



Introduction
Reducing the expression of the Indy (I'm Not Dead Yet) gene in lower organisms extends life span by mechanism resembling caloric restriction [1–3]. In D. melanogaster, mutating the Indy gene reduces body fat content, insulin-like proteins and reactive oxygen species production, extending life span [1, 4]. Interestingly, Indy mRNA is down-regulated by dietary restriction in normal flies and it was shown that Indy long-lived flies share several phenotypes with long-lived calorie restricted flies [2]. Similarly, in C.elegans, knock down of the Indy homolog CeNAC2 extends life span [5], an effect mediated at least in part via AMPK/aak2 [6]. The mammalian Indy homolog encoded protein mINDY (NaCT) is part of the SLC13 protein family, consisting of Na-carboxylate and Na-sulfate cotransporters in vertebrates, invertebrates, plants, and bacteria [7]. mINDY mediates the co-transport of citrate, succinate, and several other dicarboxylates across the plasma membrane together with sodium in an electrogenic manner [8, 9]. The amino acid sequence of the N-terminal sodium and the carboxy-binding motif is highly conserved between many species, from bacterium to rat to human [10].
Our laboratory has demonstrated that deletion of the mIndy gene (Slc13a5) protects mice from aging and high fat diet-induced adiposity, insulin resistance and hepatic steatosis [9]. Moreover, we were able to show that in rats, glucagon is a transcriptional regulator of the mIndy gene, inducing mIndy expression via a CREB-dependent mechanism [11]. The highest mRNA expression levels of mIndy in mammals are observed in the liver [9], however, liver specific contributions to its beneficial effects have not been determined so far. mIndy has been suggested to be a target for the treatment of aging- and life style induced metabolic diseases [7, 10, 12], and it is, thus, of high interest to understand which tissues need to be targeted to achieve the full beneficial effect.
Here, our aim was to investigate the effect of inducible and liver specific knock down of mIndy via the use of 2′-O-methoxyethyl chimeric antisense oligonucleotides (ASOs). We used this approach to test whether or not targeting mIndy in adult, high-fat fed rats, is sufficient to prevent hepatic steatosis and hepatic insulin resistance. Moreover, we chose this intervention because it resembles a therapeutic approach.
Results
Effect on fasting parameters and liver fat content
After 4 weeks of 2′-O-methoxyethyl chimeric ASO treatment mIndy mRNA expression was reduced by 91% (P<0.001) in the treatment group (Fig. 1A). Body weight changes over the course of the study remained similar between the two groups.
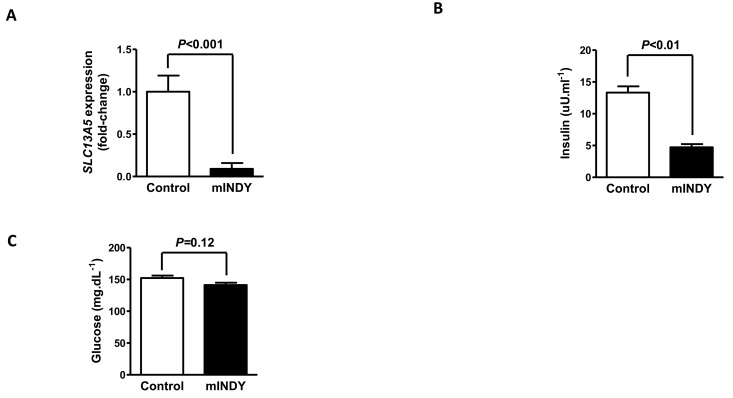
Figure 1. (A) After 4 weeks of 2′-O-methoxyethyl chimeric ASO treatment, mIndy mRNA expression was reduced by 91%. (B) Fasting plasma insulin concentrations were markedly reduced in the mIndy ASO treated rats. (C) Fasting glucose concentrations in the mIndy ASO and control ASO treated groups. All data are mean ± SEM, N=10 for each group; significances by double sided t-test.
The mIndy 2′-O-methoxyethyl chimeric ASO treated rats showed a 74% reduction in fasting plasma insulin (13.3 ± 1.0 μU.mL−1 vs. 4.7 ± 0.5 μU.mL−1, P<0.01) and their fasting plasma glucose concentration tended to be reduced (152.3 ± 3.8 mg.dL−1 for control group vs. 141.2 ± 3.7 mg.dL−1 for mIndy ASO treated, P=0.12) compared to the control group (Fig. 1B+C), suggesting improved insulin sensitivity in the mIndy ASO treated rats.
Liver triglycerides in the mIndy ASO treated group were significantly reduced (17.9 ± 1.6 mg.g−1 tissue for control group vs. 13.4 ± 1.2 mg.g−1 tissue for mIndy ASO treated, P=0.04) (Fig. 2A). Consistent with this, we observed a significant 35% reduction of plasma triglycerides in the mIndy ASO treated group compared to the control group (37.0 ± 2.6 mg.dL−1 for control group vs. 24.6 ± 1.6 mg.dL−1 for mIndy ASO treated, P<0.01) (Fig. 2B). Other plasma metabolites were determined by an unbiased metabolomic approach using GC-TOF-MS. Profiles indicated significant changes in specific amino acids and total cholesterol and are listed in Table 1.

Figure 2. (A) 4 weeks of mIndy ASO treatment resulted in a 25% reduction in liver triglycerides and (B) a 35% reduction in plasma triglycerides. All data are mean ± SEM, N=10 for each group; significances by double sided t-test
Table 1.
| Metabolite | Control (RPI) | mINDY ASO (RPI) | P – value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glucose | 5.95 ± 0.02 | 5.82 ± 0.01 | 0.012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cholesterol | 4.68 ± 0.02 | 4.58 ± 0.04 | 0.040 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Palmitic Acid | 4.66 ± 0.03 | 4.51 ± 0.05 | 0.022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Octadecanoic acid | 4.28 ± 0.04 | 4.15 ± 0.04 | 0.025 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tryptophan | 4.49 ± 0.02 | 4.39 ± 0.04 | 0.041 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glucopyranose | 5.19 ± 0.03 | 5.07 ± 0.01 | 0.010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tyrosine | 4.52 ± 0.02 | 4.43 ± 0.04 | 0.035 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Methionine | 4.12 ± 0.01 | 4.04 ± 0.02 | 0.005 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ornithine | 4.17 ± 0.02 | 4.08 ± 0.03 | 0.028 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Plasma metabolite profiles as assessed by GC-TOF-MS metabolite profiling in mIndy ASO treated rats and animals from the control group. RPI=Relative peak intensities of the metabolites; normalized by the median of 13C-sorbitol intensities of all samples by the 13C-sorbitol intensity of the respective sample and log10 transformed. All data are mean ± SEM, N=10 for each group. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Effect on insulin sensitivity
To determine the organ specific contribution to the improvement in glucose metabolism, the gold standard hyperinsulinemic-euglycemic clamp (HEC) test with stable isotope tracers was used. The glucose infusion rate during the clamp was significantly higher in mIndy ASO treated rats as compared to the control group (26.6 ± 1.4 mg.kg−1.min−1 for control group vs. 31.2 ± 1.2 mg.kg−1.min−1 for mIndy ASO treated rats, P<0.01) (Fig. 3A), confirming improved insulin sensitivity. The improvement was associated with a trend towards a reduction in basal rates of hepatic glucose production (5.0 ± 0.5 mg.kg−1.min−1 for control group vs. 4.2 ± 0.4 mg.kg−1.min−1 for mIndy ASO treated rats, P=0.07) during the HEC (Fig. 3B). Furthermore, hepatic insulin responsiveness was increased in the mIndy 2′-O-methoxyethyl chimeric ASO rats as reflected by increased suppression of hepatic glucose production during the HEC (28 ± 5% for control group vs. 52 ± 8% for mIndy ASO treated rats, P<0.05) (Fig. 3C).
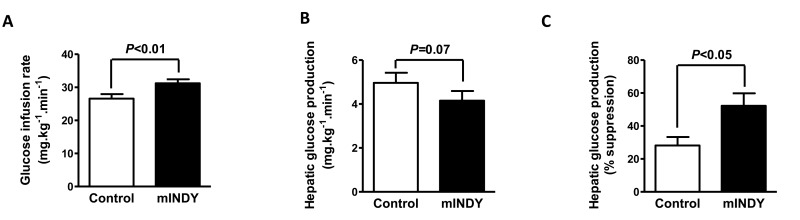
Figure 3. (A) Glucose infusion rate during the hyperinsulinemic-euglycemic clamp (HEC) is increased in mIndy ASO treated rats. (B) Trend for reduced hepatic glucose production during the HEC in the mIndy ASO treated rats. (C) Suppression of hepatic glucose production during the HEC was increased in mIndy ASO treated rats as compared to the control group. All data are mean ± SEM, N=10 for each group; significances by double sided t-test.
Discussion
Our data are the first to show that inducible liver-specific knockdown of the mammalian homolog of the longevity gene Indy (Slc13a5) using second generation ASOs in rats ameliorates diet-induced hepatic steatosis, reduces plasma insulin, lipid and amino acid levels and improves hepatic insulin sensitivity independent of body weight. These features are core components of the metabolic syndrome that develops with high calorie diets [16] and aging [17, 18]. mIndy encodes for a citrate transporter located on the plasma membrane of hepatocytes [19, 20]. Citrate is a central metabolite associated with metabolic regulation [21] and aging [22]. Cytosolic citrate is converted to acetyl-coenzyme A (acetyl-CoA), the essential precursor for fatty acid, triglyceride and cholesterol biosynthesis. In our data set, we observed that reducing mIndy in the liver reduced hepatic as well as plasma triglyceride content. It is well established that reducing ectopic lipid deposition results in improved insulin sensitivity [16, 23], as shown in our study by marked reductions in basal insulin levels, a trend to improved basal hepatic glucose output and improved insulin mediated suppression of hepatic glucose production. In line with our data, reducing the conversion of cytosolic citrate to acetyl-CoA prevents liver lipid accumulation and insulin resistance [24] in mice. Moreover, we have shown that whole body deletion of mIndy reduces citrate uptake into the liver and prevents aging- and diet-induced accumulation of hepatic triglycerides and diacylglycerols , a mechanism well known to protect from hepatic insulin resistance [9, 16, 25, 26]. Reducing mIndy in primary human hepatocytes reduces hepatocellular lipid content [27] while overexpression or stimulation of mIndy in primary hepatocytes and cell lines increases hepatocellular citrate uptake and citrate derived lipid content [11, 28, 29].
Previous data indicated that reducing Indy/mIndy in lower organisms and mice lead to cellular and molecular processes that mediate a healthy aging process and longevity [1], i.e. increased mitochondrial biogenesis [3, 4, 9], reduced rate of reactive oxygen species production per mitochondrion [3], reduced body fat content [2, 5, 6, 9], increased PGC-1α expression [3, 4] and activated AMPK [6, 9]. Our data presented here add novel aspects. The insulin/igf1 pathway has been shown to be involved in mammalian regulation of lifespan and tumor growth [18, 30]. Moreover, restriction of specific amino acids, such as methionine [31], has been shown to extend lifespan in different species [32, 33]. Interestingly, our data indicate that selective hepatic reduction of mIndy markedly reduces fasting insulin levels, and our unbiased metabolomic data show that specific amino acid levels, i.e. methionine, tyrosin and ornithine, are reduced. Decreased levels of circulating amino acids have also been put into context of improved insulin sensitivity [34]. In humans, key serum amino acids levels change with aging [35]. Reduced amino acid concentrations might therefore resemble a phenotype going along with a healthy aging process. Cross-sectional and prospective analyses of large patient cohorts revealed that elevated plasma amino acid levels, especially of branched chain amino acids, tyrosine, alanine and phenylalanine, may contribute to an increased incidence of diabetes [36] and are also related to life span [37].
mIndy ASO treated rats further show reduced total cholesterol as well as stearic acid and palmitic acid levels in the metabolome profile. It is well established that serum free fatty acids (FFAs) are important contributing factors for the development of insulin resistance [16]. Along these lines, increased de novo lipid synthesis occurs via insulin-stimulated elongation of palmitic acid, contributing to increased stearic acid levels [38]. In humans, circulating levels of palmitic acid and stearic acid are correlated to insulin resistance while reduced levels of FFAs improve insulin resistance [38].
In contrast to whole body mIndy−/− mice [9], mIndy ASO treatment did not affect body weight. Differences between the two models include a shorter timeframe of mIndy knockdown in the study presented here, the fact that knockdown is less effective than deletion and that other organs besides the liver may contribute to the body weight effect. In this context, it is important to note that mutations in the human SLC13A5 gene have been reported in neonatal childhood epilepsy with teeth hypoplasia or hypodontia in a recent study [39], a feature not observed in the whole body Slc13a5 knockout mice so far. Whether or not these mutations are causative is not entirely clear. From our data presented here, we can now conclude that selective hepatic reduction of mIndy is able to ameliorate hepatic steatosis and to improve diet-induced insulin resistance. Neuronal contribution seems not to be needed. Our data suggest that hepatic mIndy is an interesting target for the treatment of NAFLD and type 2 diabetes and first selective mINDY (SLC13A5) inhibitors have shown promise in this regard (40)Further studies need to address the important question whether or not the knockdown of mIndy in mammals will also promote longevity.
Methods
Animals
All protocols were approved by the Yale University School of Medicine Animal Care and Use Committee. Male Sprague-Dawley rats were purchased from Charles River Laboratories at about 400 g. After the rats acclimated for at least one week, rats received i.p. injections over a period of 4 weeks of either 2′-O-methoxyethyl chimeric anti-sense oligonucleotides targeted against mIndy or a control ASO targeted against a sequence that does not match any known transcript in the rat. During the 4 weeks of ASO treatment, animals were fed a 60% high-fat diet based on safflower oil (Dyets, Bethlehem, Pennsylvania, USA) and body weights were monitored biweekly. All animals had ad libitum access to 6% sucrose water. After the treatment, rats underwent surgery under isoflurane anesthesia and catheters were inserted in the jugular vein and internal carotid artery. All animals were allowed to recover for at least 1 week before any further experiments were performed.
Measurement of liver and plasma triglycerides
Liver triacylglycerides were extracted by the method of Bligh and Dyer [13] and measured spectrophotometrically with a commercial triglyceride reagent (Diagnostic Chemicals Limited [DCL], Oxford, Connecticut). Plasma was collected from the jugular vein of overnight fasted rats and assayed for plasma triglyceride concentrations using a commercial DCL triglyceride reagent.
Hyperinsulinemic-euglycemic clamp
For the hyper-insulinemic-euglycemic clamp, after an overnight fast, awake, unrestrained rats first underwent a primed-continuous basal infusion of [6,6-2H2] glucose (5 mg.kg−1.min−1 prime for 5 min, 1 mg.kg−1.min−1 continuous infusion) through a catheter in the carotid artery. Blood samples (300 μl whole blood) were taken from a catheter in the jugular vein after 100, 110, and 120 min of infusion. This was followed by a HEC with regular insulin (40 mU.kg−1 bolus, 4 mU.kg−1.min−1) continuous infusion). Blood samples were taken at 15, 30, 45, 60, 75, 90, 100, 110, and 120 min of the clamp and glucose measured using a YSI Glucose Analyzer. Variable [6,6-2H2] glucose was given to maintain euglycemia (100-110 mg.dL−1) during the clamp. Glucose uptake in skeletal muscle was assessed after administration of a 40 μCi bolus of [14C]2-deoxyglucose at the end of the clamp and measured as described earlier [14].
mRNA quantification by Real-Time PCR
Liver total RNA was isolated using the RNeasy kit per manufacturer's instructions (Qiagen, Valencia, California, USA), and qPCR was performed as described earlier [15]. Actin was used as a housekeeping gene.
Plasma metabolite extraction, measurement, alignment and normalization
30 μl of the murine plasma was extracted with ice cold (−20°C) 400 μl 100% Methanol (13C-sorbitol added as an internal standard to control for technical variation). After shaking and centrifugation, the supernatant was vacuum-dried. GC-TOF-MS metabolite profiling was performed on a Leco Pegasus 3 time-of-flight mass spectrometer (Leco, St.Joseph, MI, USA). The Direct Thermal Desorption injector (ATAS GL International, The Netherlands) was coupled to an HP 5890 gas chromatograph and an autosampler with automatic derivatisation and liner exchange. This eliminates the impact of potential degradation or synthesis artifacts and sample carryover. During the derivatisation a retention time index standard mixture was also added. For detailed information refer to [9]. Chromatogram acquisition parameters were those described previously [9]. The results were exported from Leco Chroma TOF software (version 3.25) as cdf-files. Peak detection, retention time alignment, and library matching were performed with the R-script “Target Search”. Relative peak intensities of the metabolites were normalized by the median of 13C-sorbitol intensities of all samples by the 13C-sorbitol intensity of the respective sample and log10 transformed.
Statistical analysis
All results are presented as mean ± SEM. Group comparisons (N=10 for each group) were performed by the 2-tailed unpaired Student's t-test or ANOVA where appropriate using Prism 6 for Windows software (GraphPad, Inc., La Jolla, CA, USA). A P-value of 0.05 or less was considered statistically significant.
Abbreviations
ASO: Anti-sense oligonucleotide; HEC: hyperinsulinemic-euglycemic clamp; NAFLD: nonalcoholic fatty liver disease.
Acknowledgments
We thank Mario Kahn, John Stack, Gina Butrico, Ali Nasiri, Jianying Dong and Maria Batsu for technical assistance.
Funding
This project was supported by grants from the Austrian Science Fund (FWF), project number J 3267, the German Research Foundation (BI1292/4-2), the German Center for Diabetes Research (DZD) and the National Institutes of Health (R01 DK-40936, P30 DK-45735).
Conflicts of Interest
SB is an employee of Isis Pharmaceuticals and may own stock in the company. ALB owns shares of Eternygen GmbH. The remaining authors declare that there is no duality of interest associated with this manuscript.
References
- 1. Rogina B, et al. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science. 2000; 290: 2137 -2140. [PubMed] .
- 2. Wang PY, et al. Long-lived Indy and calorie restriction interact to extend life span. Proc Natl Acad Sci U S A. 2009; 106: 9262 -9267. [PubMed] .
- 3. Neretti N, et al. Long-lived Indy induces reduced mitochondrial reactive oxygen species production and oxidative damage. Proc Natl Acad Sci U S A. 2009; 106: 2277 -2282. [PubMed] .
- 4. Rogers RP and Rogina B. Increased mitochondrial biogenesis preserves intestinal stem cell homeostasis and contributes to longevity in Indy mutant flies. Aging (Albany NY). 2014; 6: 335 -350. [PubMed] .
- 5. Fei YJ, et al. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem J. 2004; 379: 191 -198. [PubMed] .
- 6. Schwarz F, et al. Knockdown of Indy/CeNac2 extends Caenorhabditis elegans life span by inducing AMPK/aak-2. Aging (Albany NY). 2015; 7: 553 -567. [PubMed] .
- 7. Willmes DM and Birkenfeld AL. The Role of INDY in Metabolic Regulation. Comput Struct Biotechnol J. 2013; 6: e201303020 [PubMed] .
- 8. Gopal E, et al. Transport of nicotinate and structurally related compounds by human SMCT1 (SLC5A8) and its relevance to drug transport in the mammalian intestinal tract. Pharm Res. 2007; 24: 575 -584. [PubMed] .
- 9. Birkenfeld AL, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011; 14: 184 -195. [PubMed] .
- 10. Mancusso R, et al. Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature. 2012; 491: 622 -626. .
- 11. Neuschafer-Rube F, et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes. 2014; 63: 1048 -1057. [PubMed] .
- 12. Rogers RP and Rogina B. The role of INDY in metabolism, health and longevity. Front Genet. 2015; 6: 204 [PubMed] .
- 13. Bligh E.G and Dyer W.J. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959; 37: 911 -917. [PubMed] .
- 14. Erion DM, et al. Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metab. 2009; 10: 499 -506. [PubMed] .
- 15. Birkenfeld AL, et al. Influence of the hepatic eukaryotic initiation factor 2alpha (eIF2alpha) endoplasmic reticulum (ER) stress response pathway on insulin-mediated ER stress and hepatic and peripheral glucose metabolism. J Biol Chem. 2011; 286: 36163 -36170. [PubMed] .
- 16. Birkenfeld AL and Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 Diabetes. Hepatology. 2014; 59: 713 -723. [PubMed] .
- 17. North BJ and Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. 2012; 110: 1097 -1108. [PubMed] .
- 18. de Cabo R, et al. The search for antiaging interventions: from elixirs to fasting regimens. Cell. 2014; 157: 1515 -1526. [PubMed] .
- 19. Gopal E, et al. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am J Physiol Gastrointest Liver Physio. 2007; 292: G402 -408. .
- 20. Knauf F, et al. The life-extending gene Indy encodes an exchanger for Krebs-cycle intermediates. Biochem J. 2006; 397: 25 -29. [PubMed] .
- 21. Alves TC, et al. Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab. 2015; 22: 936 -947. [PubMed] .
- 22. Cheng S, et al. Distinct metabolomic signatures are associated with longevity in humans. Nat Commun. 2015; 6: 6791 [PubMed] .
- 23. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014; 371: 1131 -1141. [PubMed] .
- 24. Wang Q, et al. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology. 2009; 49: 1166 -1175. [PubMed] .
- 25. Jornayvaz FR, et al. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc Natl Acad Sci U S A. 2011; 108: 5748 -5752. [PubMed] .
- 26. Lee HY, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology. 2011; 54: 1650 -1660. [PubMed] .
- 27. Li L, et al. SLC13A5 is A Novel Transcriptional Target of the Pregnane X Receptor and Sensitizes Drug-Induced Steatosis in Human Liver. Mol Pharmacol. 2015; 87: 674 -682. [PubMed] .
- 28. Inoue K, et al. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem J. 2003; 374: 21 -26. [PubMed] .
- 29. Neuschafer-Rube F, et al. Arylhydrocarbon receptordependent mIndy (Slc13a5) induction as possible contributor to benzo[a]pyrene-induced lipid accumulation in hepatocytes. Toxicology. 2015; 337: 1 -9. [PubMed] .
- 30. Rozing MP, et al. Human insulin/IGF-1 and familial longevity at middle age. Aging (Albany NY). 2009; 1: 714 -722. [PubMed] .
- 31. Ruckenstuhl C, et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014; 10: e1004347 [PubMed] .
- 32. Emran S, et al. Target of rapamycin signalling mediates the lifespan-extending effects of dietary restriction by essential amino acid alteration. Aging (Albany NY). 2014; 6: 390 -398. [PubMed] .
- 33. Solon-Biet SM, et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014; 19: 418 -430. [PubMed] .
- 34. Palmer ND, et al. Metabolomic profile associated with insulin resistance and conversion to diabetes in the Insulin Resistance Atherosclerosis Study. J Clin Endocrinol Metab. 2015; 100: E463 -468. [PubMed] .
- 35. Bancel E, et al. Effect of the age and the sex on plasma concentration of amino acids. Ann Biol Clin (Paris). 1994; 52: 667 -670. [PubMed] .
- 36. Tillin T, et al. Diabetes risk and amino acid profiles: crosssectional and prospective analyses of ethnicity, amino acids and diabetes in a South Asian and European cohort from the SABRE (Southall And Brent REvisited) Study. Diabetologia. 2015; 58: 968 -979. [PubMed] .
- 37. Edwards C, et al. Mechanisms of amino acid-mediated lifespan extension in Caenorhabditis elegans. BMC Genet. 2015; 16: 8 [PubMed] .
- 38. Chu X, et al. Sterol regulatory element-binding protein-1c mediates increase of postprandial stearic acid, a potential target for improving insulin resistance, in hyperlipidemia. Diabetes. 2013; 62: 561 -571. [PubMed] .
- 39. Hardies K, et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. 2015; 138: 3238 -3250. [PubMed] .
- 40. Huard K, et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci Rep. 2015; 5: 17391 [PubMed] .