



Cancer, like most other chronic diseases, is a signal transduction disease par excellence, a consequence of aberrant transmission of environmental cues to the interior of the cell via a complex network of signaling hubs. Heterotrimeric G proteins are one such major signaling hub that are essential components of the signal gating machinery in healthy eukaryotic cells. They serve as molecular switches for signal transmission via 7 transmembrane domain G protein coupled receptors (GPCRs) to intracellular effectors [39]. Activation of G proteins is tightly regulated by a network of modulators: guanine nucleotide exchange factors (GEFs) trigger activation, GTPase activating proteins (GAPs) enhance inactivation, and finally, guanidine dissociation inhibitors (GDIs) uncouple the trimer and maintain the G protein in an inactive (GDP-bound) conformation [40]. These modulators function coordinately to maintain finiteness of signal transduction via G proteins [41], mostly by ensuring that activation of G proteins is spatially and temporally restricted, i.e., triggered exclusively at the plasma membrane (PM) by agonist activation of GPCRs via a process that is completed within a few hundred milliseconds [42].
The importance of maintaining the critical balance between G protein activation and deactivation and the loss of such balance in cancer has been highlighted by studies on several cancer-associated mutants of trimeric G protein α-subunits and GPCRs (reviewed in [43, 44]). These mutations trigger malignant transformation and oncogenesis by rendering the G proteins constitutively active in the GTP-bound conformation either by impairing its intrinsic ability to hydrolyze GTP (i.e., GTPase-deficient) or by reducing its sensitivity to the action of GAPs (i.e., GAP-insensitive). Thus, it has now been established that “hyperactivation of G proteins” is a bona-fide basis for oncogenic signaling via trimeric G proteins.
Despite the insights gained, the rare oncogenic driver mutations in G proteins in a handful of cancers do not explain the basis for deregulated G protein signaling in the vast majority of cancers that do not harbor mutant G or GPCR proteins. A growing body of work by us and others [24, 45, 46] have indicated that genetic or epigenetic factors that deregulate the intricate network of G protein regulatory proteins are just as significant as those that directly affect the G proteins /GPCRs, if not more. More specifically, a recently identified family of non-receptor GEFs, called rheostats [35] best exemplify the wide prevalence and broad significance of deregulated G protein regulatory network in cancers. Rheostats like GIV (Gα-Interacting Vesicle-associated; a.k.a Girdin) [24] and other members of this family, are non-receptor GEFs for trimeric G proteins; they derive their name based on their ability to 'adjust' the duration of G protein signaling depending on the abundance of functional copies of the GEF in cells [35]. Studies on GIV-GEF have led to the rapid emergence of a new paradigm in non-canonical activation of trimeric G proteins that has distinctive temporal and spatial features. Such activation appears to be less constrained and less restricted than canonical G protein activation by receptor GEFs (i.e., GPCRs) in three major ways (summarized in [24]): 1) G proteins can be transactivated by diverse classes of receptors, e.g., growth factor RTKs, TLRs, integrins and GPCRs--many of which are typically not known to bind or activate G proteins; 2) G proteins both at the PM and on internal membranes that are discontinuous with the PM can be activated; and 3) Activation continues for prolonged periods of time (as opposed to milliseconds). While the molecular mechanisms that govern such non-canonical G protein activation and the variety of pathways it modulates (summarized in Figure 1) are still unfolding, the relevance of this new paradigm in cancer and other diseases is clear (summarized in [24]).
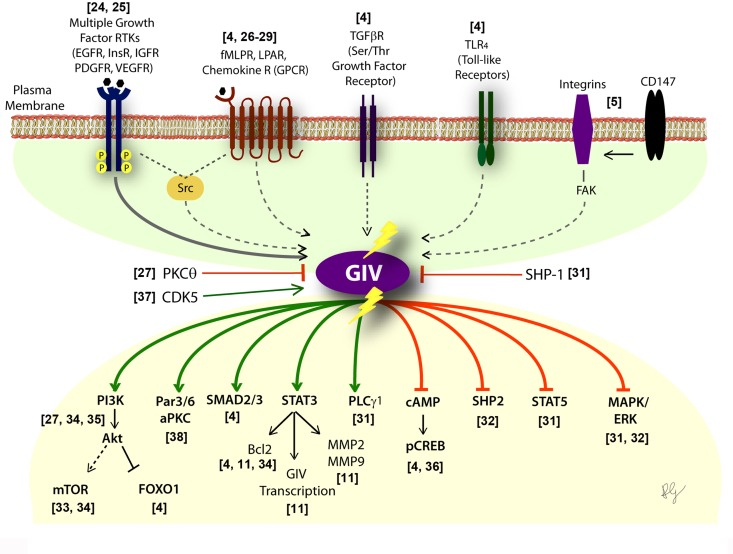
Figure 1. Activation of G proteins by GIV-GEF modulates multi-receptor signaling and broadly impacts the downstream signaling network Schematic showing the diverse classes of receptors (upper half) which sense a variety of chemical signals, that converge on GIV. Lower part shows the consequence of non-canonical transactivation of G proteins by GIV (when GIV-GEF is functionally intact or turned "ON") on the multitude of downstream pathways within the signaling network. Green = enhancement; Red = suppression. Shown in the middle are three known ways to either inhibit (PKCθ selectively phosphoinhibits GIV-GEF [27]; SHP-1 dephosphorylates tyrosine-phosphorylated GIV [30]) or activate (CDK5 phosphoactivates GIV-GEF [37]) GIV-dependent signaling.
Most of these diseases, if not all, are characterized by cellular processes (migration, proliferation, apoptosis/survival, autophagy, secretion, etc) that are driven by more than one receptor or one class of receptors, and most often require synergistic signaling of diverse classes of receptors. GIV appears to serve as a platform on which crosstalk between diverse classes of receptors either directly (in the case of RTKs [24]) or indirectly (via mechanisms that are still unclear) converge; GIV's intrinsic GEF activity subsequently translates the converging signals into activation of Gαi in the vicinity of activated receptors. The impact of such transactivation on downstream signals is equally diverse (Figure 1). When GIV-GEF is transcriptionally upregulated [11, 38] and/or turned "ON" by phospho-activation [37], Gαi is activated and multiple signaling pathways are either enhanced or suppressed, thereby affecting an entire network, not just individual pathways. Conversely, when GIV-GEF is turned "OFF" by selective phosphoinhibition [27] or alternative splicing [31, 35], Gαi activation cannot be coupled to incoming signals; consequently, the network assumes a yin-yang mirror image pattern (Figure 1).
Given the broad landscape of signaling pathways that GIV modulates, and its ubiquitous nature of expression, it is not surprising that deregulation of GIV-GEF drives several pathophysiologic conditions (Table). In the context of cancer, it is clear that high copies of GIV means unrestricted G protein signaling and propagation of signals that enhance tumorigenesis (like invasiveness, stemness and chemoresistance; [24], Table) regardless of the receptor of origin. Given the nature of receptor classes modulates and the prometastatic nature of the signaling pathways enhanced, GIV's expression at high levels carries a poor prognosis across a broad range of solid tumors (Figure 2). Although prometastatic signaling is the most well understood role of GIV, the striking yin-yang effect of GEF-ON versus GEF-OFF states has also been described in the context of some other cellular processes and diseases, e.g., fibrosis, wound healing, diabetes [24], most, if not all these diseases are also multi-receptor in origin (Table; green columns). It is possible that a similar yin-yang effect modulates all other diseases where GIV's role has been defined but the role of its GEF function has not (Table; red columns).
| Disease/Pathology Investigated | Effect of GIV's GEF function | Receptor(s) Studied | Citation | |
|---|---|---|---|---|
| Cancer Progression | Migration/Invasion | “ON” = Enhances “OFF” = Inhibits | IGF1R, EGFR, Multi-receptor* | [8, 11, 36, 51, 52] |
| Stemness | Not examined | -- | [19] | |
| Chemoresistance | Not examined | -- | [53] | |
| Tumor-Stroma Interactions | Not examined | PDGFR, TGFβR, CXCR4 | [2] | |
| Tumor angiogenesis | Not examined | VEGFR | [54] | |
| Organ Fibrosis (Liver) | Myofibroblast transdifferentiation, collagen production, chemotaxis, mitosis, anti-apoptotic signaling | “ON” = Enhances “OFF” = Inhibits | PDGFR, CCR1, TGFβR | [4] |
| Dermal Wound Healing | Wound closure | “ON” = Enhances “OFF” = Inhibits | Multi-receptor* | [52] |
| Nephrotic Syndrome | Podocyte survival after glomerular injury | “ON” = Enhances survival “OFF” = Inhibits survival | VEGFR | [34] |
| Insulin Resistance, Type II Diabetes | Metabolic insulin response in the skeletal muscle | Not examined | InsR | [55] |
| Disorders of Blood Vessels | Neonatal vascular development; Pathologic neovascularization; vein repair; vein graft | Not examined | PDGF, Angiotensin II, VEGF | [56-59] |
| Neuronal Plasticity, Memory formation | Synaptic plasticity | Not examined | NMDA | [60] |
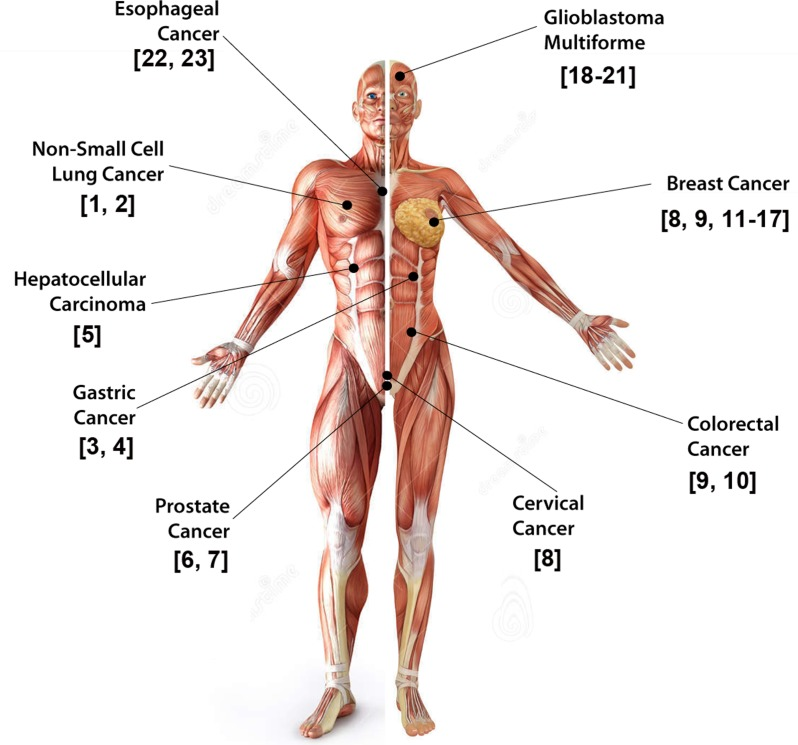
Figure 2. GIV is a bona-fide prometastatic protein Schematic summarizing the variety of solid tumors in which elevated expression of GIV/Girdin in tumor cells has been linked to its role in imparting stemness, invasiveness, prometastatic and anti-apoptotic signaling, aggressiveness and poor clinical outcome has been studied.
Finally, the ability to manipulate a broad signaling network and reset it from an unstable pathologic to a stable physiologic state downstream of multiple receptors by fine-tuning GIV's GEF function is an attractive concept because of many reasons: 1) Eliminates the need to block physiologic signaling via cell surface receptors; 2) Overcomes the limitations of unknown upstream and downstream components; 3) Preserves the utility of biomarkers/therapeutic targets despite re-wiring of signaling pathways during the course of disease progression. Recently, in a proof-of-concept study [47] using cell permeable peptides exogenous modulation of GIV's GEF function allowed resetting pathologic signaling networks and phenotypes in diverse cell types, while sparing individual receptors. Such a strategy represents a fundamental deviation from the current strategy of individual pathway/receptor-blockade, that sooner or later fails due to a switch in addiction of the tumor from the targeted pathway to other pathways [48]. An appropriate analogy for such individual receptor/pathway blockade is the futility of severing the heads of a Hydra; for each head severed, two more grows in its place. While the studies on GIV-GEF indicate that it may be an unusual hub for convergent multi-receptor signaling for the broad modulation of the "disease network", and raise our hope that the GIV●Gαi-interface may serve as an effective target for therapy, several hurdles lie ahead of us before such a possibility can be realized. For example, targeting a protein like GIV, which is expressed ubiquitously and serves a long list of roles in normal tissues [24] may carry an insurmountable risk of side effects. Even if targeting the targeted therapy selectively to the tumor cells overcomes the first challenge, the second challenge is that inhibition of the GIV●Gαi-interface may inadvertently disrupt also signaling via other members of this family [49, 50] that share a similar structural basis for activating G proteins.
In conclusion, through the studies on GIV, we have obtained a sneak preview of just how large the footprint of oncogenic signaling via trimeric G proteins could be. Because GIV is just one of the members of the rheostat family, we are likely seeing only the proverbial tip of the iceberg, and a lot more must be known before any of these findings can be translated to transformative and impactful therapies and/or biomarkers for personalized care.
Funding
This work was funded by NIH (R01CA160911 and DK099226) to P.G.
Conflicts of Interest
The author has no conflict of interests to declare.
References
- 1. Song JY, et al. Clinical significance of Girdin expression detected by immunohistochemistry in non-small cell lung cancer. Oncol Lett. 2014; 7: 337 -341. [PubMed] .
- 2. Yamamura Y, et al. Akt-Girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer Res. 2015; 75: 813 -823. [PubMed] .
- 3. Wang C, et al. Expression and clinical significance of girdin in gastric cancer. Mol Clin Oncol. 2014; 2: 425 -428. [PubMed] .
- 4. Lopez-Sanchez I, et al. GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat Commun. 2014; 5: 4451 [PubMed] .
- 5. Wang Y, et al. A chimeric antibody targeting CD147 inhibits hepatocellular carcinoma cell motility via FAK-PI3K-Akt-Girdin signaling pathway. Clin Exp Metastasis. 2015; 32: 39 -53. [PubMed] .
- 6. Tomiyama L, et al. Loss of Dlg5 expression promotes the migration and invasion of prostate cancer cells via Girdin phosphorylation. Oncogene. 2015; 34: 1141 -1149. [PubMed] .
- 7. Zhao W, et al. In Vitro Antimetastatic Effect of Phosphatidylinositol 3-Kinase Inhibitor ZSTK474 on Prostate Cancer PC3 Cells. Int J Mol Sci. 2013; 14: 13577 -13591. [PubMed] .
- 8. Jiang P, et al. An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 2008; 68: 1310 -1318. [PubMed] .
- 9. Garcia-Marcos M, et al. Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB J. 2011; 25: 590 -599. [PubMed] .
- 10. Jun BY, et al. Expression of girdin in human colorectal cancer and its association with tumor progression. Dis Colon Rectum. 2013; 56: 51 -57. [PubMed] .
- 11. Dunkel Y, et al. STAT3 protein up-regulates Galpha-interacting vesicle-associated protein (GIV)/Girdin expression, and GIV enhances STAT3 activation in a positive feedback loop during wound healing and tumor invasion/metastasis. J Biol Chem. 2012; 287: 41667 -41683. [PubMed] .
- 12. Jin F, et al. Clinical implications of Girdin and PI3K protein expression in breast cancer. Oncol Lett. 2013; 5: 1549 -1553. [PubMed] .
- 13. Lin C, et al. Tyrosine phosphorylation of the Galpha-interacting protein GIV promotes activation of phosphoinositide 3-kinase during cell migration. Sci Signal. 2011; 4 -ra6. [PubMed] .
- 14. Liu C, et al. Girdin protein: a new potential distant metastasis predictor of breast cancer. Med Oncol. 2012; 29: 1554 -1560. [PubMed] .
- 15. Nishimae K, et al. The impact of Girdin expression on recurrence-free survival in patients with luminal-type breast cancer. Breast Cancer. 2013; .
- 16. Wang A, et al. Expression of tumor necrosis factor receptor-assicated factor 4 correlates with expression of Girdin and promotes nuclear translocation of Girdin in breast cancer. Mol Med Rep. 2015; 11: 3635 -3641. [PubMed] .
- 17. Peng WT, et al. Elevated expression of Girdin in the nucleus indicates worse prognosis for patients with estrogen receptor-positive breast cancer. Ann Surg Oncol. 2014; 21: S648 -656. [PubMed] .
- 18. Gu F, et al. Girdin, an actin-binding protein, is critical for migration, adhesion, and invasion of human glioblastoma cells. J Neurochem. 2014; 131: 457 -469. [PubMed] .
- 19. Natsume A, et al. Girdin maintains the stemness of glioblastoma stem cells. Oncogene. 2012; 31: 2715 -2724. [PubMed] .
- 20. Zhang B, et al. Reduction of Akt2 inhibits migration and invasion of glioma cells. Int J Cancer. 2009; 125: 585 -595. [PubMed] .
- 21. Ni W, et al. Girdin regulates the migration and invasion of glioma cells via the PI3KAkt signaling pathway. Mol Med Rep. 2015; Epub ahead of print .
- 22. Cao K, et al. Talen-mediated girdin knockout downregulates cell proliferation, migration and invasion in human esophageal carcinoma ECA109 cells. Mol Med Rep. 2014; 10: 848 -854. [PubMed] .
- 23. Shibata T., et al. Girdin, a regulator of cell motility, is a potential prognostic marker for esophageal squamous cell carcinoma. Oncol Rep. 2013; 29: 2127 -2132. [PubMed] .
- 24. Ghosh P. G protein-Coupled Growth Factor Receptor Tyrosine Kinases: No Longer an Oxymoron. Cell Cycle. 2015; Epub ahead of print .
- 25. Garcia-Marcos M, et al. GIV/Girdin transmits signals from multiple receptors by triggering trimeric G protein activation. J Biol Chem. 2015; 290: 6697 -6704. [PubMed] .
- 26. Ghosh P, et al. Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol. 2008; 182: 381 -393. [PubMed] .
- 27. Lopez-Sanchez I, et al. Protein kinase C-theta (PKCtheta) phosphorylates and inhibits the guanine exchange factor, GIV/Girdin. Proc Natl Acad Sci U S A. 2013; 110: 5510 -5515. [PubMed] .
- 28. Garcia-Marcos M, et al. A structural determinant that renders G alpha(i) sensitive to activation by GIV/girdin is required to promote cell migration. J Biol Chem. 2010; 285: 12765 -12777. [PubMed] .
- 29. Garcia-Marcos M, et al. GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc Natl Acad Sci U S A. 2009; 106: 3178 -3183. [PubMed] .
- 30. Mittal Y, et al. Src homology domain 2-containing protein-tyrosine phosphatase-1 (SHP-1) binds and dephosphorylates G(alpha)-interacting, vesicle-associated protein (GIV)/Girdin and attenuates the GIV-phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway. J Biol Chem. 2011; 286: 32404 -32415. [PubMed] .
- 31. Ghosh P, et al. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell. 2010; 21: 2338 -2354. [PubMed] .
- 32. Lin C, et al. Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol Biol Cell. 2014; 25: 3654 -3671. [PubMed] .
- 33. Garcia-Marcos M, et al. A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol Biol Cell. 2011; 22: 673 -686. [PubMed] .
- 34. Wang H, et al. GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J Am Soc Nephrol. 2015; 26: 314 -327. [PubMed] .
- 35. Ghosh P, et al. GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh Migr. 2011; 5: 237 -248. [PubMed] .
- 36. Midde KK, et al. Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proc Natl Acad Sci U S A. 2015; 112: E937 -46. [PubMed] .
- 37. Bhandari D, et al. Cyclin Dependent Kinase 5 Phosphorylates and Activates Guanidine Exchange Factor GIV/Girdin. Proc Natl Acad Sci U S A. 2015; in Press .
- 38. Sasaki K, et al. Regulation of epithelial cell polarity by PAR-3 depends on Girdin transcription and Girdin-Galphai3 signaling. J Cell Sci. 2015; 128: 2244 -2258. [PubMed] .
- 39. Oldham WM, et al. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008; 9: 60 -71. [PubMed] .
- 40. Siderovski DP, et al. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005; 1: 51 -66. [PubMed] .
- 41. Morris AJ, et al. Physiological regulation of G protein-linked signaling. Physiol Rev. 1999; 79: 1373 -1430. [PubMed] .
- 42. Lohse MJ, et al. Kinetics of G-protein-coupled receptor signals in intact cells. Br J Pharmacol. 2008; 153: S125 -132. [PubMed] .
- 43. Dorsam RT, et al. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007; 7: 79 -94. [PubMed] .
- 44. O'Hayre M, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013; 13: 412 -424. [PubMed] .
- 45. Liebmann C. G protein-coupled receptors and their signaling pathways: classical therapeutical targets susceptible to novel therapeutic concepts. Curr Pharm Des. 2004; 10: 1937 -1958. [PubMed] .
- 46. Hurst JH, et al. Hooks, Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochem Pharmacol. 2009; 78: 1289 -1297. [PubMed] .
- 47. Ma GS, et al. Therapeutic effects of cell-permeant peptides that activate G proteins downstream of growth factors. Proc Natl Acad Sci U S A. 2015; 112: E2602 -2610. [PubMed] .
- 48. Chong CR, et al. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013; 19: 1389 -1400. [PubMed] .
- 49. Aznar N, et al. Daple is a Novel Non-Receptor GEF Required for Trimeric G protein Activation in Wnt Signaling. eLife. 2015; .
- 50. Garcia-Marcos M, et al. G Protein binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of G(alpha)i-regulatory motifs. J Biol Chem. 2011; 286: 28138 -28149. [PubMed] .
- 51. Enomoto A, et al. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005; 9: 389 -402. [PubMed] .
- 52. Ma GS, et al. Therapeutic effects of cell-permeant peptides that activate G proteins downstream of growth factors. Proc Natl Acad Sci U S A. 2015; 19: E2602 -2610. [PubMed] .
- 53. Zhang YJ, et al. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World J Gastroenterol. 2014; 20: 8229 -8236. [PubMed] .
- 54. Kitamura T, et al. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008; 10: 329 -337. [PubMed] .
- 55. Hartung A, et al. The Akt substrate Girdin is a regulator of insulin signaling in myoblast cells. Biochim Biophys Acta. 2013; 1833: 2803 -2811. [PubMed] .
- 56. Ito T, et al. Girdin and its phosphorylation dynamically regulate neonatal vascular development and pathological neovascularization in the retina. Am J Pathol. 2013; 182: 586 -596. [PubMed] .
- 57. Miyachi H, et al. Role of Girdin in intimal hyperplasia in vein grafts and efficacy of atelocollagen-mediated application of small interfering RNA for vein graft failure. J Vasc Surg. 2014; 60: 479 -489 e5. [PubMed] .
- 58. Miyake H, et al. The actin-binding protein Girdin and its Akt-mediated phosphorylation regulate neointima formation after vascular injury. Circ Res. 2011; 108: 1170 -1179. [PubMed] .
- 59. Miyachi H, et al. A Novel Approach against Vascular Intimal Hyperplasia Through the Suppression of Girdin. Ann Vasc Dis. 2015; 8: 69 -73. [PubMed] .
- 60. Nakai T, et al. Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. J Neurosci. 2014; 34: 14995 -15008. [PubMed] .