or 72 h. Panel A shows morphometric features of the cells revealed by measurement of nuclear DNA (DAPI) fluorescence reporting on the bivariate distributions (scatterplots) nuclear area versus intensity of maximal pixel of fluorescence, respectively. Intensity of maximal pixel is correlated with chromatin condensation and in the untreated cells has the highest value and marks mitotic (M) and immediately post-mitotic (pM) G1 cells, which also have low value of DAPI area [41]. In the senescing cells, while nuclear area increases, the intensity of maximal pixel decreases [39,40,64]. These morphometric changes reflect enlargement of the projected nuclear area and decreased DAPI local staining per unit area, due to “flattened” cellular appearance, the hallmark of cellular senescence [42,43]. The insets show DNA content frequency histograms of cells from the respective cultures. Panel B: Bar plots reporting mean values (+SD) of nuclear (DNA, DAPI) area, DNA (DAPI) maximal pixel, and ratio of maximal pixel to nuclear area, respectively, of cells from control and Mxt treated cultures. Panel C: Bivariate distributions (DNA content vs p21) reporting expression of p21WAF1 with respect to the cell cycle phase; the figures show the n-fold increase in mean expression of p21 of G1 and G2M cells from the Mxt-treated cultures with respect to respective cells in Ctrl. Panel D: Bivariate distributions of DNA content versus senescence-associated galactosidase (SA-β-gal) activity. Figures indicate percent of SA-β-gal positive (above the threshold marked by the horizontal lines) cells. Insets show the frequency distribution of SA-β-gal positive cells; the figures in insets show the n-fold increase in the mean activity of SA-β-gal in Mxt-treated cultures over Ctrl (1.0). Panel E: Images of cells growing in the absence (left) and presence of 2 nM Mxt for 72 h (right) stained to detect activation of SA-β-gal activity recorded by laser scanning cytometer (Research Imaging Cytometer iCys); 50 μm bars mark the length scale.
Induction of senescence of A549 cells growing in the presence of Mxt is distinctly suppressed, in a concentration-dependent fashion, by BRB (Fig. 2). Thus, the morphometric parameter reporting the nuclear change (Mp:area) which in the presence of Mxt alone is decreased from 1.0 to 0.28, in cultures treated with Mxt and 5 μM BRB is decreased to only 0.56, which is a 100% reduction compared to Mxt alone. At higher BRB concentration this effect is more pronounced, and at 60 μM BRB the reduction is 182% vis-à-vis Mxt alone. The attenuation of the Mxt-induced senescence by BRB, when measured by the induction of SA-β-gal activity, is also quite evident, and BRB-concentration dependent. The BRB-induced decrease of SA-β-gal activity in Mxt-treated cells was 26% at 5 μM BRB and further decreased to 75% at 60 μM concentration (Fig. 2).
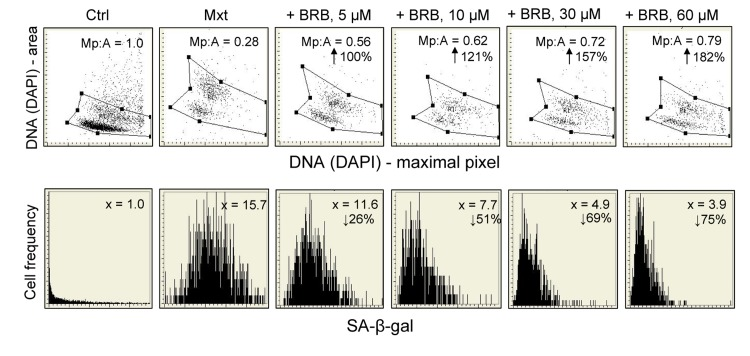
Figure 2. Attenuation of Mxt-induced senescence of A549 cells by berberine (BRB) as measured by cell morphometric features and SA-β-gal activity Exponentially growing A549 cells were untreated (Ctrl) or treated with 2 nm Mxt in the absence and presence of BRB at concentration as shown, for 5 days. Top panels: Morphometric analysis, reporting changes in nuclear area (DNA-DAPI) versus maximal pixel of DAPI fluorescence. The ratio of maximal pixel to nuclear area (Mp:A) is expressed as a fraction of that of the untreated cells; shown is the percent increase in Mp:A in the BRB-treated cultures with respect to cells growing with Mxt alone, with the arrows. Bottom panels: The frequency histograms reporting SA-β-gal activity. The figures present the increase (n-fold) in the enzyme activity with respect to the Ctrl (1.0), measured as the mean intensity of SA-β-gal absorption of cells in the respective cultures.
Fig. 3 illustrates the effect of BRB on expression of p21WAF1, γH2AX and ribosomal protein (rpS6) phosphorylated on Ser235/236 (rpS6P) in A549 cells, detected by phospho-specific Ab, induced to senescence by treatment with Mxt for 5 days. The Mxt-induced senescence of these cells manifests in dramatic increase in expression of p21 (34.5-fold), which is more pronounced than after 3 days of treatment with Mxt (Fig. 1). However, the increase in expression of p21 is markedly reduced in cells treated with Mxt in the presence of BRB and the reduction is BRB-dose dependent, starting with 56% at 5 μM and decreasing by 94% at 60 μM concentration. The induction of p21 by Mxt is paralleled by cell arrest in G1 and G2M phases of the cell cycle, and the arrest in G2M is to some extent reduced at 5 and 10 μM BRB (top panels, insets). The Mxt-induced senescence is also marked by 3.5-fold rise in expression of γH2AX and this rise is also attenuated, in a concentration-dependent fashion, by BRB, varying from 10% to 52% at 5 to 60 μM concentration of this isoquinoline. Phosphorylation of rpS6 on Ser235/236 is reduced by 11% in cells induced to senescence by Mxt alone. However, there is a dramatic further reduction in expression of this phosphorylated protein in cells growing in the presence of Mxt and BRB compared with Mxt alone, also in the dose-dependent mode, from 34% at 5 μM, to as much as 81% at 60 μM BRB concentration.
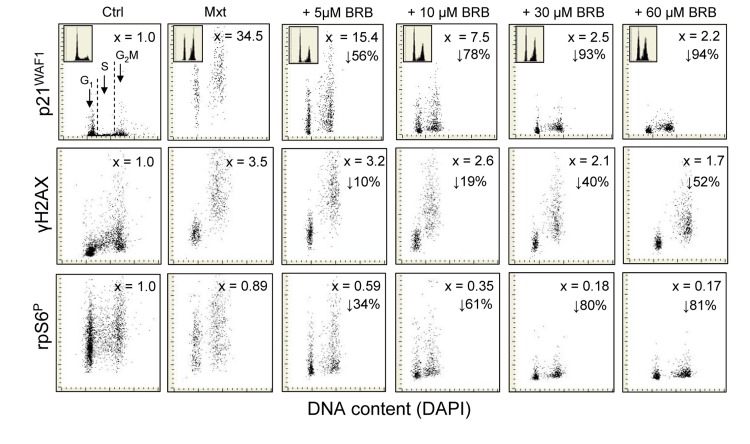
Figure 3. Attenuation of Mxt-induced senescence of A549 cells by BRB as measured by reduction in expression of p21WAF1, γH2AX and rpS6P Exponentially growing A549 cells were untreated (Ctrl) or treated with Mxt in the absence and presence of BRB at concentrations as shown, for 5 days. Top panels: bivariate distributions of p21 versus cellular DNA content; the figures (x) present the increase (n-fold) in the mean expression for all cells of p21 with respect to the untreated cells, the percent reduction in p21 in cultures with BRB with respect to Mxt alone, is shown with the arrows. Mid-panels: expression of γH2AX versus DNA content; the figures (x) represent the increase (n-fold) in the mean expression of γH2AX with respect to untreated cells (1.0); the percent reduction in expression of γH2AX in cultures grown with BRB with respect to cells growing in the presence of Mxt alone is presented with the arrows. Bottom panels: expression of rpS6P versus DNA content. The figures illustrate the change (n-fold) with respect to the untreated cells; the percent reduction in expression of rpS6P in cultures with BRB with respect to cells treated with Mxt alone is shown with the arrows.
The results shown in Figs. 1-3 demonstrate that the induction of premature cellular senescence of A549 cell by treatment with Mxt is distinctly attenuated by BRB and the attenuation is already evident at its 5 μM concentration. In our prior study we observed that treatment of several cell types including A549, TK6, WI-38 cells and normal proliferating lymphocytes with different potential gero-suppressive agents reduced both, the mTOR- as well as DNA damage- signaling [33]. Among these agents was BRB which at 60 μM concentration was seen to markedly suppress the level of constitutive phosphorylation of mTOR, rpS6 and 4EBP1 as well as of H2AX. These findings were consistent with the notion that BRB had potential gero-suppressive properties combined with the ability protect DNA from endogenous oxidants [33]. In light of the current observation that 5-60 μM BRB suppresses induction of the premature senescence we have tested its ability at these lower concentrations to affect the level of constitutive mTOR signaling in cells not induced to premature senescence. Such low BRB concentrations are relevant in terms of the drug pharmacokinetics and its in vivo effects [45-48]. To this end we treated human lymphoblastoid TK6 cells, the cells which we explored in the prior study [33], with the range of BRB concentration as used for A549 cells. As is evident from the data in Fig. 4 growth of these cells in the presence of 5 – 60 μM BRB led to a distinct reduction of rpS6 phosphorylation. The reduction was evident already at 5 μM BRB and at that concentration its extent showed distinct cell cycle phase specificity. Namely at 5 μM the reduction was more pronounced in G2M- and S- phase cells, lowering expression of rpS6P in these cells by 60% and 49%, respectively, compared with 39% for G1 cells. The degree of suppression of rpS6 phosphorylation was BRB-concentration dependent, reaching over 80% for the G2M and S-phase cells at its 60 μM concentration. The BRB concentration-dependency was quite apparent from the rpS6P frequency histograms (Fig. 4 top panels, insets).
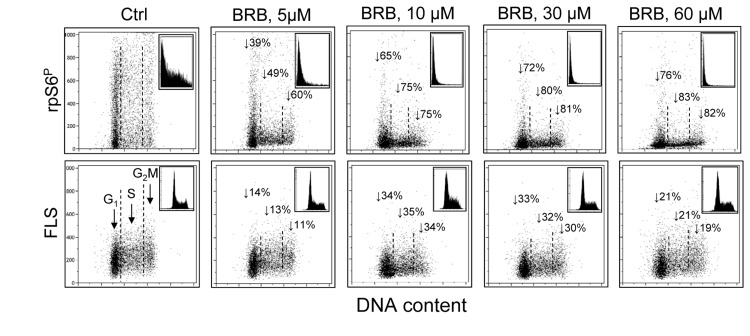
Figure 4. Suppression of rpS6 phosphorylation and reduction of size of human lymphoblastoid TK6 cells grown in the presence of BRB at 5 μM - 60 μM concentration Exponentially growing TK6 cells were untreated (Ctrl) or treated with BRB at concentrations as shown, for 24 h. Top panels: the bivariate distributions of rpS6P expression versus DNA content. Figures show percent decrease in expression of the mean rpS6P for cells at G1, S and G2M phases of the cell cycle, respectively, growing in the presence of BRB vis-à-vis to the untreated cells. Insets show the frequency histograms of rpS6P expression for all cells in culture. Bottom panels: Bivariate distributions of cellular forward light scatter (FLS) versus DNA content. Percent decrease in of mean value of FLS, considered to represent cellular size [49,50], of G1, S and G2M of cells growing in the presence of BRB with respect to the untreated cells is shown with the arrows. Insets illustrate DNA content frequency histograms of cells from the respective cultures.
Interestingly, growth of TK6 cells in the presence of 5 - 60 μM of BRB for 24 h led to reduction of their size (Fig. 4, bottom panels). The reduction was estimated from analysis of the forward light scatter (FLS), which when measured by flow cytometry is considered to be a marker of cell size [49,50]. The diminished size of TK6 cells was evident already at 5 μM concentration of BRB and it was approximately of similar extent for cells in different phases of the cell cycle. The cell size reduction was more extensive at 10 μM and 30 μM than at 5 μM BRB concentration. No significant changes in the cell cycle distribution were seen at these BRB concentrations, as reflected by the DNA content frequency histograms (insets).
BRB is fluorescent and its fluorescence and localization in mitochondria in live cells was initially reported by Borodina and Zelenin in 1977 [51]. Its fluorescence is maximally induced by UV light at 421-431 nm and emission is within a wide range of green to yellow (514-555 nm) wavelength [52]. In binding to mitochondria BRB has an affinity to respiratory electron transport chain and the extent of its binding appears to correlate with mitochondrial potential ΔΨm [53-55]. This is in analogy to another mitochondrial probe rhodamine123 (Rh123) which is a widely used marker of mitochondrial energizing [56,57]. We have presently observed that 60 min exposure of A549 cells to BRB distinctly labels mitochondria (Fig. 6). This can be seen however for only for short period of time after exposure to blue light (< 3 min) or even shorter (< 1 min) to UV light. With longer exposures the BRB fluorescence disappears from mitochondria and undergoes translocation to nuclei and nucleoli (Fig. 5). It should be noted that BRB fluorescence was not apparent in cells that were fixed and subsequently rinsed, and thus it did not interfere with the subsequent measurement of fluorescent markers reporting mTOR/S6- and DNA damage- signaling.
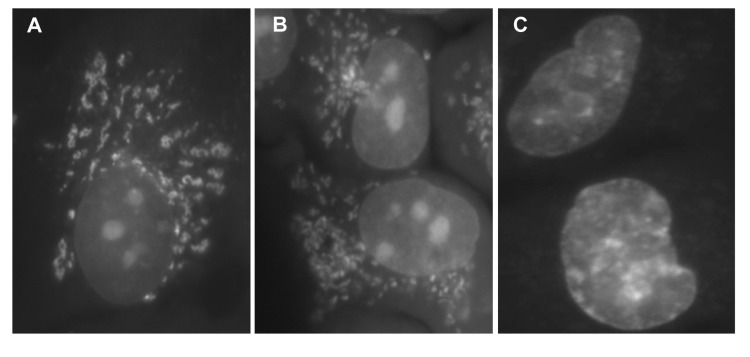
Figure 5. Detection of BRB fluorescence in live A549 cells exposed to this isoquinoline Exponentially growing A549 cells were treated with 5 μM BRB for 60 min, rinsed with PBS and examined under fluorescence microscopy. Immediately after illumination (the first min after exposure to UV) the BRB yellow fluorescence was localized almost exclusively in mitochondria (A). With extended time of illumination (2 min) intensity of fluorescence of mitochondria declined while nuclear and nucleolar fluorescence become more apparent (B). After 5 min exposure to UV no mitochondrial fluorescence was evident whereas intensity of nuclear and nucleolar fluorescence was distinctly increased (C). Images taken with the Nikon Microphot FXA; objective Fluor 40X.
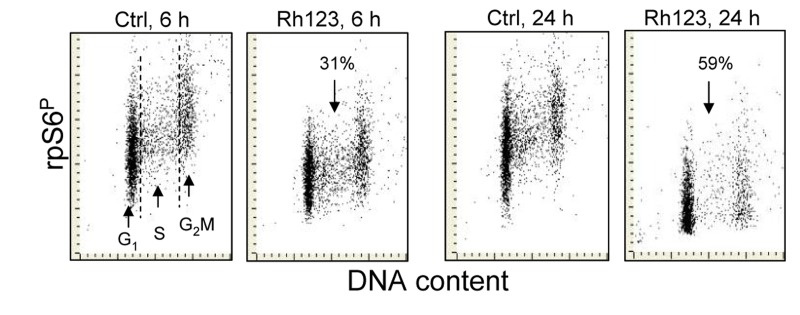
Figure 6. Effect of treatment of A549 cells with Rh123 on the level of expression of phosphorylated rpS6 Exponentially growing A549 cells were treated for 6 h or 24 h with 1 μM Rh123. Expression of rpS6P in cytoplasm was detected by phosphospecific Ab and measured by the iCys laser scanning cytometry. The figures indicate percent decree in expression of RP-S6P in the Rh123 treated cells vis-à-vis the respective control (Ctrl) cells.
The evidence in literature [53-55] and our observation (Fig. 5) indicate that in live cells BRB localizes in mitochondria where most likely it interferes with the electron transport chain. It may be suspected therefore that its presently measured effect namely attenuation of phosphorylation of rpS6 may be mediated by targeting electron transport in mitochondria. We have tested therefore the effect of a classical mitochondrial potential probe Rh123 [56,57] on rpS6 phosphorylation. As is evident from the data in Fig. 6 exposure of A549 cells in the presence of Rh123 leads to a decline in the level of constitutive phosphorylation of rpS6. The effect is not cell cycle-phase specific but related to time of exposure to Rh123, as 31% decrease is noted after 6 h and 59% after 24 h of treatment with Rh123, respectively.
Discussion
Potential gero-suppressive properties of berberine
Several elements of stress-induced premature cellular senescence are relevant to aging. The key element, considered to be the major driving force of aging, is persistent activation of mTOR and its downstream target rpS6 kinase (p70S6 kinase; p70S6K), a serine/threonine kinase that phosphorylates rpS6. Phosphorylation of rpS6 induces protein synthesis at the ribosome [27,36,38]. When equilibrium between protein synthesis and its degradation (including by autophagy) is broken and synthesis prevails, cells become hypertrophic acquiring senescent (“growth imbalance”) phenotype, the phenomenon described nearly five decades ago [59]. There is an extensive and rapidly accumulating evidence in support of this mechanism as the primary inducer of premature cellular senescence as well as major contributor to organismal aging [20-28,59-63]. As mentioned in the Introduction we have tested seven different potential gero-suppressive agents in terms of their ability to attenuate the level of constitutive mTOR/S6 signaling in normal cell types and cell lines. Each of these agents was quite effective in causing the decline of this signaling [33]. Interestingly, they all also reduced the level of constitutive DNA damage response, considered to be a reporter of DNA damage by endogenous oxidants [30-32]. In continuation of these studies we initiated to assess whether these agents can modulate the induction of premature cellular senescence. The senescence was instigated by low level of persistent DNA damage maintained by cell exposure to DNA-targeting drugs mitoxantrone [39] or mitomycin C [40]. Such treatment was seen to induce replication stress manifesting by low level of DNA replication activity combined with the induction of DNA damage signaling viz. ATM activation and H2AX phosphorylation, and development of characteristic features of cellular senescence [39,40,64,65]. In the pilot experiments we observed that one of the investigated gero-suppressive agents, namely BRB, was particularly effective in attenuation of premature cell senescence. The present studies were designed to investigate this phenomenon in more detail, using BRB concentrations that are relevant to its potential pharmacological in vivo doses [45-48].
The present results clearly indicate that administration of BRB into cultures of A549 cells undergoing premature senescence reduced the development of senescent phenotype as revealed by analysis of cells morphometric features, activation of SA-β-gal and induction of CDK inhibitor p21WAF1. BRB also attenuated the level of mTOR/S6 signaling by lowering the level of phosphorylation of rpS6, as well as expression of γH2AX. All these effects were BRB-concentration-dependent and already evident at its lowest, 5 μM concentration. In parallel experiments, we observed that BRB also decreased rpS6 phosphorylation and reduced the size of TK6 cells (Fig. 4). In the prior study 60 μM BRB was seen to attenuate the level of constitutive mTOR/S6 and DNA damage signaling, and to reduce both the mitochondrial potential (ΔΨm) as well as the abundance of ROS [33]. Since as mentioned, the mechanisms of stress-induced cellular senescence and aging have much in common, collectively these data suggest that BRB can be effective at an in vivo achievable concentration [45-48,66,67] as a gero-suppressive agent.
The mechanism by which BRB exerts these effects may be through targeting mitochondria. Its localization in mitochondria was reported before [53-55] and presently shown in the case of A549 cells (Fig. 5). BRB in mitochondria is photolabile; even the short exposure to UV light results in a loss of its mitochondrial localization and apparent translocation into nuclei. The specific target appears to be the respiratory electron transport chain [53,54]; inhibition of the electron transport results in a decrease of ATP production which leads to an increase of AMP to ATP ratio which in turn triggers the AMP-activated protein kinase (AMPK) [68]. The inhibition of mTOR/S6 signaling, the event presently observed (Fig. 3), is one of the key effects of AMPK activation [27,28,36-38]. This mechanism is essentially identical to that induced by metformin, which also targets electron transport in complex 1 of mitochondria and in this way activates AMPK [69]. In fact, among its many clinical applications BRB, similar to metformin, has been promoted as an anti-diabetic supplement [70-72]. Since metformin was shown to extend lifespan of C. elegans [73,74] and even rodents [15,75,76] (although not of Drosophila [77]), it is reasonable to expect that BRB may demonstrate gero-suppressive properties as well.
In the present study we observed that exposure of A549 cells to the classic cationic mitochondrial probe Rh123, which also targets electron transport chain and monitors mitochondrial electrochemical potential ΔΨm [56,57], led to reduction of rpS6 phosphorylation (Fig. 6). It is thus possible that inhibition of the mitochondrial respiratory chain by other modalities, via similar mechanism of activation of AMPK and mTOR inhibition as in the case of metformin or BRB, may have potential gero-suppressive properties as well. It should be noted however, that these potential gero-suppressive agents may differ in other properties and may have different side effects. Thus, for example, whereas rapamycin extends lifespan of various organisms including vertebrates [16,78], it does not ameliorate some traits of animal aging [79,80]. The possibility of induction of autophagy as an additional mechanism counteracting cellular senescence and providing anti-aging benefits should be estimated in parallel with the inhibitory activity on the mTOR/S6 signaling [81-83]. The choice of an agent or perhaps a combination of several agents differing in primary binding site and/or mechanism of action that, would have maximal gero-suppressive properties and minimal side effects, has to be explored to assess the advantages of their use for attenuation of aging processes. Their analysis in vitro such as exploring potential in preventing the premature, stress-induced, cellular senescence as presently shown in the case of BRB, may offer an advantage over in vivo experiments testing animals' longevity, by yielding the data rapidly and economically.
BRB has a long history of medicinal use in both Ayurvedic and old Chinese medicine. More recently, BRB has found wide application to treat a variety of different maladies. However, because it has been used primarily as a dietary nutritional supplement the evidence of its clinical toxicity or side effects such as is generally being obtained from well monitored clinical trials is scarce but forthcoming [72,84]. Interestingly, many applications at which BRB was reported to have positive health effects relate to age-related diseases [84-89], including metabolic and cardiovascular risks [84], type 2 diabetes [45-47,70-72], atherosclerosis [68], senile osteoporosis [87], Alzheimer's disease [88], hypercholesterolemia [89] and diabetes-induced renal inflammation [90]. It is tempting to speculate that this diversity of medical benefits reportedly provided by BRB, having one common denominator namely organismal aging, stems from the gero-suppressive properties of this isoquinoline, as presently detected. It should be noted, however that BRB [91,92], similar as metformin [93-95], was shown to exert anticancer properties, suppressing growth and/or sensitizing cancer cells to various other anticancer modalities.
In vitro assessment of gero-suppressive agents
Our prior studies [32-35,64] and the present results indicate that evaluation of effectiveness of potential anti-aging modalities can be achieved in vitro by monitoring their effect on mTOR/S6- and ROS-DNA damage signaling pathways that leads to attenuation of the stress-induced cellular senescence. Quantification of these effects is carried on using phospho-specific Abs that detect critical phosphorylation of the mTOR targets such as rpS6 and 4EBP1 concurrently with the level of ROS, ΔΨm, constitutive expression of γH2AX and activation of ATM, all measured in individual cells by multiparametric flow- or laser scanning- cytometry.
Fig. 7 presents key pathways associated with cellular senescence and aging linking mTOR/S6- and DNA damage- signaling, the targets of potential gero-suppressive agents that can be assessed this way. Reduction of signals activating m-TOR (raptor) pathway such as mitogens, growth factors and amino acids, triggering MAPK, Rsgs, MAP4K3, RaIA and hVps34, provide the outmost target for gero-suppression. These signals are suppressed by 2-deoxyglucose and other calorie restriction mimetics as well as by inhibitors of growth factors, primarily of IGF-1. Downstream of these signals, the mTOR activation is directly suppressed by its specific inhibitor rapamycin, and indirectly by activation of AMP-PK. Among effective activators of AMP-PK is metformin, as well as BRB. While the gero-suppressive effects of metformin are well documented [15,16] the present data demonstrate strongly suppressive effect of BRB on induction of the stress-induced premature cellular senescence. As mentioned, activation of AMP-PK can be achieved by targeting mitochondrial energy production, as it was shown with Rh123 (Fig. 6).
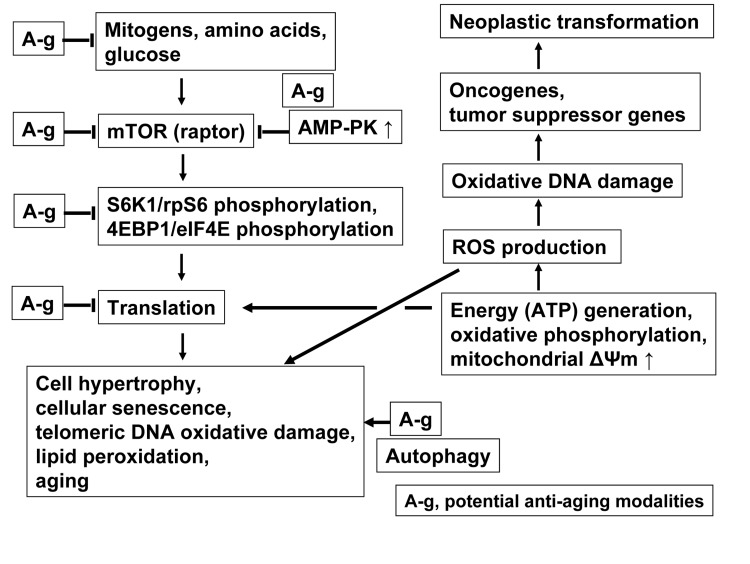
Figure 7. Schematic presentation of key pathways associated with cellular senescence and aging linking mTOR- and DNA damage- signaling The ongoing translation particularly during perturbed cell cycle progression (replication stress), is considered to be the major factor leading to senescence. Suppression of translation that may have anti-aging effect can be achieved at several steps along the mTOR signaling pathway (marked A-g), as discussed in the text. Activation of autophagy provides an additional gero-suppressive effect. The translation requires production of ATP and thus generates ROS that cause oxidative DNA damage, which when occurs at sites of oncogenes and tumor suppressor genes, may lead to neoplasia. The damage of telomeric DNA and lipid peroxidation by ROS further contributes to the senescent phenotype. The in vitro model of stress-induced cellular senescence as presently described can be used to evaluate potential gero-suppressive agents in terms of their effect in reduction of mTOR/S6- and DNA- damage signaling. See text for further details.
Since the increased rate of translation driven by rpS6 phosphorylation is the primary motor of cell growth in size (hypertrophy, “growth imbalance”, which is a characteristic phenotype of senescence) it is expected that inhibition of the translation rate through other means, downstream of mTOR activation, can also have gero-suppressive effects. For example, mimetics of amino acids that are not incorporated into protein but lower the rate of translation may have similar gero-suppressive effect. Likewise, reduction of ribosomal activity by such means as modification of the mRNA with the mRNA 5'-cap analogs [97, 98] or other approaches [99] may be gero-suppressive as well. Attenuation of the senescence phenotype that may be independent of translation is mediated by autophagy (Fig. 7) [100-102]. Specific inducers of autophagy, therefore, may have an additive effect with the mTOR inhibitors in terms of suppression of aging. However, most mTOR inhibitors are also activators of autophagy [100-102].
Our observation that the gero-suppressive agents inhibit constitutive level of mTOR activation concurrently with reduction of DNA damage signaling [33] provides evidence of a direct linkage between mTOR activation and oxidative DNA damage (Fig. 7). This is expected since translation requires production of energy (ATP) through oxidative phosphorylation which generates ROS; the increased ROS production (translation) leads to a greater oxidative DNA damage. It should be noted however that when ROS induce damage to telomeric sections of DNA this has an effect in promoting replicative senescence (Fig. 7). Likewise, the ROS-mediated lipid peroxidation is another marker of cellular senescence and aging.
Whereas mTOR/S6 signaling is the primary basis for induction of premature cell senescence and aging the endogenous ROS when cause DNA damage at sites coding for oncogenes or tumor suppressor genes predispose to neoplastic transformation (Fig. 7). Anti-oxidants (ROS scavengers) therefore are expected to be more effective in terms of chemo-prevention rather than as anti-aging modalities, and this indeed appears to be the case [103-106]. The attempts to attenuate aging processes including the increase in organismal longevity by antioxidants were largely unsuccessful [reviewed in 20]. On the other hand, most gero-suppressive agents were shown to have chemopreventive properties as well [106-112].
Materials and Methods
Cells, Cell Treatment
Human non-small cell lung carcinoma A549 cells, obtained from American Type Culture Collection (ATCC; Manassas, VA), were grown in Ham's F-12K Nutrient Mixture (Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (GIBCO/BRL; Grand Island, NY) in 25 ml FALCON flasks (Becton Dickinson Co., Franklin Lakes, NJ) at 37.5 °C in an atmosphere of 95 % air and 5% CO2. The cells were maintained in exponential and asynchronous phase of growth by repeated trypsinization and reseeding prior to reaching sub-confluency. The cells were then trypsinized and seeded at about 1×104 cells per chamber in 2-chambered Falcon Culture Slides (Beckton Dickinson). To induce cellular senescence A549 cells were treated with 2 nM mitoxantrone (Mxt; Sigma-Aldrich, St. Louis, MO) as described before [39]. Concurrently with Mxt berberine (BRB; Sigma-Aldrich) was included into cultures at a final concentration as shown in Figure legends. At the end of incubation medium from each chamber was aspirated, 1 ml of 1% methanol-free formaldehyde in phosphate buffered saline (PBS) was added and the cells fixed by gently rocking the slides at room temperature for 15 min. Following aspiration of the formaldehyde the chamber slides were disassembled and the slides submerged in 70% ethanol. The fixed slides were stored at 4°C before staining and analysis. Human lymphoblastoid TK6 cells, kindly provided by Dr. Howard Liber [96], were maintained in suspension in RPMI 1640 medium (GIBCO/Life Technologies) supplemented with L-glutamine (2 mM) and fetal bovine serum (10%), as described [33-35]. These cells were also exposed to 1 μM rhodamine 123 (Rh123; Sigma-Aldrich) to be assessed for expression of rpS6P and forward light scatter by flow cytometry. Other details on cultures' treatment are presented in figure legends.
Detection of γH2AX, rpS6P, p21WAF1 and activity of senescence-associated β-galactosidase
After fixation the cells were washed twice in PBS and with 0.1% Triton X-100 (Sigma-Aldrich) in PBS for 15 min and with a 1% (w/v) solution of bovine serum albumin (BSA; Sigma-Aldrich in PBS for 30 min to suppress nonspecific antibody (Ab) binding. The cells were then incubated in 1% BSA containing a 1:300 dilution of phospho-specific (Ser139) γH2AX mAb (Biolegend, San Diego, CA) and/or with a 1: 200 dilution of phosphospecific (Ser235/236), rpS6 Ab (Epitomics, Burlingame, CA) or 1;100 dilution of p21WAF1 Ab (Cell Signaling, Danvers, MA) at 4°C overnight. The secondary Ab was tagged either with AlexaFluor 488 or 647 fluorochrome (Invitrogen/Molecular Probes, used at 1:100 dilution in 1% BSA). The incubation was at room temperature for 45 min. Cellular DNA was counterstained with 2.8 μg/ml 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) at room temperature for 15 minutes. Activity of senescence-associate β-galactosidase (SA-β-gal) was detected using the protocol provided by Chemicon's® Cellular Senescence Assay Kit (Millipore, Billerica, MA). After adding of Antifade (for fluorescence-labeled cells) or 50% glycerol in PBS (for (SA-β-gal) the slides were subjected to quantitative study by laser scanning cytometry. The protocol for measuring the chromatic (SA-β-gal) dye staining was provided by CompuCyte (Westwood, MA). The fixation, rinsing and labeling of A549 cells was carried out on slides, and TK6 cells in suspension. Other details have been described previously [64,112].
Analysis of Cells by Cytometry
A549 cells: Cellular immunofluorescence representing the binding of the respective phospho-specific Abs as well as the blue emission of DAPI stained DNA was measured by laser scanning cytometer [39-41] iCys (CompuCyte) utilizing standard filter settings; fluorescence was excited with 488-nm argon, helium neon (633 nm) and violet (405 nm) lasers. Intensities of maximal pixel and integrated fluorescence were measured and recorded for each cell. At least 3,000 cells were measured per sample. Gating analysis was carried out as described in Figure legends. TK6cells: Intensity of cellular fluorescence was measured using a MoFlo XDP (Beckman-Coulter, Brea, CA) high speed flow cytometer/sorter. DAPI fluorescence was excited with the UV laser (355-nm), AlexaFluor 488, DCF and Rh123 with the argon ion (488-nm) laser. Although BRB is fluorescent [Fig. 5] it was not retained by the cells following their fixation and repeated washings and control experiments excluded the possibility that its fluorescence significantly contributed to analysis of the measured cells that could lead to a bias. Analysis of forward light scatter by flow cytometry provides information on cell size [49,50]. All experiments were repeated at least three times, representative data are presented. Other details of the particular experimental procedures were described before [64,112].
Acknowledgments
Supported by: NCI, CA RO1 28704 and Robert A. Welke Cancer Research Foundation.
Conflicts of Interest
The authors have no conflicting interest to declare.
References
- 1. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37: 614 -636. [PubMed] .
- 2. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345: 458 -460. [PubMed] .
- 3. Kuilman T, Michaloglou C, Mooi W, Peeper DS. The essence of senescence. Genes Dev. 2010; 24: 2463 [PubMed] .
- 4. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5: 741 [PubMed] .
- 5. Sherr CJ and DePinho RA. Cellular senescence: Mitotic clock or culture shock? Cell. 2000; 102: 407 [PubMed] .
- 6. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593 [PubMed] .
- 7. Chen Z, Trotman LC, Shaffer D, Lin H-K, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005; 436: 725 [PubMed] .
- 8. Gerwitz DA, Holt SE, Elmore LW. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol. 2008; 76: 947 [PubMed] .
- 9. Litwiniec A, Grzanka A, Helmin-Basa A, Gackowska L, Grzanka D. Features of senescence and cell death induced by doxorubicin in A549 cells: Organization and level of selected cytoskeletal proteins. J Canc Res Clin Oncol. 2010; 36: 717 .
- 10. Banito A and Gil J. Induced pluripotent stem cells and senescence: Learning the biology to improve the technology. EMBO Reports. 2010; 11: 353 [PubMed] .
- 11. Sikora E, Arendt T, Bennett M, Narita M. Impact of cellular senescence signature on ageing research. Ageing Res Rev. 2011; 10: 146 -152. [PubMed] .
- 12. Zou H, Stoppani E, Volonte D, Galbiati F. Caveolin-1, cellular senescence and age-related diseases. Mech Ageing Dev. 2011; 132: 533 -542. [PubMed] .
- 13. Salminen A and Kaarniranta K. Control of p53 and NF-κB signaling by WIP1 and MIF: role in cellular senescence and organismal aging. Cell Signal. 2011; 23: 747 [PubMed] .
- 14. Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging. Oncogene. 2013; Epub Feb 18. .
- 15. Anisimov VN, Berstein LM, Popovich IG, Zabezhinski MA, Egormin PA, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Kovalenko IG, Poroshina TE. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY). 2011; 3: 148 -157. [PubMed] .
- 16. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092 [PubMed] .
- 17. Barzilai A and Yamamoto K. DNA damage responses to oxidative stress. DNA repair (Amst). 2004; 3: 1109 [PubMed] .
- 18. Beckman KB and Ames BN. Oxidative decay of DNA. J Biol Chem. 1997; 272: 13300 -13305. .
- 19. Karanjawala ZE and Lieber MR. DNA damage and aging. Mech Ageing Dev. 2004; 125: 405 [PubMed] .
- 20. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 21. Blagosklonny MV. mTOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 [PubMed] .
- 22. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 [PubMed] .
- 23. Cabreiro F, Ackerman D, Doonan R, Araiz C, Back P, Papp D, Braeckman BP, Gems D. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med. 2011; 51: 1575 [PubMed] .
- 24. Lapointe J and Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2009; 67: 1 -8. [PubMed] .
- 25. Hands SL, Proud CG, Wyttenbach A. mTOR's role in ageing: protein synthesis or autophagy? Aging (Albany). 2009; 1: 586 .
- 26. Blagosklonny MV and Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany). 2009; 1: 357 .
- 27. Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signaling networks. Biochem J. 2012; 441: 1 [PubMed] .
- 28. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2010; 12: 21 [PubMed] .
- 29. Burhans WC and Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 33: 7545 [PubMed] .
- 30. Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006; 29: 495 [PubMed] .
- 31. Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006; 5: 1940 [PubMed] .
- 32. Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, Darzynkiewicz Z. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007; 71A: 905 -914. [PubMed] .
- 33. Halicka HD, Zhao H, Li J, Lee Y-S, Hsieh T-C, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive DNA mage and mTOR- signaling. Aging (Albany NY). 2012; 4: 952 -965. [PubMed] .
- 34. Halicka HD, Zhao H, Li J, Traganos DF, Studzinski G, Darzynkiewicz Z. Attenuation of constitutive DNA damage signaling by1,25-dihydroxyvitamin D3. Aging (Albany). 2012; 4: 270 -278. .
- 35. Halicka HD, Zhao H, Li J, Traganos F, Zhang S, Lee M, Darzynkiewicz Z. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging (Albany). 2011; 3: 1028 .
- 36. Hay N and Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004; 1926 -1945. [PubMed] .
- 37. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471 [PubMed] .
- 38. Ma XM and Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009; 10: 307 [PubMed] .
- 39. Zhao H, Halicka HD, Jorgensen E, Traganos F, Darzynkiewicz Z. New biomarkers probing the depth of cell senescence assessed by laser scanning cytometry. Cytometry A. 2010; 77A: 999 -1007. [PubMed] .
- 40. McKenna E, Traganos F, Zhao H, Darzynkiewicz Z. Persistent DNA damage caused by low levels of mitomycin C induces irreversible senescence of A549 cells. Cell Cycle. 2012; 12: 3132 [PubMed] .
- 41. Pozarowski P, Holden E, Darzynkiewicz Z. Laser scanning cytometry: Principles and applications. An update. Meth Molec Biol. 2013; 913: 187 .
- 42. Rodier F and Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192: 547 [PubMed] .
- 43. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010; 24: 2463 [PubMed] .
- 44. Kapuscinski J and Darzynkiewicz Z. Interactions of antitumor agents ametantrone and mitoxantrone (novantrone) with double-stranded DNA. Biochem Pharmacol. 1985; 34: 4203 [PubMed] .
- 45. Guo Y, Pope C, Cheng X, Zhou H, Klaassen CD. Dose-response of berberine on hepatic cytochromes P450 mRNA expression and activities in mice. J Ethnopharmacol. 2011; 138: 111 [PubMed] .
- 46. Liu L, Yu YL, Yang JS, Li Y, Liu YW, Liang Y, Liu XD, Xie L, Wang GJ. Berberine suppresses intestinal disaccharidases with beneficial metabolic effects in diabetic states, evidences from in vivo and in vitro study. Naunyn Schmiedebergs Arch Pharmacol. 2010; 381: 371 [PubMed] .
- 47. Cicero AF and Tartagani E. Antidiabetic properties of berberine: from cellular pharmacology to clinical effects. Hosp Pract (Minneap). 2012; 40: 56 .
- 48. Xu LN, Lu BN, Hu MM, Xu YW, Han X, Qi Y, Peng JY. Mechanisms involved in the cytotoxic effects of berberine on human colon cancer HCT-8 cells. Biocell. 2012; 36: 113 -20. [PubMed] .
- 49. Wlodkowic D, Telford W, Skommer J, Darzynkiewicz Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol. 2011; 103: 55 [PubMed] .
- 50. Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992; 13: 795 [PubMed] .
- 51. Borodina VM and Zelenin AV. Fluorescence microscopy demonstration of mitochondria in tissue culture cells using berberine. Tsitologiia. 1977; 19: 1067 [PubMed] .
- 52. Díaz MS, Freile ML, Gutiérrez MI. Solvent effect on the UV/Vis absorption and fluorescence spectroscopic properties of berberine. Photochem Photobiol Sci. 2009; 8: 970 -974. [PubMed] .
- 53. Mikes V and Dadák V. Berberine derivatives as cationic fluorescent probes for the investigation of the energized state of mitochondria. Biochim Biophys Acta. 1983; 723: 231 [PubMed] .
- 54. Mikes V and Yaguzhinskij LS. Interaction of fluorescent berberine alkyl derivatives with respiratory chain of rat liver mitochondria. J Bioenerg Biomembr. 1985; 17: 23 [PubMed] .
- 55. Turner N, Li J-Y, Gosby A, To SWC, Cheng Z, Myoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, Hu L-H, Li J, Ye J-M. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I. Diabetes. 2008; 57: 1414 [PubMed] .
- 56. Darzynkiewicz Z, Staiano-Coico L, Melamed MR. Increased mitochondrial uptake of rhodamine 123 during lymphocyte stimulation. Proc Natl Acad Sci USA. 1981; 78: 2383 [PubMed] .
- 57. Darzynkiewicz Z, Traganos F, Staiano-Coico L, Kapuscinski J, Melamed MR. Interactions of rhodamine 123 with living cells studied by flow cytometry. Cancer Res. 1982; 42: 799 [PubMed] .
- 58. Cohen LS and Studzinski GP. Correlation between cell enlargement and nucleic acid and protein content of HeLa cells in unbalanced growth produced by inhibitors of DNA synthesis. J Cell Physiol. 1967; 69: 331 [PubMed] .
- 59. Blagosklonny MV. mTOR-driven quasi-programmed aging as a disposable soma theory: Blind watchmaker vs. intelligent designer. Cell Cycle. 2013; 12: 1842 [PubMed] .
- 60. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 [PubMed] .
- 61. Demidenko ZN and Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009; 8: 1901 [PubMed] .
- 62. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009; 8: 1896 [PubMed] .
- 63. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 [PubMed] .
- 64. Zhao H and Darzynkiewicz Z. Biomarkers of cell senescence assessed by imaging cytometry. Meth Molec Biol. 2013; 965: 83 .
- 65. Seifrtova M, Havelek R, Soukup T, Filipova A, Mokry J, Rezacova M. Mitoxantrone ability to induce premature senescence in human dental pulp stem cells and human dermal fibroblasts. J Physiol Pharmacol. 2013; 64: 255 [PubMed] .
- 66. Zhou Y, He P, Liu A, Zhang L, Liu Y, Dai R. Drug-drug interactions between ketoconazole and berberine in rats: pharmacokinetic effects benefit pharmacodynamic synergism. Phytother Res. 2012; 26: 772 [PubMed] .
- 67. Zhou H and Mineshita S. The effect of berberine chloride on experimental colitis in rats in vivo and in vitro. J Pharmacol Exp Ther. 2000; 294: 822 [PubMed] .
- 68. Wang Q, Zhang M, Liang B, Shirwany N, Zhu Y, Zou MH. Activation of AMP-activated protein kinase is required for berberine-induced reduction of atherosclerosis in mice: the role of uncoupling protein 2. PLoS One. 2011; e25436 [PubMed] .
- 69. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000; 348: 607 [PubMed] .
- 70. Lee YS, Kim WS, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, Hohnen-Behrens C, Gosby A, Kraegen EW, James DE, Kim JB. Berberine, a natural plant product, activates AMP-Activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006; 55: 1006: 2256 -2264. [PubMed] .
- 71. Shen N, Huan Y, Shen ZF. Berberine inhibits mouse insulin gene promoter through activation of AMP activated protein kinase and may exert beneficial effect on pancreatic β-cell. Eur J Pharmacol. 2012; 694: 120 [PubMed] .
- 72. Dong H, Wang N, Zhao L, Lu F. Berberine in the treatment of type 2 diabetes mellitus: a systemic review and meta-analysis. Evid Based Complement Alternat Med. 2012; 2012:591654. Epub 2012 Oct 15. .
- 73. Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene ND, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013; 153: 228 [PubMed] .
- 74. Onken B and Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE. 2010; 5: e8758 [PubMed] .
- 75. Anisimov VN, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Egormin PA, Yurova MV, Rosenfeld SV, Semenchenko AV, Kovalenko IG, Poroshina TE, Berstein LM. Gender differences in metformin effect on aging, life span and spontaneous tumorigenesis in 129/Sv mice. Aging (Albany NY). 2010; 2: 945 [PubMed] .
- 76. Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY). 2010; 2: 760 [PubMed] .
- 77. Slack C, Foley A, Partridge L. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS One. 2012; 7: e47699 Epub 2012 Oct 16. [PubMed] .
- 78. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460: 392 [PubMed] .
- 79. Wilkinson E, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012; 11: 675 -82. [PubMed] .
- 80. Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schröder S, Adler T, Afonso LC, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013; pii: 67674. [Epub ahead of print]. .
- 81. Nair S and Ren J. Autophagy and cardiovascular aging lesson learned from rapamycin. Cell Cycle. 2012; 11: 2092 [PubMed] .
- 82. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011; 146: 682 [PubMed] .
- 83. Leontieva OV and Blagosklonny MV. Hypoxia and gerosuppression: the mTOR saga continues. Cell Cycle. 2012; 11: 3926 -3931. [PubMed] .
- 84. Derosa G, Maffioli P, Cicero AF. Berberine on metabolic and cardiovascular risk factors: an analysis from preclinical evidences to clinical trials. Expert Opin Biol Ther. 2012; 12: 1113 [PubMed] .
- 85. Yu HH, Kim KJ, Cha JD, Kim HK, Lee YE, Choi NY, You YO. Antimicrobial activity of berberine alone and in combination with ampicillin or oxacillin against methicillin-resistant Staphylococcus aureus. Journal of Medicinal Food. 2005; 8: 454 [PubMed] .
- 86. Kuo CL, Chi CW, Liu TY. The anti-inflammatory potential of berberine in vitro and in vivo. Cancer Lett. 2004; 203: 127 [PubMed] .
- 87. Li H, Miyahara T, Tezuka Y, Tran OL, Seto H, Kadota S. Effect of berberine on bone mineral density in SAMP6 as a senile osteoporosis model. Biol Pharma Bull. 2003; 26: 130 .
- 88. Ki HF and Shen L. Berberine: a potential multipotent natural product to combat Alzheimer's Disease. Molecules. 2011; 16: 6732 [PubMed] .
- 89. Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, Wang Y, Wang Z, Si S, Pan H, Wang S, Wu J, Wang Y, Li Z, Liu J, Jiang J-D. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nature Medicine. 2004; 10: 1344 -1351. .
- 90. Xie X, Chang X, Chen L, Huang K, Huang J, Wang S, Shen X, Liu P, Huang H. Berberine ameliorates experimental diabetes-induced renal inflammation and fibronectin by inhibiting the activation of RhoA/ROCK signaling. Mol Cell Endocrinol. 2013; Epub Jul 26 .
- 91. Chappell WH, Abrams SL, Franklin RA, LaHair MM, Montalto G, Cervello M, Martelli AM, Nicoletti F, Candido S, Libra M, Polesel J, Talamini R, Milella M, Tafuri A, Steelman LS, McCubrey JA. Ectopic NGAL expression can alter sensitivity of breast cancer cells to EGFR, Bcl-2, CaM-K inhibitors and the plant natural product berberine. Cell Cycle. 2012; 11: 4447 -4461. [PubMed] .
- 92. Cai Y, Xia Q, Luo R, Huang P, Sun Y, Shi Y, Jiang W. Berberine inhibits the growth of human colorectal adenocarcinoma in vitro and in vivo. J Nat Med. 2013; [Epub ahead of print] .
- 93. Würth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, Fenoglio D, Sambuceti G, Filaci G, Daga A, Barbieri F, Florio T. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle. 2013; 12: 145 [PubMed] .
- 94. Menendez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2012; 11: 2782 [PubMed] .
- 95. Pan J, Chen C, Jin Y, Fuentes-Mattei E, Velazquez-Tores G, Benito JM, Konopleva M, Andreeff M, Lee MH, Yeung SC. Differential impact of structurally different anti-diabetic drugs on proliferation and chemosensitivity of acute lymphoblastic leukemia cells. Cell Cycle. 2012; 11: 2314 [PubMed] .
- 96. Schwartz JL, Jordan E, Evans HH, Lenarczyk M, Liber H. The TP53 dependence of radiation-induced chromosome instability in human lymphoblastoid lines. Radiat Res. 2003; 159: 730 [PubMed] .
- 97. Zuberek J, Kubacka D, Jablonowska A, Jemielity J, Stepinski J, Sonenberg N, Darzynkiewicz E. Weak binding affinity of human 4EHP for mRNA cap analogs. RNA. 2007; 13: 691 [PubMed] .
- 98. Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, Biffo S, Merrick WC, Darzynkiewicz E, Pillai RS, Filipowicz W, Duchaine TF, Sonenberg N. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science. 2007; 317: 1764 [PubMed] .
- 99. Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenohabditis elegans. Ageing Cell. 2007; 6: 111 .
- 100. Blagosklonny MV. Hypoxia, mTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013; 9: 260 [PubMed] .
- 101. Nair S and Ren J. Autophagy and cardiovascular aging: lesson learned from rapamycin. Cell Cycle. 2012; 11: 2092 [PubMed] .
- 102. Bennetzen MV, Mariño G, Pultz D, Morselli E, Færgeman NJ, Kroemer G, Andersen JS. Phosphoproteomic analysis of cells treated with longevity-related autophagy inducers. Cell Cycle. 2012; 11: 1827 [PubMed] .
- 103. Blagosklonny MV. Rapalogs in cancer prevention: anti-aging or anticancer? Cancer Biol Ther. 2012; 13: 1349 -1354. [PubMed] .
- 104. Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, Pavlides S, Lamb R, Sneddon S, Howell A, Sotgia F, Lisanti MP. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle. 2012; 11: 4402 [PubMed] .
- 105. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle. 2012; 11: 4402 [PubMed] .
- 106. Thapa D and Ghosh R. Antioxidants for prostate cancer chemoprevention: challenges and opportunities. Biochem Pharmacol. 2012; 83: 1319 [PubMed] .
- 107. Li D. Metformin as an antitumor agent in cancer prevention and treatment. J Diabetes. 2011; 3: 320 [PubMed] .
- 108. Buitrago-Molina LE and Vogel A. mTOR as a potential target for the prevention and tratment of hepatocellular carcinoma. Curr Cancer Drug Target. 2012; Epub .
- 109. Kajser J. Will an aspirin a day keep cancer away? Science. 2012; 337: 1471 -1473. [PubMed] .
- 110. Anis KV, Rajeshkumar NV, Kuttan R. Inhibition of chemical carcinogenesis by berberine in rats and mice. J Pharm Pharmacol. 2001; 53: 763 [PubMed] .
- 111. Zhu Z, Jiang W, McGinley JN, Thompson HJ. 2-Deoxyglucose as an energy restriction mimetic agent: effects on mammary carcinogenesis and on mammary tumor cell growth in vitro. Cancer Res. 2005; 65: 7023 [PubMed] .
- 112. Juan ME, Alfaras I, Planas JM. Colorectal cancer chemoprevention by trans-resveratrol. Pharmacol Res. 2012; 65: 584 [PubMed] .
- 113. Darzynkiewicz Z, Zhao H, Halicka HD, Rybak P, Dobrucki J, Wlodkowic D. DNA damage signaling assessed in individual cells in relation to the cell cycle phase and induction of apoptosis. Crit Rev Clin Lab Sci. 2012; 49: 199 -217. [PubMed] .