



Introduction
Aging leads to a progressive decline of cell and tissue function and is the primary risk factor for many ailments, including the prevalent neurodegenerative disorders Alzheimer's disease (AD) and Parkinson's disease (PD). Mitochondria are the central hub of cellular metabolism and mitochondrial dysfunction, especially in stem cells [1], has been shown to cause the development of premature aging phenotypes in mice [2]. Paradoxically, slight inhibition of mitochondrial respiration can also lead to small increases in reactive oxygen species (ROS) production and extend the lifespan of yeast, C. elegans, Drosophila, and mice [3-6]. Even in young animals, roughly 0.15% of electrons passing through the mitochondrial electron transport chain (ETC) combine with molecular oxygen to form superoxide [7, 8]. Mitochondrial ROS production increases with age and leads to progressive damage of cellular macromolecules as outlined in the mitochondrial free radical theory of aging [9].
Dietary restriction (DR) increases the lifespan of many organisms including C. elegans [10]. As interest in the molecular mechanisms responsible for the effect of DR on lifespan have expanded, so has the discovery of the pathways involved and the search for DR mimetic compounds that promote survival and stress resistance [11, 12]. The ketone body beta-hydroxybutyrate (βHB) has been described as a DR mimetic compound [13], in part because it increases in the plasma during DR and when administered exogenously leads to decreased levels of oxidative stress [14]. In mammals, βHB is produced in the liver, primarily from the catabolic breakdown of fatty acids, and is used as an alternative energy source when blood glucose is low. This is especially important in the brain where only a very limited amount of fatty acid beta-oxidation takes place [15]. In mitochondria βHB is catabolized to acetoacetate by βHB dehydrogenase 1 (BDH1). The reaction is linked to the reduction of NAD to NADH, which fuels mitochondrial ETC complex I. The resulting acetoacetate is catabolized to acetoacetyl-CoA and then to acetyl-CoA, which is metabolized as part of the TCA cycle. The βHB dehydrogenase 2 (BDH2) enzyme localizes to the cytoplasm, but no changes in ketone body metabolism were found in BDH2 knockout mice suggesting BDH2 plays a limited role, if any, in ketone body metabolism [16].
Researchers are investigating DR mimetics not only for their possible lifespan extending capabilities, but also for their potential ability to delay the onset and progression of age-associated diseases such as AD. [17, 18]. Phenotypes of AD brain include extracellular senile plaques containing Aß peptide as well as intracellular neurofibrillary tangles consisting of hyper-phosphorylated tau protein [19]. Intracellular Aß can inhibit mitochondrial ETC complex IV and increase ROS production [20]. βHB has shown some efficacy in the protection against AD-mediated neurodegeneration in animal models and human trials. βHB protected cultured hippocampal neurons from Aß 1–42 toxicity [21]. βHB or a ketogenic diet has shown mixed effects on disease phenotypes in mouse models of AD. For example in one study, a ketogenic diet lowered Aß levels, but did not affect cognitive impairment [22]. In another study a ketogenic diet improved motor function, but did not affect cognition or tau or Aß pathology [23]. However, a further study found that supplementation of βHB methyl ester to AD mice was able to restore cognitive function and decrease Aß levels, likely due to the fact that the methyl ester is transported through the blood-brain barrier more efficiently than the free acid [24]. There is also evidence for the clinical use of ketone bodies to treat neurodegenerative disorders as oral ingestion of medium chain triglycerides, which are catabolized in part to ketone bodies, increased plasma levels of βHB and led to improved cognitive function in human patients with AD [25].
PD, another aging-associated disorder, is characterized by an accumulation of Lewy bodies in the substantia nigra region of the brain. The alpha-synuclein protein is a major component of Lewy bodies and can also localize to mitochondrial membranes [26] causing decreased ETC complex I activity with an accompanying increase in ROS production [27]. This may be partly responsible for the increased mitochondrial oxidative damage that has been observed in brains from autopsied PD patients [28, 29]. βHB has also been shown to be efficacious in several animal models of PD. Mice treated with βHB showed partial protection against neurodegeneration and motor deficiency induced by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which induces PD-like symptoms [30]. Additionally, βHB protected cultured neurons from toxicity induced by the structurally related ETC complex I inhibitor 1-methyl-4-phenylpyridinium (MPP(+)) [21]. In human clinical trials, PD patients treated with a ketogenic diet for one month improved their Unified Parkinson's Disease Rating Scale scores by a mean of 43% [31].
Although much is known about the effects of βHB on neurodegenerative and other aging-associated diseases, not much is known about its effects on aging. Moreover, the mechanisms through which βHB are protective are not entirely clear. However, recent evidence suggests that βHB protects against oxidative stress through its action as a class I and class IIa histone deacetylase inhibitor to increase expression of stress response genes such as FoxO3A and MT2 [14]. In this report we determined the effect of βHB on lifespan in C. elegans and determined the cytoprotective signaling pathways required for this effect. We then determined the effects of βHB on proteotoxicity in nematode models of AD, PD, and amyotrophic lateral sclerosis (ALS).
Results
D-beta-hydroxybutyrate extends the lifespan of C. elegans
Addition of 2, 10, or 20 mM DL-beta-hydroxybutyrate (βHB) to the culture medium of C. elegans feeding on heat-killed E. coli increased lifespan with 20 mM having the greatest effect, increasing mean lifespan by 26%, from 17.2 to 21.7 days (Figure 1A). 50 mM and 100 mM concentrations decreased lifespan (Supplementary Table 1). Therefore a 20 mM concentration was used in further experiments. When C. elegans were fed live E. coli, 20 mM βHB only extended mean lifespan by 14%, from 16.0 to 18.3 days. This is likely due to catabolism of a portion of the βHB by the bacteria. To determine if the lifespan extension was due to D-βHB or L-βHB, we performed lifespan experiments with each isomer separately and found that only D-βHB addition resulted in lifespan extension (Figure 1B).
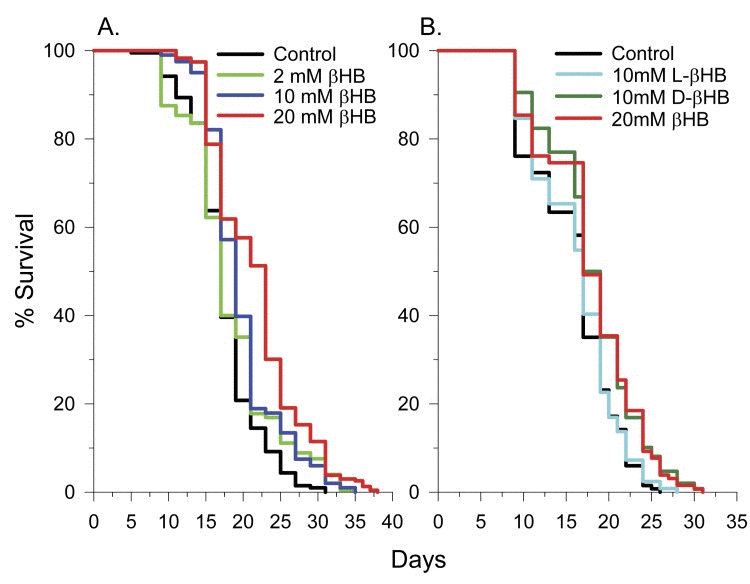
Figure 1. D-βHB extends the lifespan of N2 C. elegans worms. (A) Concentration dependency of βHB-mediated extension of lifespan. (B) D-βHB, but not L-βHB addition led to lifespan extension. When no D or L prefix is present, βHB refers to DL-βHB.
βHB or butyrate individually, but not when combined, extend the lifespan of C. elegans
The histone deacetylase (HDAC) inhibitors sodium butyrate and valproic acid have been shown to extend lifespan in C. elegans [32, 33]. Since βHB has a similar chemical structure as butyrate and since βHB has been shown to inhibit class I and IIa histone deacetylases (HDACs 1, 3, and 4) in mammals with a Ki of 2-5 mM [13], we determined if βHB could further extend the lifespan of sodium butyrate treated worms. As shown in Figure 2A, and as previously found by others [33], sodium butyrate extended lifespan, but strikingly the combination of sodium butyrate and βHB led to a slightly decreased lifespan. This data is consistent with the possibility that βHB is functioning as an HDAC inhibitor as HDAC inhibitors such as valproic acid are known to cause decreased lifespan at higher concentrations in C. elegans ([32] and Supplementary Table 1). The combination of sodium butyrate and βHB likely has an additive inhibitory effect on HDAC activity, thereby decreasing lifespan. βHB addition also decreased the lifespan of worms treated with valproic acid (Supplementary Table 1), likely through a similar mechanism.
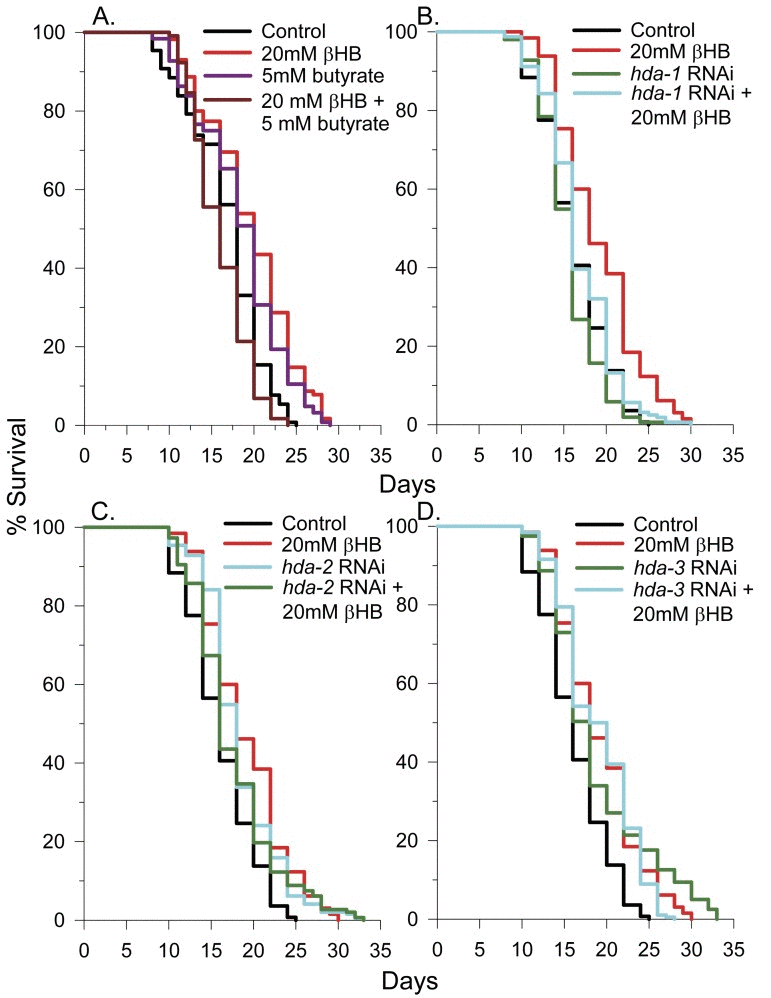
Figure 2. βHB-mediated HDAC inhibition plays a role in lifespan extension. (A) Survival of N2 worms in the presence of βHB, butyrate, or both compounds together. (B) Effects of hda-1, (C)hda-2, or (D)hda-3 RNAi knockdown on C. elegans lifespan in the presence or absence of 20 mM βHB.
Inhibition of HDA-2 and HDA-3 play a role in βHB-mediated lifespan extension
Many general HDAC inhibitors inhibit both class I and class II HDAC enzymes [13, 34]. The C. elegans genome has 3 class I HDACs, hda-1, hda-2, and hda-3. In addition there are 5 class II HDACs, hda-4, hda-5, hda-6, hda-10, and hda-11, with hda-4 being the only member of class IIa [35]. To determine if HDAC inhibition plays a role in βHB-mediated lifespan extension, we knocked down the 3 class I HDACs in C. elegans by RNAi in the presence or absence of βHB and determined the effect on lifespan. RNAi knockdown of hda-1 had no effect on lifespan and βHB addition extended lifespan, but to a lesser extent than in the absence of knockdown (Fig. 2B). However, RNAi knockdown of either hda-2 (Fig. 2C) or hda-3 (Fig. 2D) extended C. elegans lifespan, by 13% and 16% respectively, and prevented βHB from further extending lifespan. Therefore βHB likely extends lifespan in part through inhibition of hda-2 and hda-3. We also performed lifespan analysis using hda-2(ok1479), hda-3(ok1991), hda-4(ok518), and hda-10(ok3311) mutant worms (Supplementary Figure 1A-D). All of the HDAC mutant strains had roughly 30% decreased mean lifespans indicating that a partial knockdown, but not full knockout of HDA-2 or HDA-3 activity promotes lifespan extension. Consistent with the RNAi knockdown results, βHB addition did not extend the lifespan of the hda-2 or hda-3 mutant animals. However, βHB addition did lead to lifespan extension in the hda-4 and hda-10 mutants, suggesting βHB primarily extends lifespan through inhibiting the class I HDACs in C. elegans.
F55E10.6 is likely a D-βHB inducible βHB dehydrogenase gene
There is no strong homolog of human mitochondrial BDH1 in C. elegans. However, a BLASTP search identified 4 candidate genes with 38-41% protein sequence identity with BDH1 (dhs-2, dhs-20, dhs-16 (a 3-hydroxysteroid dehydrogenase [36]), and F55E10.6 (similar to human microsomal retinol dehydrogenase [37] and hydroxysteroid dehydrogenases [38]). DHS-2, DHS-16, and DHS-20 have been predicted to have a mitochondrial localization [39] like BDH1, however DHS-20 has also been predicted to have an ER localization [40] as has F55E10.6 [41]. Therefore, we knocked down each of the 4 candidates individually in the worms grown in the absence or presence of D-βHB and measured D-βHB dehydrogenase activity in the worm extracts. None of the knockdowns showed decreased basal D-βHB dehydrogenase activity.
However, we found that adding D-βHB to the growth medium resulted in a 2-fold increase in D-βHB dehydrogenase activity and knockdown of F55E10.6 largely prevented this increased D-βHB dehydrogenase activity (Fig. 3A), suggesting that F55E10.6 likely encodes the D-βHB inducible D-βHB dehydrogenase activity. Knockdown of either dhs-2 or dhs-16 increased the ability of D-βHB to upregulate D-βHB dehydrogenase activity. We also found that adding L-βHB to the culture medium resulted in a roughly 3-fold increase in D-βHB dehydrogenase activity in the worm extracts that was independent of the expression of F55E10.6, dhs-2, dhs-16, or dhs-20 (Figure 3B), suggesting L-βHB induces a separate D-βHB dehydrogenase enzyme. Consistent with this data, when worms were cultured with a racemic mixture of 20 mM βHB (DL-βHB) we found an almost additive 4.5-fold increase in D-βHB dehydrogenase activity (data not shown).
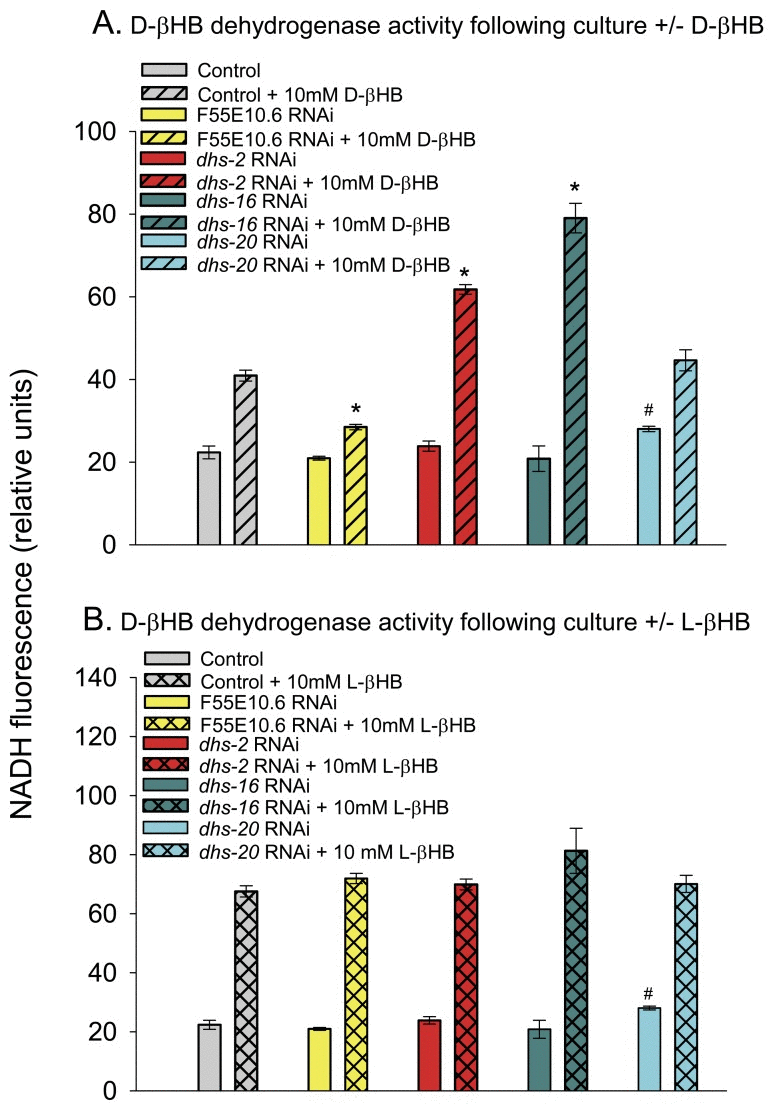
Figure 3. D-βHB dehydrogenase activity in worm extracts following RNAi-mediated gene knockdown. (A) D-βHB dehydrogenase activity following worm culture in the absence or presence of 10 mM D-βHB. Conditions in the legend refer to the culture conditions. The genes F55E10.6, dhs-2, dhs-16, or dhs-20 were knocked down by RNAi feeding (* p < 0.05 compared to Control + 10 mM D-βHB; # p < 0.05 compared to Control). (B) D-βHB dehydrogenase activity following worm culture in the absence or presence of 10 mM L-βHB. The assay conditions were the same as panel A (# p < 0.05 compared to Control).
We also measured L-βHB dehydrogenase activity in the worm extracts (Supplementary Figure 2). There was roughly 5-fold lower basal L-βHB dehydrogenase activity than D-βHB dehydrogenase activity in the worm extracts (data not shown). Adding D-βHB to the culture medium yielded a roughly 50% increase in L-βHB dehydrogenase activity in control worm extracts that was almost completely blocked by RNAi knockdown of F55E10.6. Therefore, the F55E10.6 βHB dehydrogenase activity can likely utilize either D-βHB or L-βHB as substrates, but the activity with D-βHB appears roughly 10-fold higher than with L-βHB. Interestingly, knockdown of dhs-2, dhs-16, or dhs-20 decreased basal L-βHB dehydrogenase activity in the extracts (Supplementary Figure 2). Determining whether any of these genes play a direct role in L-βHB metabolism, or if a gene encoding an enzyme with L-βHB dehydrogenase activity is down-regulated by knockdown of these genes awaits further investigation.
F55E10.6 is required for βHB-mediated longevity
To determine if βHB-mediated upregulation of F55E10.6 was essential for the effect of βHB on longevity, lifespan was monitored in worms in which expression of F55E10.6 was knocked down by RNAi feeding. RNAi knockdown of F55E10.6 increased lifespan by 7% and unexpectedly completely prevented lifespan extension induced by βHB supplementation (Figure 4A). F55E10.6 is a SKN-1 transcriptional target [42]. SKN-1 is a homolog of mammalian Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and a transcriptional regulator that induces the expression of genes involved in antioxidant defense and xenobiotic metabolism to promote longevity. Therefore, addition of βHB to the culture media activates SKN-1, which induces expression of F55E10.6. F55E10.6 could either metabolize βHB or metabolize another endogenous substrate leading to lifespan extension.
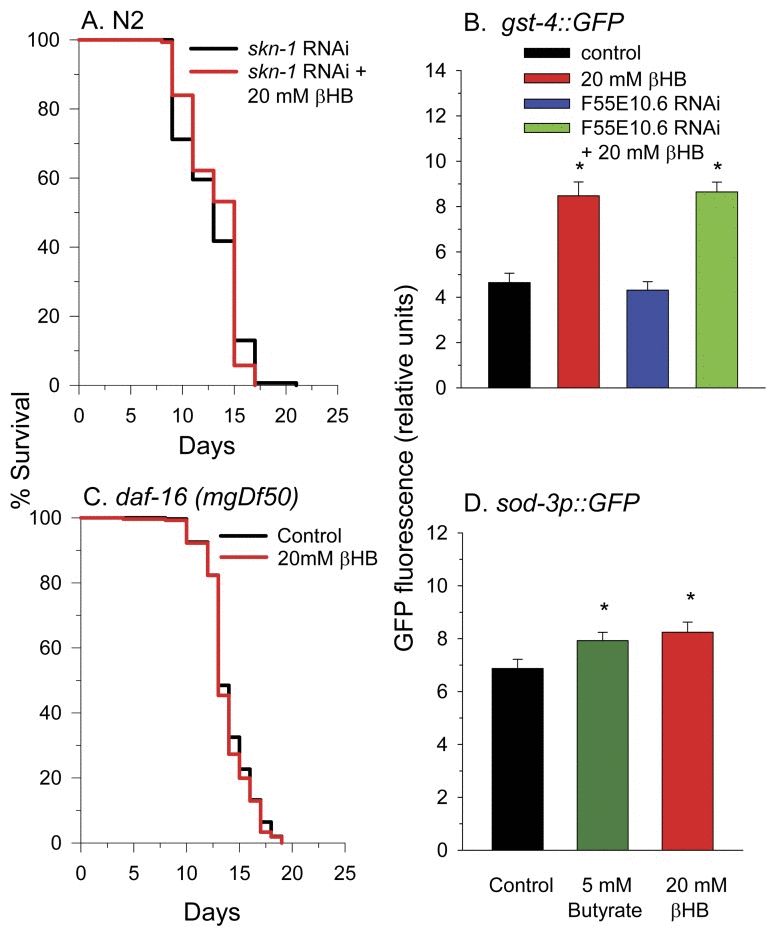
Figure 4. F55E10.6 is required for βHB-mediated lifespan extension, but not for βHB-induced oxygen consumption. (A) Treatment with βHB did not increase the lifespan of N2 worms fed RNAi to F55E10.6. (B) The addition of N-acetyl-L-cysteine (NAC) did not decrease the lifespan of βHB treated worms. (C) The effect of 20 mM βHB and RNAi knockdown of F55E10.6 on oxygen consumption (* p < 0.05 vs. untreated; # p < 0.05 vs. Control). (D) The effect of 20 mM βHB treatment on ATP levels in day 4 N2 worms (p = 0.202). Data are represented as mean +/− SEM.
It is possible that metabolism of βHB, either dependent or independent of F55E10.6, is required for lifespan extension. This increased βHB metabolism may increase TCA cycle and electron transport chain (ETC) activity increasing ROS production, which has been shown to lead to lifespan extension in C. elegans [43]. Therefore we determined if administration of the antioxidant and glutathione precursor N-acetylcysteine (NAC) prevented the lifespan extension induced by βHB. As shown in Figure 4B, NAC by itself moderately increased lifespan, but NAC supplementation did not prevent lifespan extension mediated by βHB. Therefore, βHB is likely extending lifespan through a mechanism that does not require increased ROS production.
Knockdown of F55E10.6 does not prevent the βHB-mediated increase in oxygen consumption
Since F55E10.6 expression was essential for βHB-mediated lifespan extension, we wished to determine if supplemented βHB was being utilized as a respiratory substrate by the worms and whether knocking down F55E10.6 would decrease βHB-induced respiratory metabolism. Therefore, we determined the effect of βHB supplementation on worm oxygen consumption (Figure 4C). βHB supplementation increased oxygen con-sumption by 2.3 fold indicating that βHB is being metabolized by the worms. Unexpectedly, we found that RNAi knockdown of F55E10.6 in the absence of βHB also increased oxygen consumption by around 2.3 fold, suggesting that F55E10.6 represses mitochondrial biogenesis or respiratory function. But RNAi knock-down of F55E10.6 did not decrease the βHB-mediated increase in oxygen consumption. The results suggest that the metabolism of βHB by F55E10.6 does not play a significant role in the use of βHB as an energy substrate for respiration, and so other mechanisms likely explain the requirement of F55E10.6 for lifespan extension.
We also determined the effect of βHB addition on worm ATP levels (Figure 4D). ATP levels were not significantly altered by βHB addition, although oxygen consumption rates increased suggesting that βHB either stimulates energy utilization pathways or decreases the coupling efficiency of oxidative phosphorylation. In this regard, we have previously shown that growth of C. elegans in the presence of the TCA cycle metabolites malate or fumarate resulted in a partial uncoupling of mitochondria [44].
SKN-1 and DAF-16 are required for βHB-mediated longevity
To determine other molecular pathways through which βHB functions to extend lifespan, we performed lifespan experiments using worms deficient in common longevity pathways. In C. elegans the SKN-1 transcriptional activator is normally sequestered in the cytoplasm by WDR-23 and the DDB1/CUL4 ubiquitin ligase complex until the presence of specific xenobiotics or reactive oxygen species leads to a disruption of the interaction. This allows nuclear translocation of SKN-1 leading to the activation of a phase II detoxification transcriptional response and lifespan extension [45, 46]. We next determined if βHB extended the lifespan of worms in which SKN-1 levels were knocked down by RNAi feeding. Consistent with a role for SKN-1 and SKN-1 transcriptional targets such as F55E10.6 in βHB-mediated longevity, βHB did not extend the lifespan in these SKN-1 RNAi worms (Figure 5A). Additionally βHB was able to increase GFP fluorescence in the gst-4::gfp SKN-1 reporter strain of worms (Figure 5B) supporting this assertion. βHB was also able to induce expression of this reporter strain following knockdown of the F55E10.6 gene suggesting that F55E10.6 functions downstream of SKN-1 in the longevity pathway, as is expected for a SKN-1 transcriptional target.
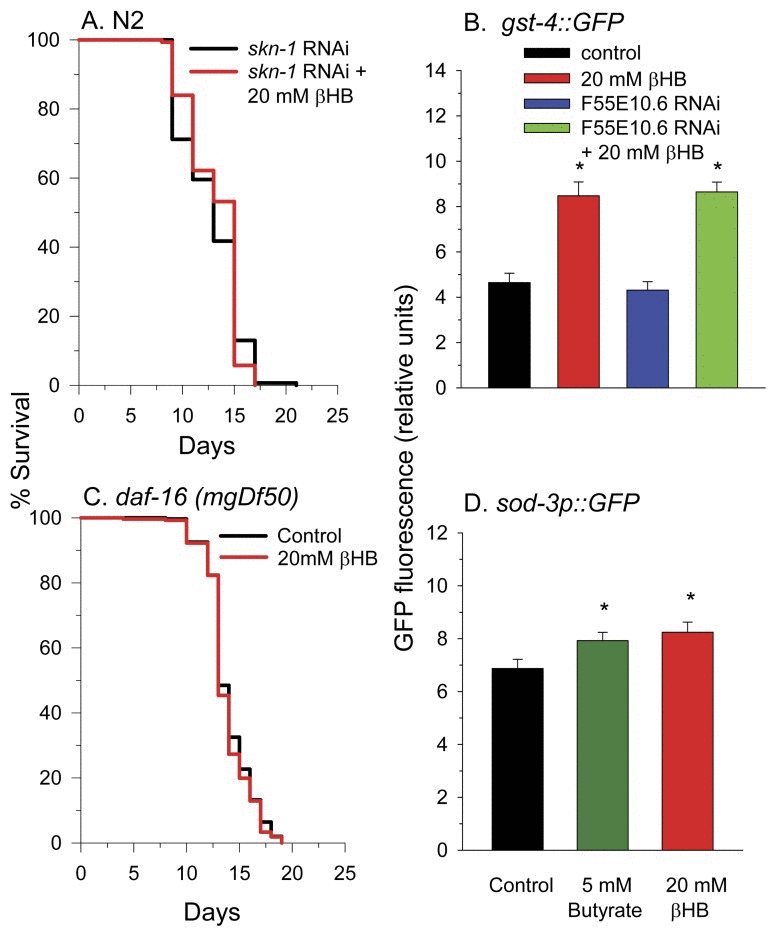
Figure 5. SKN-1 and DAF-16 are required for βHB-mediated lifespan extension. (A) βHB addition did not increase the lifespan of N2 worms fed RNAi to knockdown expression of skn-1. (B) βHB addition increased fluorescence of the gst-4::gfp SKN-1 reporter strain. Data are represented as mean +/− SEM (* p < 0.05). (C) βHB addition did not increase lifespan in daf-16(mgDf50) mutant worms. (D) βHB or butyrate increased fluorescence when administered to the sod-3::gfp DAF-16 reporter strain. Data are represented as mean +/− SEM (* p < 0.05).
Disruption of the DAF-2 insulin receptor signaling pathway is known to extend lifespan through activation of the DAF-16 transcriptional activator. DAF-16 is homologous to mammalian FOXO genes. βHB supplementation to worms homozygous for the daf-16(mgDf50) null allele did not lead to lifespan extension (Figure 5C). Furthermore, βHB supplementation slightly increased fluorescence of the sod-3::gfp DAF-16 reporter strain of worms (Figure 5D), supporting the ability of βHB treatment to activate DAF-16 activity for lifespan extension. Butyrate treatment also led to a similar small increase in GFP fluorescence of the sod-3::gfp worms suggesting a similar mechanism through which βHB and butyrate extend lifespan.
Another transcriptional regulator linked to longevity is hypoxia inducible factor-1 (HIF-1) [47]. We hypothesized that βHB catabolism would increase the concentration of TCA cycle metabolites that inhibit the alpha-ketoglutarate-dependent degradation of HIF-1 by the proteasome [48, 49] initiated by the EGL-9 prolyl hydroxylase [50]. However, we found that βHB supplementation did not increase fluorescence in the nhr-57::gfp reporter strain [47] for HIF-1 transcriptional activity (Supplementary Figure 3A). In addition supplementation with 10 mM pyruvate, or the TCA cycle metabolites citrate, succinate, fumarate, malate, or oxaloacetate also failed to induce GFP expression (Supplementary Figure 3B) suggesting that C. elegans HIF-1 may be regulated slightly differently than mammalian HIF-1.
βHB increases thermotolerance
Since lifespan extension, and DAF-16 and SKN-1 activation in particular, has been linked to stress resistance, we determined the effect of βHB supplementation on thermotolerance in C. elegans. As shown in Figure 6, βHB administration extended the mean survival time of the worms after they were shifted to an elevated temperature by 22%. Due to the increased thermotolerance we hypothesized that heat shock proteins were induced by βHB supplementation. Therefore we monitored GFP fluorescence in 4 heat shock reporter strains of worms following βHB treatment (Supplementary Figure 4A-D). We used the strains hsp-6::gfp and hsp-60::gfp to monitor the mitochondrial unfolded protein response [51], hsp-4::gfp to monitor ER stress, and hsp-16.2::gfp to monitor heat shock factor-1 (HSF-1)-mediated gene expression [52]. βHB supplementation did not induce expression of any of these four reporter strains. Therefore βHB supplementation does not induce a broad heat shock response, even though thermo-tolerance was increased.
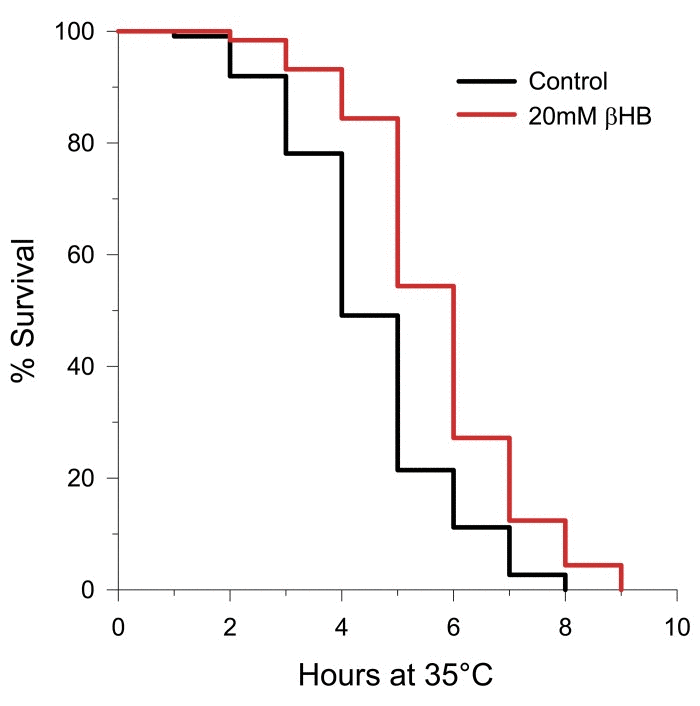
Figure 6. Treatment with βHB increases thermotolerance (log-rank p < 0.001) in N2 worms when upshifted from 20°C to 35°C. βHB mean survival time = 5.7 hours. Control mean survival time = 4.5 hours.
Decreased protein synthesis rates likely contribute to βHB-mediated lifespan extension
Recently it has been recognized that several of the common longevity pathways converge to decrease the rate of translation initiation to extend lifespan [53, 54]. This can occur through several mechanisms including preventing phosphorylation of ribosomal protein S6 by S6 kinases of the TOR signaling pathway, blocking phosphorylation of eukaryotic initiation factor 4E (eIF4E) binding proteins (4E-BPs) by the TOR kinase, or by activation of general control nonderepressible 2 (GCN2) kinase. GCN2 activation can occur in the presence of uncharged tRNAs due to amino acid restriction [55] or during times when mitochondria produce high levels of reactive oxygen species [56]. C. elegans appears to lack close functional homologs of mammalian 4E-BPs [57] (although one distant homolog has been reported [58]), so they likely control the rate of translation initiation mainly through the p70 S6 kinase homolog RSKS-1 and the worm GCN2 homolog GCN-2. Therefore, we obtained the mutant strains rsks-1(ok1255) and gcn-2(ok871) and performed lifespan analysis in the absence or presence of βHB. The untreated lifespan of the rsks-1 mutant was greater than that of the N2 control (Figure 7A) as expected, while the untreated lifespan of the gcn-2 mutant was less than the N2 control (Fig. 7B). With either strain, we found that βHB-mediated lifespan extension was greatly blunted compared to the effect on the N2 control strain.
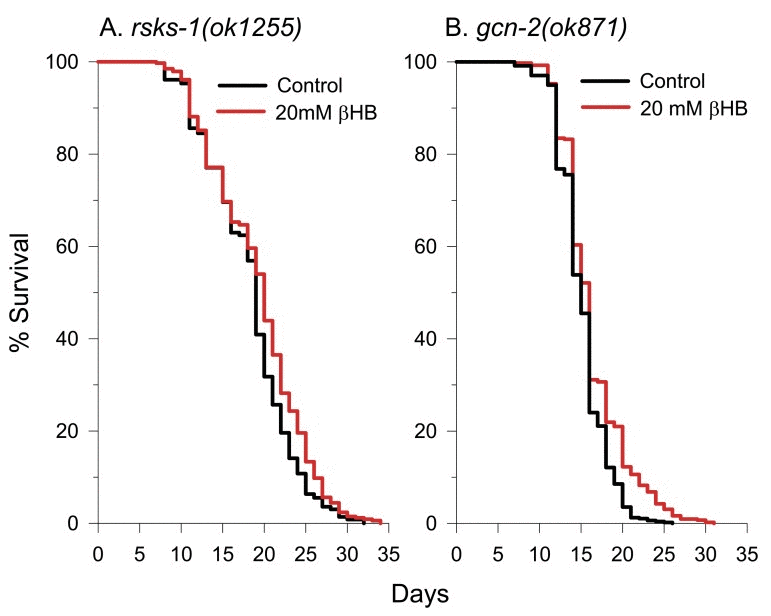
Figure 7. A decreased rate of protein synthesis contributes to βHB-mediated longevity. (A) βHB-mediated lifespan extension was blunted in rsks-1(ok1255) mutant worms. (B) βHB-mediated lifespan extension was also blunted in gcn-2(ok871) mutant worms.
There was a 5% mean lifespan extension in the βHB-treated rsks-1 mutant and a 8% mean lifespan extension in the βHB-treated gcn-2 mutant compared to the 26% lifespan extension in the βHB-treated N2 control. Therefore, the ability to decrease translation rates through both the TOR/RSKS-1 and GCN-2 pathways likely allows for full βHB-mediated lifespan extension in the wild-type N2 animals.
ETC Complex I function is needed for full βHB-mediated lifespan extension
Following mitochondrial βHB dehydrogenase function, acetoacetate is converted to acetoacetyl-CoA with the concurrent conversion of succinyl-CoA to succinate as a byproduct of the succinyl-CoA: 3-ketoacid CoA transferase reaction. Because of this succinate production, it has been suggested that βHB protected a PD cell model by increasing mitochondrial ETC complex II (succinate dehydrogenase) activity, bypassing the ETC complex I deficits present in the disease [30]. To determine if normal mitochondrial ETC complex I or complex II activity is required for βHB-mediated lifespan extension, we determined the effect of βHB supplementation on the lifespan of short-lived complex I-defective gas-1(fc-21) mutants [59] (Figure 8A) and short-lived complex II defective mev-1(kn1) mutants [60] (Figure 8B). βHB extended the lifespan of the gas-1 mutant by 11%, but not to the full 26% extent observed in wild-type worms. Therefore, normal ETC complex I function is necessary for the full effect of βHB on longevity. βHB supplementation fully extended the lifespan of mev-1 mutants indicating that βHB does not require normal ETC complex II function to extend lifespan.
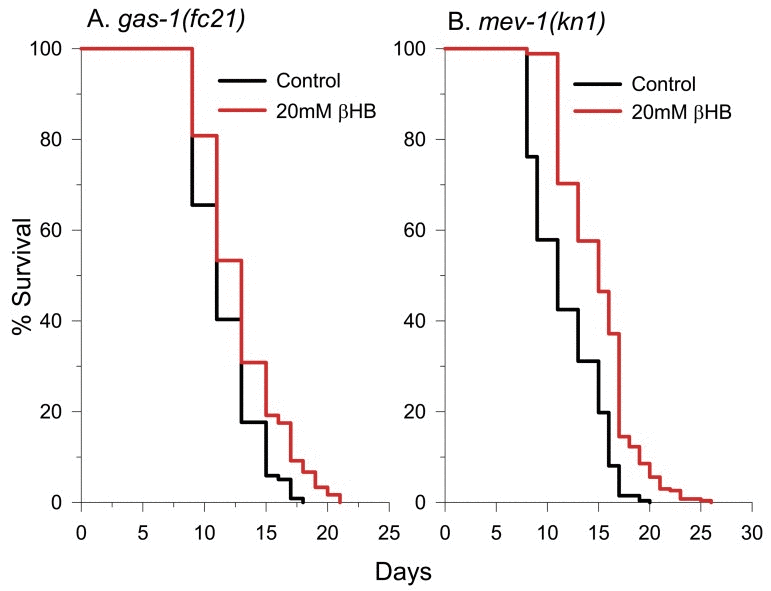
Figure 8. βHB extends the lifespan of short-lived mitochondrial ETC complex I and complex II mutants. (A)gas-1(fc21) survival in the absence and presence of βHB. (B)mev-1(kn1) survival in the absence and presence of βHB.
βHB-mediated longevity requires AAK-2, SIR-2.1, CBP-1, and may occur in a similar manner as in DR
To identify if other important longevity regulators are required for βHB-mediated longevity, βHB was supplemented to AMP kinase (AMPK) aak-2(TG38) mutant worms (Figure 9A) and sir-2.1(ok434) NAD-dependent protein deacetylase mutant worms (Figure 9B) and lifespan was monitored. βHB addition did not extend the lifespan of either strain suggesting that both proteins play a role in βHB-mediated longevity. Since ketone body levels rise during caloric restriction (CR) in mammals and increased βHB levels may be responsible for some portion of the increased stress and disease resistance conferred by CR, we determined the effect of βHB supplementation on lifespan in the nematode eat-2(ad1116) model of dietary restriction (DR) in which pharyngeal pumping is slowed (Figure 9C). We found that treatment with βHB had no significant effect on the longevity of eat-2 worms suggesting that βHB extended lifespan using some of the same downstream effectors activated in DR. The CREB binding protein-1 (CBP-1) transcriptional co-activator and protein acetyltransferase has been shown to be essential for DR-mediated longevity in C. elegans [33]. Therefore we determined the effect of βHB on lifespan in worms where cbp-1 expression was knocked down by RNAi (Fig. 9D). Consistent with βHB extending lifespan in a manner similar to DR, knocking down cbp-1 prevented lifespan extension induced by βHB treatment.
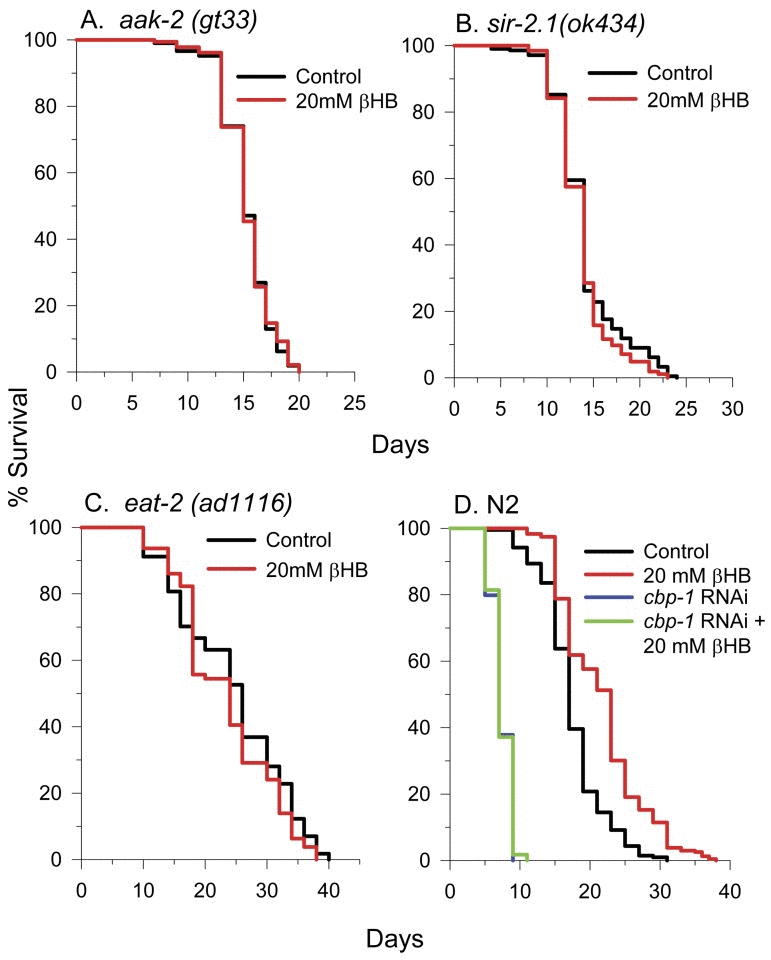
Figure 9. βHB extends lifespan in a similar manner as DR and requires AAK-2, SIR-2.1, and CBP-1. (A) βHB does not extend lifespan of AMPK mutant aak-2(TG38) worms, (B) sir-2.1(ok434) worms, or (C)eat-2(ad1116) worms. (D) Additionally, βHB does not extend the lifespan cbp-1 RNAi knockdown N2 worms.
Many of the same compounds that extend lifespan in C. elegans in a CBP-1 dependent manner also protect against glucose toxicity [61]. It's been shown that C. elegans shows a reduced lifespan when grown in a high glucose containing media [62-64]. When we grew worms in 50 mM glucose, lifespan was decreased by roughly 30% (Figure 10A). βHB supplementation to the glucose-containing media partially restored the lifespan, resulting in a lifespan reduction of only 21% compared to the non-glucose treated controls.
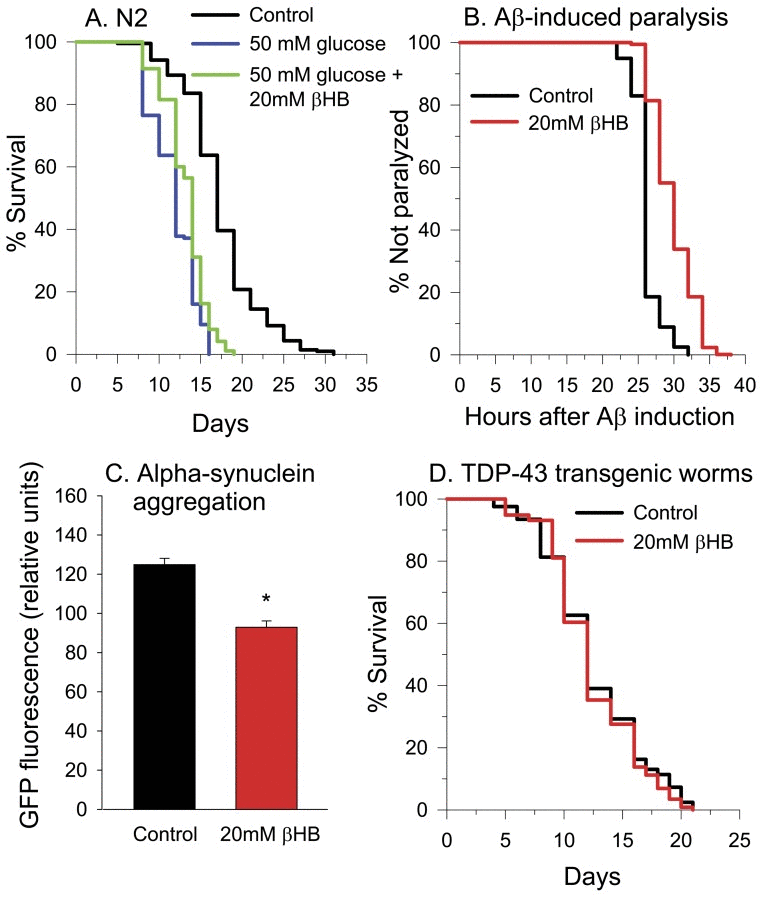
Figure 10. βHB protects against glucose toxicity and proteotoxicity. (A) Treatment with βHB partially protects against 50 mM glucose-induced reduction of lifespan in N2 worms. (B) Survival of the CL4176 strain of C. elegans expressing Aβ in muscle following temperature upshift. Treatment with βHB increases the time to paralysis (log-rank p < 0.001). βHB-treated mean lifespan = 29 hours, untreated control mean lifespan = 26 hours. The curves are generated from the results of six assays (n > 500 for both groups). (C) Treatment with βHB decreases α-synuclein-GFP aggregation in the NL5901 strain GFP fluorescence readings were taken on day 8 of worm lifespan. Data are represented as mean +/− SEM. (log-rank p < 0.001) (D) 20 mM βHB did not protect against the shortened lifespan induced by human TDP-43 overexpression when the worms were grown at 16°C.
βHB delays Aß-induced paralysis and decreases alpha-synuclein aggregation
We next performed experiments using a strain of worms engineered to express human AD-associated Aß peptide within body wall muscle upon temperature upshift from 16° to 25°C, which leads to paralysis of all worms by 32 hours after upshift [65]. Figure 10B shows the paralysis over time in these worms in the presence and absence of βHB treatment. βHB increased the mean paralysis time following Aß induction by 15%, from approximately 26 to 30 hours. Since βHB supplementation was beneficial in this model of proteotoxicity, we next determined the effects of βHB administration on a PD-model worm strain expressing human α-synuclein fused to yellow fluorescent protein (YFP) in the body wall muscle [66]. Alpha-synuclein protein is prone to aggregation and is the major protein constituent of Lewy bodies in PD brain [67]. YFP aggregation and fluorescence was decreased by 35% in worms treated with βHB for 8 days, indicating a protective decrease in the levels of alpha-synuclein aggregates (Figure 10C).
A ketogenic diet has been shown to delay loss of motor performance and loss of spinal cord motor neurons in the SOD1-G93A mouse model of amyotrophic lateral sclerosis (ALS) [68]. So lastly, we performed experiments using worms overexpressing human TDP-43 [69], which forms insoluble aggregates in the nervous system of patients with ALS and other neuro-degenerative disorders [70] and when expressed in the nervous system of worms [69]. TDP-43 expression caused a greatly reduced lifespan in C. elegans both when grown at 20°C and when grown at 16°C (Supplementary Table 1). 20 mM βHB supplementation was unable to prevent the reduction in lifespan (Figure 10D). Concentrations of βHB from 2 mM to 200 mM were also tested (Supplementary Table 1). Only 30 mM βHB was found to be effective at delaying toxicity and the increase in longevity at this concentration was only 5%.
Discussion
Administering βHB to C. elegans extended lifespan and delayed proteotoxicity and glucose toxicity. βHB extended C. elegans lifespan in a SIR-2.1 and AMPK-dependent manner that also required the stress-responsive transcription factors DAF-16 and SKN-1. Since βHB did not extend lifespan in eat-2 pharyngeal pumping mutants, βHB likely acts as a dietary restriction mimetic, as previously hypothesized for its effects in mammals [71]. Even though protective effects of D-βHB on rodent disease models are known, this is the first report to identify D-βHB as a positive modulator of organismal longevity in wild-type animals. We also identified many of the signaling pathways and genes required for this effect. A key finding is that D-βHB-mediated lifespan extension requires SKN-1 and its transcriptional target F55E10.6, a short-chain dehydrogenase/reductase with βHB dehydrogenase activity, although inhibition of HDACS HDA-2 and HDA-3 are also required for the increased longevity.
The role of F55E10.6 in βHB-mediated lifespan extension
The βHB dehydrogenase enzyme assay data suggest that F55E10.6 is a D-βHB-inducible βHB-dehydrogenase enzyme. However, D-βHB may not be the preferred physiological substrate for the enzyme or the substrate required for lifespan extension. Since knockdown of F55E10.6 did not affect the increased oxygen consumption following βHB supplementation, the enzyme does not likely possess a mitochondrial localization producing NADH for ETC complex I function. In this regard in addition to sharing homology with mitochondrial BDH1, F55E10.6 shares homology with microsomal retinol dehydrogenases [37] and microsomal hydroxysteroid dehydrogenases [38]. Due to these homologies and the role that SKN-1 plays in controlling the ER stress response [41], F55E10.6 has been putatively assigned an ER localization [41], although no signal peptide was found [40]. Key to its localization, F55E10.6 is predicted to have a transmembrane domain [40].
It is of interest that knockdown of F55E10.6, a SKN-1 target gene increased C. elegans oxygen consumption, suggesting that SKN-1 signaling may decrease mitochondrial biogenesis or function. Expression of F55E10.6 has been shown to decline with aging [72], likely due to the aging-related decline in activity of SKN-1 [42]. The mammalian SKN-1 homolog Nrf2 is also known to play a role in mitochondrial biogenesis. When overexpressed Nrf2 was shown to be a negative modulator of mitochondrial mass and membrane potential in a high throughput screen using C2C12 myoblast cells [73]. However, when upregulated under physiological conditions Nrf2 was found to be a positive regulator of mitochondrial biogenesis by inducing nuclear respiratory factor-1 (NRF-1) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) expression in heart [74], liver [75], and lung [76].
Full βHB-mediated lifespan extension requires mitochondrial ETC complex I function
βHB-induced lifespan extension was partially blocked in ETC complex I mutant worms and was unaffected in mitochondrial ETC complex II mutant worms. This likely suggests that the lifespan extension is driven partly by βHB metabolism-independent effects and partly by metabolism-dependent effects. Normal complex I activity may be needed to maintain a high NAD/NADH ratio beneficial for maximal lifespan extension [77]. In this regard, it has been shown that rotenone can induce a roughly 10-fold reduction in C. elegans respiration, but only a 2-fold reduction occurred in the presence of 10 mM βHB [78]. Therefore, βHB either stabilized complex I function in the presence of rotenone or stimulated complex II-dependent respiration to bypass this block of complex I function. In mammals βHB has been shown to stabilize and increase the efficiency of ETC complex I [68, 79]. The increased rate of NADH oxidation in the presence of βHB led to decreased ROS levels in mouse neocortical neurons following glutamate excitotoxicity [79]. βHB may also enhance complex I activity in worms, but it may not be able to fully do so in the gas-1 mutants preventing full lifespan extension.
Proposed mechanism for βHB-mediated lifespan extension
We propose 2 possible mechanisms for lifespan extension mediated by βHB supplementation. In the first mechanism (see Supplementary Figure 5), we propose that βHB directly inhibits HDACs to increase histone acetylation [14] causing gene expression changes leading to SKN–1 activation, independent of βHB catabolism. The metabolism-independent activation of SKN-1 is consistent with our previous data showing that stimulation of metabolism by supplementation of several TCA cycle metabolites did not activate SKN-1 transcriptional activity [44]. Next, SKN-1 activity induces expression of F55E10.6 [42, 45], required for proper execution of the SKN-1 longevity program. SKN-1 activation has been shown to repress expression of the insulin-like peptides DAF-28 and INS-39, decreasing DAF-2 insulin receptor signaling to activate DAF-16 [80]. βHB catabolism also likely increases the level of specific TCA cycle intermediates, which may contribute to the DAF-16-mediated lifespan extension. We and others have shown that supplementation of the TCA cycle meta-bolites fumarate, malate, and oxaloacetate activated nuclear translocation of DAF-16 to extend lifespan in an AMPK and SIR-2.1-dependent manner [44, 81]. Others have also found that the TCA cycle metabolite alpha-ketoglutarate extends lifespan through a TOR kinase-dependent mechanism [82]. Although this model is consistent with our data, is also possible that βHB-mediated HDAC inhibition causes a direct transcriptional upregulation of DAF-16, as βHB-mediated HDAC inhibition directly upregulates expression of the DAF-16 homolog FOXO3A in mammals [14].
In addition, βHB metabolism may increase acetyl-CoA levels that serve as a substrate for histone acetyl-transferases to increase histone acetylation [83], which could strengthen the effects of HDAC inhibition to extend lifespan. However, increased cytoplasmic acetyl-CoA levels have also been shown to inhibit autophagy [84], which could potentially dampen lifespan extension. However, the acetyl group from mitochondrial acetyl-CoA can be transferred to carnitine to form acetylcarnitine and exported from mitochondria to the nucleus, where acetyl-CoA is reformed and used for nuclear histone acetylation [85]. This mechanism may allow for increased histone acetylation without decreased rates of autophagy.
The second proposed model of how βHB may extend lifespan is through inhibition of the insulin signaling pathway. In mammals, it has been shown, contrary to expectations, that βHB administration or a ketogenic diet blocks the insulin signaling pathway in muscle leading to insulin resistance [86]. This adaptation likely evolved to allow the brain preferential use of the bloodstream glucose during starvation. However, a ketogenic diet has also been shown to be effective at lowering blood glucose in patients with type II diabetes due to the decreased carbohydrate intake [87]. In mouse studies, βHB administration yielded a 50% reduced phosphorylation and activity of Akt/protein kinase B downstream of the insulin receptor decreasing insulin signaling [86]. The mechanism for this βHB-mediated inhibition of Akt and the insulin signaling pathway was not fully elucidated, but it relied upon administration of D-βHB and not L-βHB, suggesting that mitochondrial metabolism of D-βHB may be involved. In C. elegans, inhibition or decreased expression of Akt or other proteins of the insulin signaling pathway has been shown to activate both DAF-16 and SKN-1 leading to lifespan extension [88], thereby providing a potential mechanism for the effect of βHB on longevity.
βHB does not induce a broad heat shock response, but still increases thermotolerance
Although βHB did not induce expression of four specific heat shock proteins monitored (Supplementary Fig. 3), it did activate the DAF-16 and SKN-1 signaling pathways, which are both likely responsible for the increased thermotolerance observed following βHB treatment. Previous research has shown that RNAi knockdown of either skn-1 or daf-16 decreased thermo-tolerance [89]. DAF-16 is known to induce expression of several heat shock proteins including hsp-12.6, sip-1, and hsp-16.1, which may play a role in the increased thermotolerance. The factors that SKN-1 induce to confer thermotolerance are less clear, although SKN-1 function has been implicated in the induction of hsp-4 expression and activation of the ER stress response [41]. However, we did not find βHB to induce hsp-4::GFP expression, but a positive trend was observed (p = 0.18).
Neuroprotective effects of ketone bodies
In an AD cell model, βHB has been shown to protect hippocampal neurons from Aß toxicity [90]. The protection may have occurred through decreasing ROS levels as decreased ROS production is known to lower expression levels of beta-secretase (BACE1), a protease that contributes to toxic Aß generation [91]. This mechanism may be responsible for the ketogenic diet-induced reduction of Aß levels in a mouse model of AD [22]. The brain's ability to utilize glucose decreases in AD. To prevent deficits in brain ATP levels, βHB has been used as an alternative metabolic energy source for patients with AD [25]. Increased inflammation accompanies brain aging and may contribute to the development of AD. Increased levels of ketone bodies have been shown to reduce inflammation [71, 92] and this may result from increased mitochondrial efficiency and decreased ROS production [93].
PD is associated with aggregation of alpha-synuclein and death of dopaminergic neurons leading to motor decline. Mice treated with βHB showed partial protection against neurodegeneration and motor deficiency induced by MPTP [30]. Surprisingly, this was not due to increased NADH generation fueling complex I, but was described to be due to an increased supply of succinate, a substrate for ETC complex II [94]. As mentioned previously, the stimulation of complex II activity by βHB metabolism depends on the increased succinate produced as a byproduct of the mitochondrial succinyl-CoA: 3-ketoacid CoA transferase reaction. However, since βHB fully extended lifespan in complex II-defective mev-1 mutants, it is unlikely that this mechanism plays a substantial role in βHB-mediated lifespan extension. The mechanism of βHB-mediated protection in PD models may be similar to the mechanism by which βHB supplementation increases lifespan in the complex I-defective gas-1 worms.
Study limitations and future directions
Although we were able to dissect many of the pathways through which βHB extends lifespan in C. elegans, many questions remain. For example, is the reason that D-βHB but not L- βHB extended lifespan due to the increased ability of D- βHB to be metabolized or due to the higher efficiency of D-βHB as an HDAC inhibitor? Is HDAC inhibition required for βHB-mediated upregulation of SKN-1 or DAF-16 activity? In addition to extending lifespan as shown here, RNAi knockdown of the class I HDAC hda-3 was shown to protect against polyglutamine-mediated toxicity in a C. elegans Huntington's disease, while knockdown of most other HDACs increased toxicity [95]. Is HDAC inhibition or SKN-1 activity required for DAF-16 activation by βHB? Also is βHB catabolism required for βHB-mediated SKN-1 or DAF-16 activation? Furthermore, is the transcription factor PHA-4/FoxA, which is required for DR-mediated longevity [96], also required for βHB-mediated longevity? In this regard mammalian Foxa2 is known to induce expression of BDH1 [97]. Do βHB levels increase in DR worms and if so does this play a role in DR-mediated longevity? Lastly, are the same signaling pathways required for longevity also required for βHB-mediated protection in the C. elegans models of Aß and alpha-synuclein toxicity? Future experiments will provide answers to these questions and elucidate the molecular mechanisms responsible for the protective effects of βHB. This knowledge will allow for a broader use of βHB as a therapy for aging-related disorders.
Conclusion
βHB treatment extended lifespan and protected against metabolic, proteotoxic and thermal stress in C. elegans. βHB-mediated lifespan extension occurred through induction of the DAF-16 and SKN-1 signaling pathways and was dependent upon βHB-mediated inhibition of HDACs HDA-2 and HDA-3. Our data support the hypothesis that βHB is a DR mimetic and that βHB treatment will likely be useful in the treatment of many human aging-associated disorders.
Materials and Methods
C. elegans strains
C. elegans strains were obtained from the Caenorhabditis Genetics Center (CGC, University of Minnesota) and were cultured at 20°C in either liquid S media or NGM agar media [44] as indicated. Lifespan assays were performed using the following strains: N2 Bristol (wild-type), GR1307 [daf-16(mgDf50)], TG38 [aak-2(gt33)], DA1116 [eat-2(ad1116)], TK22 [mev-1(kn1)], CW152 [gas-1(fc21)], VC199 [sir-2.1(ok434)], RB1206 [rsks-1(ok1255)], RB967 [gcn-2(ok871)], VC983 [hda-2(ok1479)], RB1618 [hda-3(ok1991)], RB758 [hda-4(ok518)], and RB2416 [hda-10(ok3311)]. The following strains expressing GFP were used to monitor promoter activation: CL2166 [pAF15(gst-4p::GFP::NLS)], SJ4100 [hsp-6::gfp(zcIs13)], SJ4058 [hsp-60::gfp(zcIs9)], SJ4005 [hsp-4::gfp(zcIs4)], ZG449 [nhr-57p::GFP + unc-119(+)], and CL2070 [hsp-16-2::gfp(dvIs70)]. The following strains were used as disease models: NL5901 [pkIs2386 (unc-54p::alpha-synuclein::YFP + unc-119(+))], CL4176 [smg-1ts [myo-3::Aβ1–42long 3′-UTR]], and CL6049 [dvls62 (snb-1::hTDP-43 + pCL26 (mtl-2::GFP)].
Chemicals
Sodium DL-3-hydroxybutyric acid (βHB), sodium butyrate, valproic (2-propylpentanoic) acid, ethidium bromide, and potassium cyanide were purchased from Acros Organics. Sodium D-3-hydroxybutyric acid and L-3-hydroxybutyric acid were obtained from Sigma. 5-fluoro-2′-deoxyuridine (FUdR) was purchased from Research Products International Corp. and Biotang, Inc. Sodium hydroxide (Fisher Scientific) was added to metabolite stock solutions to obtain a pH of 7.0.
Lifespan Analysis
C. elegans adults were bleached as previously described [44] to yield age-synchronized eggs in S-medium. Lifespan experiments were performed suspending eggs in liquid media in 3 μM transparent cell culture inserts (BD Falcon #353181) in 12-well microplates on an orbital shaker at 135 rotations/min at 20°C [98]. 1.3 mL of S-medium containing 9×109 HT115 (DE3) E. coli per mL was placed in each well of a 12-well microplate. Then, bleach synchronized worm eggs were suspended at a concentration of 100–200 eggs/mL in the bacterial suspension in S-medium. Lastly, a cell culture insert was placed in each microplate well into which 0.25 mL of the egg/bacterial suspension (25-50 eggs) was placed (n = 3 wells per condition). Excluding RNAi lifespan experiments, bacteria were heat killed (using a Kendal model HB-S-23DHT ultrasonic cleaner) at 80°C for 60 minutes. Synchronized cultures of worms were cultured at 20°C and monitored until they reached adulthood (~72 h), at which time FUdR was added to a final concentration of 400 μM. Worm viability was scored every two days. Worms that did not respond to repeated stimulus were scored as dead and those that contained internally hatched larvae were excluded. The media containing bacteria in the wells of the microplate into which the culture inserts were submerged was removed and replaced with a new bacterial suspension every 3 days.
High glucose lifespan assays. We performed lifespans as described above with the addition 50 mM glucose to the culture medium. Animals were scored every day and inserts were transferred to fresh media every two days.
RNAi treatment
The E. coliskn-1, cbp-1, hda-1, hda-2, hda-3, F55E10.6, dhs-2, dhs-16, and dhs-20 clones from the Ahringer C. elegans RNAi library (Source BioScience LifeSciences), were grown for 16 hours and then given 1 mM IPTG to induce expression of the RNAi for 4 hours similarly as described in [99]. Lifespan experiments were performed as described above with the exception that live bacteria were used and the culture media in the microplate was replaced daily to replenish metabolite levels that may be partially depleted by bacterial metabolism.
βHB dehydrogenase assays
D-βHB or L-βHB dehydrogenase activity was assayed as in [100], slightly modified from the original method published in [101]. The reaction mix contained 100 mM Tris-HCl pH 8.0, 10 mM MgSO4, 5 mm K+ EDTA, 400 mM hydrazine hydrate, 1 μ M rotenone, 10 mM NAD, 20 mm D-βHB or L-βHB, and 10 μ L of Halt protease inhibitor cocktail (Thermo Scientific). Worms were grown with or without D-βHB or L-βHB as indicated. On day 4 of development, worms were washed 3 times with M9 buffer to free them of bacteria and condensed to 50 worms per microliter. 1 mL of concentrated worms was subjected to 3 freeze-thaws cycles in liquid nitrogen. 50 μ L of each sample (~ 2,500 worms) was added to a clear bottomed 96-well microplate, followed by addition of 100 μ L of the reaction mix above. NADH fluorescence was measured kinetically for 20 minutes using 360/40 nm excitation and 460/40 nm emission filters (n=5) on a Biotek Synergy 2 microplate reader.
Oxygen consumption measurements
N2 worms were grown in 12-well cell culture plates and fed HT115 (DE3) control or F55E10.6 RNAi knockdown-expressing E. coli as food for 4 days in the absence or presence of 20 mM βHB. Worms were washed 4 times using M9 buffer to free them of the bacteria and then resuspended in the culture media in which they were grown except without the bacteria. The average concentration of worms was obtained by taking ten 10 μL drops and counting the number of living worms in each drop. The volume of the culture was then adjusted to obtain a final concentration of 2 worms per μL. 300 μL of the worm suspension was then added to the chamber of a Clark oxygen electrode (MT200A chamber, Strathkelvin Instruments) and the respiration was monitored for 5 minutes. The respiratory rate was normalized to protein content by performing a protein assay on the worm suspension.
ATP level measurements
ATP levels were measured using CellTiter-Glo reagent (Promega) on day 4 worms grown in the absence or presence of 20 mM βHB washed free of bacteria and then lysed by freeze-thaw as described in [44].
GFP reporter strains
The GFP fluorescence of C. elegans populations was assayed using a Biotek Synergy 2 microplate reader. Strains were age synchronized and cultured in 12-well microplates as described above. At the L3 stage of larval development animals were treated with βHB or other compounds. Following 24 hours of treatment, worms were washed 3 times in S-medium and approximately 400 worms per 200 μL were added to a clear 96-well microplate and GFP fluorescence was measured using 485/20 nm excitation and 528/20 nm emission filters (n=10 per treatment group).
Microscopy and quantification
Worms used for microscopy were anesthetized in M9 media containing 1mM levamisole and transferred to agar pads with glass coverslips and analyzed using an EVOS fluorescence microscope. Comparable results were established in the absence of levamisole (data not shown). Approximately 20 worms per condition were used and experiments were repeated at least three times (n=3). ImageJ™ software was used to quantify pixel densities.
Thermotolerance assays
A synchronized population of N2 C. elegans eggs was placed on 20 mM βHB treated on non-treated NGM agar plates and allowed to hatch at 20°C. At the L4 larval stage of development animals were transferred to a 35°C incubator. Survival was scored as the number of animals responsive to gentle prodding with a worm pick. 224 βHB-treated and 250 non-treated worms were counted.
Aß-mediated paralysis assays
Paralysis assays were carried out as outlined in [65]. Briefly, second generation synchronized gravid C. elegans strain CL4176 were placed on treated or untreated 6 cm NGM plates and allowed to lay eggs for 2 hours. After two hours, adults were removed and plates were placed in a 16°C incubator for 48 hours. Following 48 hours, plates were upshifted to a 25°C incubator. Scoring for paralyzed worms began 20 hours after upshift. Animals were scored for movement every two hours. Worms were considered paralyzed if they could not complete a full body movement after stimulation with a worm pick.
Alpha-synuclein protein aggregation assays
Eggs from the NL5901 strain of C. elegans were treated with alkaline bleach, washed, and then placed in 12 well cell culture inserts as described above in the presence or absence of βHB. Following 3 days of treatment, FUdR was added to the inserts to prevent egg-laying and progeny development. Culture media in the microplates was changed every 3 days. On day 8 worms were washed 3 times with M9 media and placed on 1% agarose pads or immobilized with 10mM levamisole. Visualization of the number of inclusions expressing alpha-synuclein-YFP was captured using an EVOS fluorescence microscope. Foci larger than 2 μm2 were counted for each group (n=30) and measurements on inclusions were performed using NIH ImageJ™ software and the assay was completed at least 3 times similarly as performed in [102]. Statistical analysis was completed using GraphPad Prism software and calculation of statistical significance between various groups was carried out by Student's t-tests.
TDP-43 lifespan assays
CL6049 [dvls62((snb-1::hTDP-43 + pCL26(mtl-2::GFP))] second generation synchronized worms were placed on treated or untreated 6 cm NGM plates and allowed to lay eggs for 2 hours. After two hours, adults were removed and plates were placed in a 16°C incubator. At the L4 larval stage worms were transferred onto treated or untreated NGM plates with added 5-fluoro-2′-deoxyuridine (FUdR, 0.05 mg/mL) to inhibit egg-laying and growth of progeny. Worms were scored everyday by gentle touch with a platinum wire. Failure to respond to touch or move forward or backwards was scored as dead.
Statistical Analysis
Kaplan-Meier survival analysis and log-rank tests were performed using Sigmaplot version 11.0. Student's t-tests were used in other analyses.
Supplementary Materials
Acknowledgments
This research was funded by NIH grant AG046769 to PCB. We would like to thank Robert Buzzeo in the Core Facility of the Department of Cell Biology, Microbiology, and Molecular Biology for sharing instruments and reagents and Dr. Dominic D'Agostino for intellectual contribution. We would like to thank Dr. Bin Xue for bioinformatics analysis. We would also like to thank Dr. Christopher Link for providing the CL6049 C. elegans strain. All other nematode strains were provided by the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN, USA), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Conflicts of Interest
No conflict of interest could be disclosed for any author.
References
- 1. Ahlqvist KJ, Hamalainen RH, Yatsuga S, Uutela M, Terzioglu M, Gotz A, Forsstrom S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson NG, Wartiovaara K, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012; 15: 100 -109. [PubMed] .
- 2. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004; 429: 417 -423. [PubMed] .
- 3. Braeckman BP, Houthoofd K, De Vreese A, Vanfleteren JR. Apparent uncoupling of energy production and consumption in long-lived Clk mutants of Caenorhabditis elegans. Curr Biol. 1999; 9: 493 -496. [PubMed] .
- 4. Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007; 16: 431 -444. [PubMed] .
- 5. Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie, J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009; 19: 1591 -1598. [PubMed] .
- 6. Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS genetics. 2005; 1: 119 -128. [PubMed] .
- 7. Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997; 29: 89 -95. [PubMed] .
- 8. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002; 277: 44784 -44790. [PubMed] .
- 9. Harman D. Origin and evolution of the free radical theory of aging: a brief personal history, 1954-2009. Biogerontology. 2009; 10: 773 -781. [PubMed] .
- 10. Greer EL and Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009; 8: 113 -127. [PubMed] .
- 11. Ingram DK, Anson RM, de Cabo R, Mamczarz J, Zhu M, Mattison J, Lane MA, Roth GS. Development of calorie restriction mimetics as a prolongevity strategy. Ann N Y Acad Sci. 2004; 1019: 412 -423. [PubMed] .
- 12. Kenyon C. A conserved regulatory system for aging. Cell. 2001; 105: 165 -168. [PubMed] .
- 13. Newman JC and Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014; 25: 42 -52. [PubMed] .
- 14. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R. . Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013; 339: 211 -214. [PubMed] .
- 15. Panov A, Orynbayeva Z, Vavilin V, Lyakhovich V. Fatty acids in energy metabolism of the central nervous system. Biomed Res Int. 2014; 2014 -472459. .
- 16. Liu Z, Ciocea A, Devireddy L. Endogenous siderophore 2,5-dihydroxybenzoic acid deficiency promotes anemia and splenic iron overload in mice. Mol Cell Biol. 2014; 34: 2533 -2546. [PubMed] .
- 17. Mercken EM, Carboneau BA, Krzysik-Walker SM, de Cabo R. Of mice and men: the benefits of caloric restriction, exercise, and mimetics. Ageing Res Rev. 2012; 11: 390 -398. [PubMed] .
- 18. Pasinetti GM, Zhao Z, Qin W, Ho L, Shrishailam Y, Macgrogan D, Ressmann W, Humala N, Liu X, Romero C, Stetka B, Chen L, Ksiezak-Reding H, et al. Caloric intake and Alzheimer's disease. Experimental approaches and therapeutic implications. Interdiscip Top Gerontol. 2007; 35: 159 -175. [PubMed] .
- 19. Smith MA, Drew KL, Nunomura A, Takeda A, Hirai K, Zhu X, Atwood CS, Raina AK, Rottkamp CA, Sayre LM, Friedland RP, Perry G. Amyloid-beta, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs? Neurochem Int. 2002; 40: 527 -531. [PubMed] .
- 20. Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis. 2010; 20: Suppl 2 S535 -550. [PubMed] .
- 21. Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. d-β-Hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proceedings of the National Academy of Sciences. 2000; 97: 5440 -5444. .
- 22. Van der Auwera I, Wera S, Van Leuven F, Henderson ST. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr Metab (Lond). 2005; 2 -28. [PubMed] .
- 23. Brownlow ML, Benner L, D'Agostino D, Gordon MN, Morgan D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer's pathology. PLoS One. 2013; 8 -e75713. .
- 24. Zhang J, Cao Q, Li S, Lu X, Zhao Y, Guan JS, Chen JC, Wu Q, Chen GQ. 3-Hydroxybutyrate methyl ester as a potential drug against Alzheimer's disease via mitochondria protection mechanism. Biomaterials. 2013; 34: 7552 -7562. [PubMed] .
- 25. Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, Hyde K, Chapman D, Craft S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol Aging. 2004; 25: 311 -314. [PubMed] .
- 26. Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008; 314: 2076 -2089. [PubMed] .
- 27. Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008; 283: 9089 -9100. [PubMed] .
- 28. Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, Yamaguchi H, Shimohama S, Lee HG, Zhu X, Smith MA, Perry G. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis. 2010; 37: 731 -737. [PubMed] .
- 29. Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001; 60: 759 -767. [PubMed] .
- 30. Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003; 112: 892 -901. [PubMed] .
- 31. Jabre MG and Bejjani BP. Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology. 2006; 66 -617. author reply 617 .
- 32. Evason K, Collins JJ, Huang C, Hughes S, Kornfeld K. Valproic acid extends Caenorhabditis elegans lifespan. Aging Cell. 2008; 7: 305 -317. [PubMed] .
- 33. Zhang M, Poplawski M, Yen K, Cheng H, Bloss E, Zhu X, Patel H, Mobbs CV. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 2009; 7 -e1000245. .
- 34. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003; 133: 2485S -2493S. [PubMed] .
- 35. Choi KY, Ji YJ, Jee C, Kim DH, Ahnn J. Characterization of CeHDA-7, a class II histone deacetylase interacting with MEF-2 in Caenorhabditis elegans. Biochem Biophys Res Commun. 2002; 293: 1295 -1300. [PubMed] .
- 36. Wollam J, Magner DB, Magomedova L, Rass E, Shen Y, Rottiers V, Habermann B, Cummins CL, Antebi A. A novel 3-hydroxysteroid dehydrogenase that regulates reproductive development and longevity. PLoS Biol. 2012; 10 -e1001305. .
- 37. Baker ME. Evolution of mammalian 11beta- and 17beta-hydroxysteroid dehydrogenases-type 2 and retinol dehydrogenases from ancestors in Caenorhabditis elegans and evidence for horizontal transfer of a eukaryote dehydrogenase to E. coli. J Steroid Biochem Mol Biol. 1998; 66: 355 -363. [PubMed] .
- 38. Chetyrkin SV, Belyaeva OV, Gough WH, Kedishvili NY. Characterization of a novel type of human microsomal 3alpha-hydroxysteroid dehydrogenase: unique tissue distribution and catalytic properties. J Biol Chem. 2001; 276: 22278 -22286. [PubMed] .
- 39. Marcotte EM, Xenarios I, van Der Bliek AM, Eisenberg D. Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci U S A. 2000; 97: 12115 -12120. [PubMed] .
- 40. Suh J and Hutter H. A survey of putative secreted and transmembrane proteins encoded in the C. elegans genome. BMC Genomics. 2012; 13 -333. [PubMed] .
- 41. Glover-Cutter KM, Lin S, Blackwell TK. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013; 9 -e1003701. .
- 42. Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009; 8: 524 -541. [PubMed] .
- 43. Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012; 15: 451 -465. [PubMed] .
- 44. Edwards CB, Copes N, Brito AG, Canfield J, Bradshaw PC. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS One. 2013; 8 -e58345. .
- 45. Choe KP, Leung CK, Miyamoto MM. Unique structure and regulation of the nematode detoxification gene regulator, SKN-1: implications to understanding and controlling drug resistance. Drug Metab Rev. 2012; 44: 209 -223. [PubMed] .
- 46. Choe KP, Przybysz AJ, Strange K. The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol Cell Biol. 2009; 29: 2704 -2715. [PubMed] .
- 47. Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009; 4 -e6348. .
- 48. Hewitson KS, Lienard BM, McDonough MA, Clifton IJ, Butler D, Soares AS, Oldham NJ, McNeill LA, Schofield CJ. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem. 2007; 282: 3293 -3301. [PubMed] .
- 49. Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007; 282: 4524 -4532. [PubMed] .
- 50. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001; 107: 43 -54. [PubMed] .
- 51. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004; 117: 4055 -4066. [PubMed] .
- 52. Stringham EG, Dixon DK, Jones D, Candido EP. Temporal and spatial expression patterns of the small heat shock (hsp16) genes in transgenic Caenorhabditis elegans. Mol Biol Cell. 1992; 3: 221 -233. [PubMed] .
- 53. Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007; 6: 95 -110. [PubMed] .
- 54. Rousakis A, Vlassis A, Vlanti A, Patera S, Thireos G, Syntichaki P. The general control nonderepressible-2 kinase mediates stress response and longevity induced by target of rapamycin inactivation in Caenorhabditis elegans. Aging Cell. 2013; 12: 742 -751. [PubMed] .
- 55. Gallinetti J, Harputlugil E, Mitchell JR. Amino acid sensing in dietary-restriction-mediated longevity: roles of signal-transducing kinases GCN2 and TOR. Biochem J. 2013; 449: 1 -10. [PubMed] .
- 56. Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012; 8 -e1002760. .
- 57. Korta DZ, Tuck S, Hubbard EJ. S6K links cell fate, cell cycle and nutrient response in C. elegans germline stem/progenitor cells. Development. 2012; 139: 859 -870. [PubMed] .
- 58. Li W, DeBella LR, Guven-Ozkan T, Lin R, Rose LS. An eIF4E-binding protein regulates katanin protein levels in C. elegans embryos. J Cell Biol. 2009; 187: 33 -42. [PubMed] .
- 59. Hartman PS, Ishii N, Kayser EB, Morgan PG, Sedensky MM. Mitochondrial mutations differentially affect aging, mutability and anesthetic sensitivity in Caenorhabditis elegans. Mech Ageing Dev. 2001; 122: 1187 -1201. [PubMed] .
- 60. Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998; 394: 694 -697. [PubMed] .
- 61. Lublin A, Isoda F, Patel H, Yen K, Nguyen L, Hajje D, Schwartz M, Mobbs C. FDA-approved drugs that protect mammalian neurons from glucose toxicity slow aging dependent on cbp and protect against proteotoxicity. PLoS One. 2011; 6 -e27762. .
- 62. Lee S-J, Murphy CT, Kenyon C. Glucose Shortens the Life Span of C. elegans by Downregulating DAF-16/FOXO Activity and Aquaporin Gene Expression. Cell Metabolism. 2009; 10: 379 -391. [PubMed] .
- 63. Schlotterer A, Kukudov G, Bozorgmehr F, Hutter H, Du X, Oikonomou D, Ibrahim Y, Pfisterer F, Rabbani N, Thornalley P, Sayed A, Fleming T, Humpert P, et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes. 2009; 58: 2450 -2456. [PubMed] .
- 64. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6: 280 -293. [PubMed] .
- 65. Dostal V and Link CD. Assaying beta-amyloid toxicity using a transgenic C. elegans model. J Vis Exp. 2010; .
- 66. van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008; 4 -e1000027. .
- 67. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997; 388: 839 -840. [PubMed] .
- 68. Zhao Z, Lange DJ, Voustianiouk A, MacGrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti GM. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006; 7 -29. [PubMed] .
- 69. Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010; 19: 3206 -3218. [PubMed] .
- 70. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006; 314: 130 -133. [PubMed] .
- 71. Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009; 59: 293 -315. [PubMed] .
- 72. Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell. 2008; 134: 291 -303. [PubMed] .
- 73. Yoon JC, Ling AJ, Isik M, Lee DY, Steinbaugh MJ, Sack LM, Boduch AN, Blackwell TK, Sinclair DA, Elledge SJ. GLTSCR2/PICT1 links mitochondrial stress and Myc signaling. Proc Natl Acad Sci U S A. 2014; 111: 3781 -3786. [PubMed] .
- 74. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008; 103: 1232 -1240. [PubMed] .
- 75. Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011; 286: 16374 -16385. [PubMed] .
- 76. Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012; 53: 1584 -1594. [PubMed] .
- 77. Hashimoto T, Horikawa M, Nomura T, Sakamoto K. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology. 2010; 11: 31 -43. [PubMed] .
- 78. Ved R, Saha S, Westlund B, Perier C, Burnam L, Sluder A, Hoener M, Rodrigues CM, Alfonso A, Steer C, Liu L, Przedborski S, Wolozin B. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem. 2005; 280: 42655 -42668. [PubMed] .
- 79. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007; 145: 256 -264. [PubMed] .
- 80. Okuyama T, Inoue H, Ookuma S, Satoh T, Kano K, Honjoh S, Hisamoto N, Matsumoto K, Nishida E. The ERK-MAPK pathway regulates longevity through SKN-1 and insulin-like signaling in Caenorhabditis elegans. J Biol Chem. 2010; 285: 30274 -30281. [PubMed] .
- 81. Williams DS, Cash A, Hamadani L, Diemer T. Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging Cell. 2009; 8: 765 -768. [PubMed] .
- 82. Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014; 509: 397 -401. [PubMed] .
- 83. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science. 2009; 324: 1076 -1080. [PubMed] .
- 84. Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, et al. Regulation of autophagy by cytosolic acetyl-coenzyme a. Mol Cell. 2014; 53: 710 -725. [PubMed] .
- 85. Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics. 2009; 4: 399 -403. [PubMed] .
- 86. Yamada T, Zhang SJ, Westerblad H, Katz A. {beta}-Hydroxybutyrate inhibits insulin-mediated glucose transport in mouse oxidative muscle. Am J Physiol Endocrinol Metab. 2010; 299: E364 -373. [PubMed] .
- 87. Yancy WS Jr, Foy M, Chalecki AM, Vernon MC, Westman EC. A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutr Metab (Lond). 2005; 2 -34. [PubMed] .
- 88. Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008; 132: 1025 -1038. [PubMed] .
- 89. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15: 713 -724. [PubMed] .
- 90. Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci U S A. 2000; 97: 5440 -5444. [PubMed] .
- 91. Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008; 104: 683 -695. [PubMed] .
- 92. Gasior M, Rogawski MA, Hartman AL. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. 2006; 17: 431 -439. [PubMed] .
- 93. Henderson ST. Ketone bodies as a therapeutic for Alzheimer's disease. Neurotherapeutics. 2008; 5: 470 -480. [PubMed] .
- 94. Tieu K, Perier C, Caspersen C, Teismann P, Wu D-C, Yan S-D, Naini A, Vila M, Jackson-Lewis V, Ramasamy R. D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. Journal of Clinical Investigation. 2003; 112: 892 -901. [PubMed] .
- 95. Bates EA, Victor M, Jones AK, Shi Y, Hart AC. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006; 26: 2830 -2838. [PubMed] .
- 96. Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007; 447: 550 -555. [PubMed] .
- 97. Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004; 432: 1027 -1032. [PubMed] .
- 98. Fitzgerald VK, Mensack MM, Wolfe P, Thompson HJ. A transfer-less, multi-well liquid culture feeding system for screening small molecules that affect the longevity of Caenorhabditis elegans. Biotechniques. 2009; 47: 4 Suppl ix -xv. [PubMed] .
- 99. Timmons L and Fire A. Specific interference by ingested dsRNA. Nature. 1998; 395: 854 -854. [PubMed] .
- 100. Leong SF and Clark JB. Regional enzyme development in rat brain. Enzymes associated with glucose utilization. Biochem J. 1984; 218: 131 -138. [PubMed] .
- 101. Williamson JR and Corkey BE. Assays of intermediates of the citric acid cycle and related compounds by fluorometric enzyme methods John ML. Methods in Enzymology: Academic Press. 1969; 434 -513. .
- 102. Shukla V, Phulara SC, Yadav D, Tiwari S, Kaur S, Gupta MM, Nazir A, Pandey R. Iridoid compound 10-O-trans-p-coumaroylcatalpol extends longevity and reduces alpha synuclein aggregation in Caenorhabditis elegans. CNS Neurol Disord Drug Targets. 2012; 11: 984 -992. [PubMed] .