



Introduction
Mitochondria are the powerhouses of the cell, and a consequence of their energy production is the generation of reactive oxygen species (ROS) that may contribute to cellular damage, protein misfolding and ultimately aging [1,2]. Accompanying aging is a decline in the cell's capacity to mitigate damage from ROS and maintain protein homeostasis. Threats to mitochondrial protein homeostasis are reduced in part due to the activity of chaperones like HSP-6 and HSP-60 that are known to combat age-related dysfunction as part of the mitochondrial unfolded protein response (UPRmt) [3]. The UPRmt has received considerable attention in recent years as it has been revealed that its role may not be limited to stress response but may also regulate longevity [1,4]. Although the UPRmt is activated against damaging environmental conditions and toxins, it is not known if an organism's dietary status contributes to mitochondrial surveillance mechanisms. We previously reported that C. elegans subjected to high dietary glucose were protected against proteotoxicity and environmental stress [5], but had shortened lifespans as previously observed [6]. We wondered whether or not the UPRmt had a role in mediating longevity phenotypes associated with high dietary glucose.
The mitochondrial chaperone reporters hsp-6p::GFP and hsp-60p::GFP show increased expression in C. elegans intestinal tissue as part of the UPRmt [3]. Cultivation of worms under high dietary glucose conditions led to a dose-dependent increase in the expression of hsp-6p::GFP (Fig. 1a), but no change was observed for hsp-60p::GFP (Supp. Fig. 1). Induction of hsp-6p::GFP expression was specific to glucose, as other dietary compounds including trehalose, methionine or oleic-acid had no effect (Fig. 1b). In C. elegans the transcription factors ATFS-1 [7] and DVE-1 [8] are required for induction of the UPRmt, as is the ubiquitin-like protein UBL-5. We turned to well characterized translational reporters for dve-1 and ubl-5 [1,3,8] to investigate the possible role of dietary glucose in activation of the UPRmt. We observed that high dietary glucose increased expression of the dve-1p::dve-1::GFP reporter (Fig. 1c-d) but did not affect the expression of ubl-5p::ubl-5::GFP (Supp. Fig. 1). We next investigated if the transcription factors atfs-1 or dve-1 were functionally required for UPRmt induction. RNAi mediated knockdown of either atfs-1 or dve-1 abolished the induction of hsp-6p::GFP by high dietary glucose (Fig. 1e-f). These data suggest that a specific subset of the UPRmt acts to mitigate potential cytotoxic effects from dietary sources.
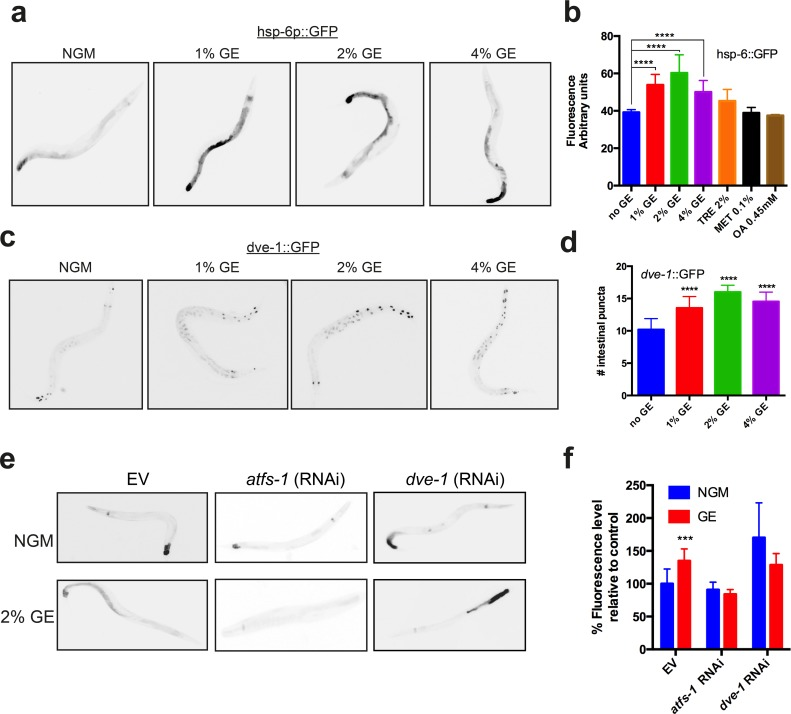
Figure 1. Glucose enrichment induces HSP-6 and DVE-1 expression (a) Glucose enrichment (GE) induces expression of a hsp-6::GFP transcriptional reporter at concentrations of 1, 2 and 4%. (b) Quantification of hsp-6p::GFP expression (mean ± SD, ****P<0.0001 compared to control). (c) GE increases expression of a dve-1::GFP translational reporter at all glucose concentrations tested. (d) Quantitative representation of DVE-1 expression after GE treatment (mean ± SD, ****P<0.0001 compared to control). (e) Knockdown of atfs-1 or dve-1 abolishes GE mediated hsp-6p::GFP expression. (f) Quantification of hsp-6p::GFP expression after atfs-1 or dve-1 (RNAi) (mean ± SD, ****P<0.0001).
Results
The UPRmt declines during aging
The induction of hsp-6p::GFP by high dietary glucose was observed in young adult animals (Fig. 1a). It has been hypothesized that aging may occur partly from an age-related decline of protein homeostasis mechanisms [9], thus we wondered if the induction of hsp-6p::GFP by glucose was maintained in aging animals. We observed that exposure to high dietary glucose at three concentrations induced the expression of hsp-6p::GFP in animals at days 1 and 3 (Fig. 2a, b) of adulthood, but this induction was lost in animals at days 5 or 9 of adulthood (Fig. 2c, d). To inspect the morphology of the mitochondrial network we used transgenic worms expressing a mitochondrial targeted signal in muscles cells [3]. Normally the mitochondrial network appears as tubular, parallel structures in muscle tissue [1,10].
We observed that these structures became increasingly disordered when the worms were exposed to high dietary glucose as well as during aging (Supp. Fig. 2). These data suggest that the ability of the UPRmt to respond to cellular toxicity decreases during aging and mitochondrial health is damaged by high dietary glucose.
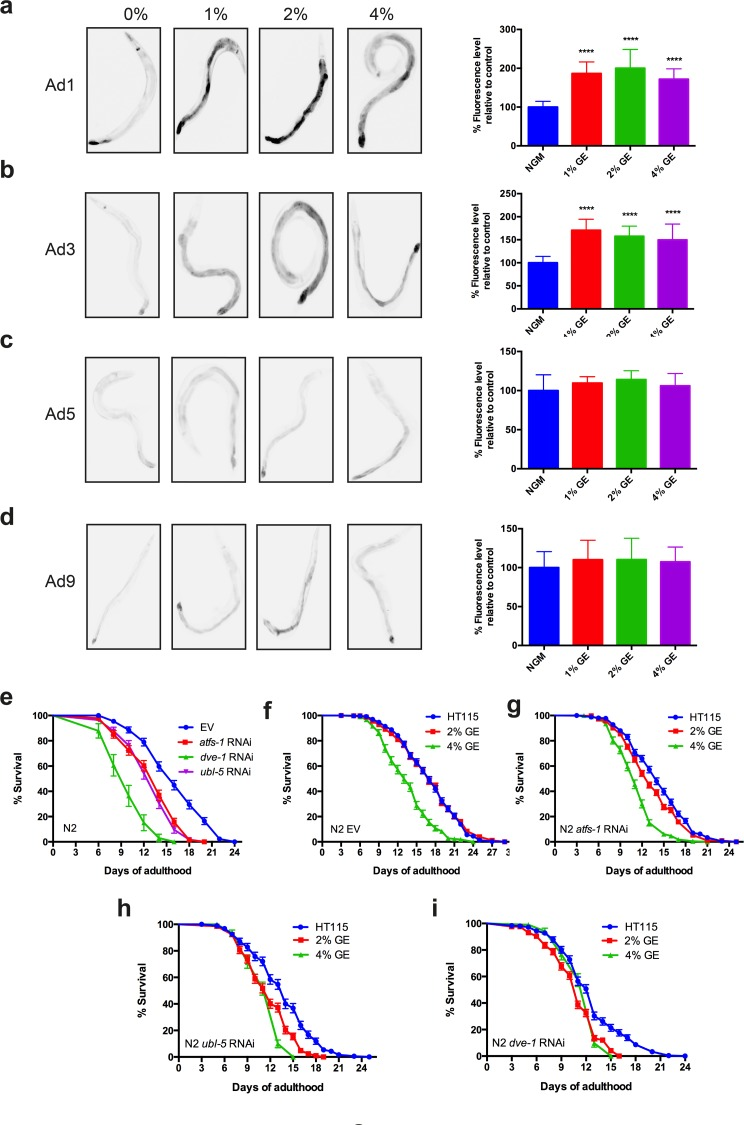
Figure 2. Glucose enrichment maintains HSP-6 expression over time (a-b) Photographs and quantification of increased hsp-6p::GFP expression in strains exposed to 1, 2 or 4% GE at days 1 and 3 of adulthood (****P<0.0001, mean ± SD). (c-d) GE failed induced expression of the hsp-6::GFP reporter in worms at days 5 or 9 of adulthood (mean ± SD). (e) RNAi against atfs-1, dve-1, or ubl-5 reduced the lifespan of N2 worms compared to controls (P<0.0001 compared to empty vector (EV) controls). (f) Worms grown on EV had reduced lifespan only at 4% GE (P<0.0001 compared to controls). Animals grown on (g) atfs-1, (h) dve-1 or (i) ubl-5 RNAi had reduced lifespan at 2% and 4% GE (P<0.0001 compared to EV control).
The UPRmt protects against glucose-mediated lifespan reduction
We next investigated whether the UPRmt protects against glucose-induced reduction of lifespan in adult animals. Wild type N2 animals exposed to RNAi against atfs-1, dve-1, or ubl-5 all showed decreased lifespan suggesting that a fully-functional UPRmt is necessary for promoting longevity (Fig 2e). Wild type N2 worms exposed to 2% glucose had lifespan similar to untreated controls, but exposure to 4% glucose reduced lifespan (Fig 2f). Interestingly, lifespan was reduced in worms exposed to either 2% or 4% glucose and treated with atfs-1, dve-1 or ubl-5 (RNAi) (Fig 2g-i). These data demonstrate that 2% glucose becomes toxic and reduces lifespan in the absence of a functional UPRmt, thus the UPRmt may protect organisms against cytotoxicity from dietary sources.
Early developmental exposure to glucose extends lifespan
The induction of hsp-6p::GFP by glucose does not indicate if the UPRmt is activated to mitigate the toxic effects of glucose, or instead mediates the protective effects of high dietary glucose. Activation of the UPRmt has been observed after inhibition of mitochondrial DNA (mtDNA) translation, or mtDNA depletion and is involved in longevity phenotypes [1]. Furthermore, the loss of UPRmt results in increased susceptibility to stress, while prolonged induction reduces lifespan (12-14). To delineate the role of the UPRmt in glucose-mediated aging phenotypes, we investigated whether activation of the UPRmt by high dietary glucose could be the cause of glucose-dependent lifespan reduction. Wild-type animals exposed to high dietary glucose for their entire life had reduced lifespan compared to untreated animals, in agreement with previous findings by us [5] and others [6] (Fig 3a). Surprisingly, animals exposed to glucose (2% and 4%) only until the pre-adult L4 larval stage, showed the opposite effect with significant lifespan extension (Fig. 3b). Furthermore, exposing worms to high dietary glucose only during adulthood reduced lifespan compared to control animals (Fig. 3c), suggesting a lifespan-promoting role of glucose during development that becomes deleterious at later adult stages. To determine if the augmented lifespan effect of early glucose exposure was dependent on the UPRmt, we investigated the contribution of the transcription factor ATFS-1, which is a master regulator of the UPRmt [7]. We exposed afts-1(gk3094) and atfs-1(et15) mutants to high dietary glucose from hatching until the L4 larval stage and observed that the enhanced longevity phenotype was lost (Fig. 3d,e). These data suggest that endogenous UPRmt activity via ATFS-1 is required for regulating glucose-dependent lifespan phenotypes.
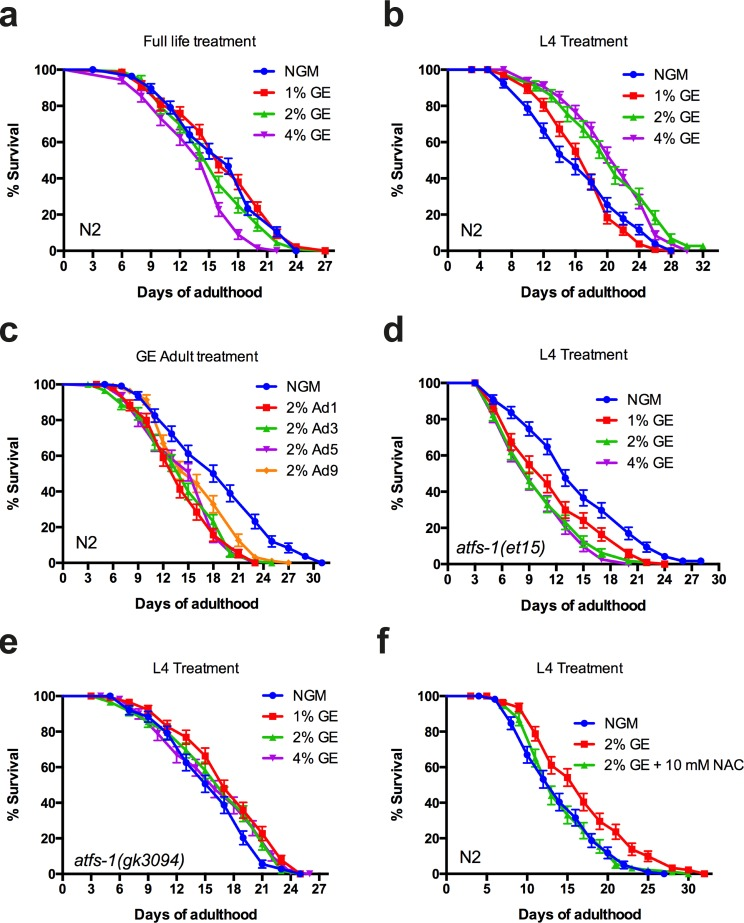
Figure 3. Early or late glucose enrichment has opposite effects on lifespan (a) GE reduced lifespan of N2 animals at 4% concentration compared to untreated animals (P<0.0001 compared to control). (b) Larval exposure to glucose extends lifespan of N2 animals at concentrations of 2% or 4% compared to untreated animals (P<0.0001 compared to controls). (c) Adult exposure to glucose reduces lifespan of wild-type animals (P<0.0001 for all conditions compared to control). (d-e) GE failed to extend lifespan in atfs-1(et15) and atfs-1(gk3094) animals when treatment ceased at the L4 larval stage. (f) GE failed to extends lifespan in N2 animals when treated with 10 mM N-acetyl cysteine.
Dietary induced mitohormesis extends lifespan
Mitochondria are the major ROS producers at the cellular level and it has long been proposed that ROS molecules and by-products are responsible for organismal aging (15). Recent work suggested that ROS rather acts as a signalling molecule with important roles in regulating longevity [11,12]. This phenomenon is known as mitohormesis [13] and the protective effects are lost in the presence of antioxidant compounds. Thus we wondered if the lifespan enhancing effect from early glucose exposure utilized a mitohormetic mechanism requiring ROS. We observed that animals treated with 10 mM N-acetyl cysteine did not have increased lifespan when exposed to high dietary glucose until the L4 stage GE (Fig. 3f). These data suggest a role for oxidative stress in glucose mediated lifespan extension when applied at early, pre-adulthood stages.
Lifespan extension by glucose requires a functional electron transport chain
Mutations that disrupt mitochondrial function can result in severe pathologies, and mitochondrial dysfunction is linked to multiple neurodegenerative disorders in humans (19-21). Surprisingly, electron transport chain (ETC) dysfunction can extend lifespan not only in C. elegans but also in Drosophila and mice [14]. Thus, we wondered if ETC function was required for the temporal effects on lifespan from high dietary glucose. We observed that the ETC complex II mutant mev-1(kn1) and the complex III mutant isp-1(qm150) strains showed no increase in lifespan when exposed to high dietary glucose until the L4 larval stage (Supp. Fig. 3). These data suggest that functional mitochondria are required for the increased lifespan from early exposure to high dietary glucose. This is consistent with the previous studies demonstrating that lifespan extending interventions can only be produced at late larval stages (L3/L4) when mitochondrial biogenesis is at it's peak [15].
Activation of the UPRmt during adulthood abolishes lifespan extension
Glucose treatment over the life of the worm is known to reduce lifespan and in this study we have shown that it is linked to increased hsp-6 expression. This suggests that activation of the UPRmt during adulthood may reduce lifespan. We wondered if early exposure only might have a similar pattern of hsp-6 expression.
Worms were treated with glucose during larval development and then grown on control NGM. At different aging stages, the animals were transferred to cytochrome c-oxydase cco-1(RNAi) plates for a 12h period. cco-1(RNAi) is known to induce the UPRmt by disrupting ETC function, and knockdown of this gene during development has been reported to mimic ETC dysfunction with similar longevity phenotypes to knockdown of other ETC genes [16]. Worms grown on NGM and placed on cco-1 RNAi as adults showed no hsp-6::GFP increased expression, consistent with the temporal activation of the UPRmt [17] (Fig. 4). Similar to untreated animals, nematodes exposed to dietary glucose early in life showed no increase in hsp-6::GFP expression. However, animals treated with glucose during development plus adult specific cco-1 RNAi had increased hsp-6::GFP expression (Fig. 4a). This effect was sustained until adult day 5 for all glucose concentrations tested (Fig. 4b, c) and was lost at adult day 9 for the lowest concentration (1%) (Fig. 4d). We wondered if there were functional consequences of additional mitochondrial stress during adulthood on lifespan. We observed that worms grown on NGM plates and exposed to cco-1(RNAi) at day 1 of adulthood had lifespans similar to untreated controls (Fig 4e). We then tested animals that had been exposed to either 2% or 4% GE during development, and then exposed them to cco-1(RNAi) at day 1 of adulthood, and observed that lifespan was reduced compared to control animals (Fig. 4f, g). Thus, early exposure to mitochondrial stress can increase lifespan, but this is a fragile phenotype since further induction of the UPRmt during adulthood results in lifespan reduction.
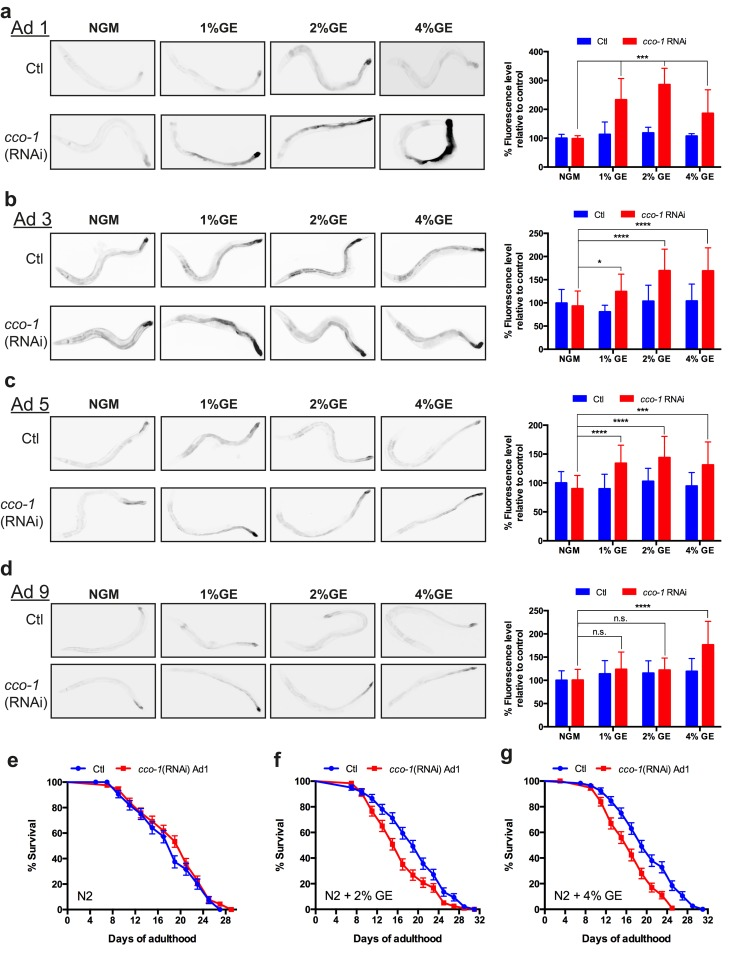
Figure 4. Early exposure to glucose sensitizes nematodes to UPRmt activation (a-d) Early exposure (L4) to glucose at different concentrations (1, 2 and 4%) had no effect on hsp-6 expression during adulthood compared to control NGM conditions. RNAi against cco-1 was able to induce hsp-6 expression in animals treated at all glucose concentrations until day 5 of adulthood, and only at 2% and 4% at day 9 (mean ± SD., *P<0.05, **P<0.01 and ****P<0.0001 compared to control). (e) The lifespan on N2 worms treated with cco-1(RNAi) during adulthood was similar to N2 control worms. The lifespans of N2 worms grown on (f) 2% or g. 4% GE plates was reduced when treated with adult-stage specific cco-1(RNAi) compared to worms treated with EV (P<0.01 for 2% GE + cco-1(RNAi) and P<0.0001 for 4% GE + cco-1(RNAi) versus controls.)
Discussion
Our study reveals a novel role of dietary glucose in regulating lifespan in C. elegans. Exposure to high dietary glucose during adulthood reduces lifespan, whereas glucose exposure restricted to developing animals has the opposite effect and extends lifespan. It is believed that mitochondrial oxidants may be responsible for mitochondria communicating to their cellular host resulting in an organism-wise shift to stress resistance and pro-longevity phenotypes [18]. High glucose concentrations are associated with a number of negative phenotypes, including decreased lifespan in C. elegans as well as mitochondrial dysfunction and oxidative stress in other organisms [6,19]. Consistently, impairing glucose metabolism extends lifespan in worms most likely via a ROS dependent signalling mechanism [12]. Altogether these studies paint a contradictory picture where both increased or decreased mitochondrial respiration are associated with increased lifespan. It may be that altered ROS signalling from dysregulated mitochondrial respiration triggers a hormetic response leading to increased lifespan [18].
Our data suggests that timing and duration may be an important factor in whether mitochondrial stress results in positive or negative effects on lifespan. High dietary glucose during development extends lifespan, requires UPRmt genes but does induce strong activation of the UPRmt. However, high dietary glucose exposure during adulthood does induce an UPRmt response and is associated with decreased lifespan. Thus glucose acts a stress at the cellular and mitochondrial level that limits lifespan if not alleviated during adulthood. These effects are consistent with the hyperfunction theory of aging that suggest cellular pathways activated during development can accelerate aging if activity is maintained during adulthood [20].
In contrast to other work [1], we found that glucose-mediated induction of the UPRmt to extend lifespan required ROS. This suggests that dietary induced mitohormesis and UPRmt may employ mechanisms different than impaired mitochondrial translation. Thus dietary mitohormesis may establish conditions in adult organisms that sensitize them to lifespan reduction via additional mitochondrial stress. Furthermore, mitochondrial stress and/or mitohormesis from dietary sources are not restricted to developmental stages in the natural world. Our data suggest that chronic engagement of the UPRmt via diet sensitizes animals to additional mitochondrial stress and may be a universal lifespan-limiting factor.
Materials and Methods
Worm strains and genetics
Worms were maintained on standard NGM plates streaked with OP50 E. coli. In some experiments D-glucose was added to NGM plates. All strains were scored at 20°C unless indicated. Strains used in this study were obtained from the C. elegans Genetics Center (University of Minnesota, Minneapolis) unless indicated and include: atfs-1(gk3094); atfs-1(et15); mev-1(kn1); isp-1(qm150); zcIs13[hsp-6::GFP]; zcIs9[hsp-60::GFP]; zcIs39[dve-1p::dve-1::GFP] ; zcIs19[ubl-5p::ubl-5::GFP]; zcIs14[myo-3::GFP(mit)] ; ccIs4251[myo-3p::GFP(NLS)::LacZ (pSAK2) + myo-3p::GFP(mit) (pSAK4) + dpy-20(+)] and sid-1(pk3321).
Fluorescence microscopy
Animals were immobilized in M9 with 5 mM levamisole and mounted on slides with 2% agarose pads. Animals were visualized with a Leica 6000 and a Leica DFC 480 camera. A minimum of 30 animals was scored per treatment over 3 trials. Mito-GFP animals were visualized at days 1, 5 and 9 of adulthood with a Leica TCS_SP5 confocal microscope and analyzed with LAS AF software. The mean and SEM were calculated for each trial and two-tailed t-tests were used for statistical analysis.
Lifespan assays
Worms were grown on NGM or NGM + 1%, 2% or 4% glucose and transferred on NGM or NGM + glucose. 30-40 animals/plate by triplicates were tested at 20°C from adult day 1 until death. Worms were declared dead if they didn't respond to tactile or heat stimulus.
RNAi experiments
RNAi-treated strains were fed with E. coli (HT115) containing an Empty Vector (EV), atfs-1 (ZC376.7), ubl-5 (F46F11.6), cco-1 (F26E4.9) RNAi clones from the ORFeome RNAi library (2) and dve-1(ZK1193.5) clone from Arhinger RNAi library. RNAi experiments were performed at 20°C. Worms were grown on either NGM or NGM + 2% glucose both enriched with 1 mM Isopropyl-b-D-thiogalactopyranoside (IPTG).
Statistics
For paralysis and stress-resistance tests, survival curves were generated and compared using the Log-rank (Mantel-Cox) test, and a 60-100 animals were tested per genotype and repeated at least three times.
Supplementary Materials
Acknowledgments
We thank S. Peyrard for technical assistance.
Funding
A.T. was supported by a CIHR and Huntington's Society of Canada fellowship. This work was supported by a NSERC Discovery Grant to J.A.P.
Conflicts of Interest
The authors declare no conflict of interests.
References
- 1. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knoautt G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013; 497: 451 -457. https://doi.org/10.1038/nature12188 [PubMed] .
- 2. Yang W and Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. Tissenbaum HA. PLoS Biol. 2010; 8: e1000556 https://doi.org/10.1371/journal.pbio.1000556 [PubMed] .
- 3. Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006; 174: 229 -239. https://doi.org/10.1534/genetics.106.061580 [PubMed] .
- 4. Liu Y, Samuel BS, Breen PC, Ruvkun G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature. 2014; 508: 406 -410. https://doi.org/10.1038/nature13204 [PubMed] .
- 5. Tauffenberger A, Vaccaro A, Aulas A, Vande Velde C, Parker JA. Glucose delays age-dependent proteotoxicity. Aging Cell. 2012; 11: 856 -866. https://doi.org/10.1111/j.1474-9726.2012.00855.x [PubMed] .
- 6. Lee S-J, Murphy CT, Kenyon C. Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 2009; 10: 379 -391. https://doi.org/10.1016/j.cmet.2009.10.003 [PubMed] .
- 7. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012; 337: 587 -590. https://doi.org/10.1126/science.1223560 [PubMed] .
- 8. Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007; 13: 467 -480. https://doi.org/10.1016/j.devcel.2007.07.016 [PubMed] .
- 9. Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell. 2015; 161: 919 -932. https://doi.org/10.1016/j.cell.2015.03.032 [PubMed] .
- 10. Burkewitz K, Morantte I, Weir HJM, Yeo R, Zhang Y, Huynh FK, et al. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell. 2015; 160: 842 -855. https://doi.org/10.1016/j.cell.2015.02.004 [PubMed] .
- 11. Van Raamsdonk JM and Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. Kim SK. 2009; 5: e1000361 https://doi.org/10.1371/journal.pgen.1000361 [PubMed] .
- 12. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6: 280 -293. https://doi.org/10.1016/j.cmet.2007.08.011 [PubMed] .
- 13. Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med. 2014; 20: 709 -711. https://doi.org/10.1038/nm.3624 [PubMed] .
- 14. Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001; 1: 633 -644. [PubMed] .
- 15. Tsang WY and Lemire BD. Mitochondrial genome content is regulated during nematode development. Biochem Biophys Res Commun. 2002; 291: 8 -16. https://doi.org/10.1006/bbrc.2002.6394 [PubMed] .
- 16. Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007; 5: e259 https://doi.org/10.1371/journal.pbio.0050259 [PubMed] .
- 17. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011; 144: 79 -91. https://doi.org/10.1016/j.cell.2010.12.016 [PubMed] .
- 18. Yun J and Finkel T. Mitohormesis. Cell Metab. 2014; 19: 757 -766. https://doi.org/10.1016/j.cmet.2014.01.011 [PubMed] .
- 19. Sharma K. Mitochondrial hormesis and diabetic complications. Diabetes. 2015; 64: 663 -672. https://doi.org/10.2337/db14-0874 [PubMed] .
- 20. Gems D and la Guardia de Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. Mary Ann Liebert, Inc 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA 2013; 19: 321 -329. https://doi.org/10.1089/ars.2012.4840 [PubMed] .