Specific premature epigenetic aging of cartilage in osteoarthritis
Abstract
Osteoarthritis (OA) is a disease affecting multiple tissues of the joints in the elderly, but most notably articular cartilage. Premature biological aging has been described in this tissue and in blood cells, suggesting a systemic component of premature aging in the pathogenesis of OA. Here, we have explored epigenetic aging in OA at the local (cartilage and bone) and systemic (blood) levels. Two DNA methylation age-measures (DmAM) were used: the multi-tissue age estimator for cartilage and bone; and a blood-specific biomarker for blood. Differences in DmAM between OA patients and controls showed an accelerated aging of 3.7 years in articular cartilage (95% CI = 1.1 to 6.3, P = 0.008) of OA patients. By contrast, no difference in epigenetic aging was observed in bone (0.04 years; 95% CI = -1.8 to 1.9, P = 0.3) and in blood (-0.6 years; 95% CI = -1.5 to 0.3, P = 0.2) between OA patients and controls. Therefore, premature epigenetic aging according to DNA methylation changes was specific of OA cartilage, adding further evidence and insight on premature aging of cartilage as a component of OA pathogenesis that reflects damage and vulnerability.
Introduction
OA is the most common chronic disease affecting the joints with about a 45% lifetime risk of developing OA of the knee [1,2]. It can affect any joint, but it occurs most often in knees, hips, spine, or hands. Symptoms include pain and stiffness, bony enlargement, crepitus with movement and decreased function of the joint. OA pathogenesis is complex and includes multiple risk factors that are still incompletely known, but old age is a critical contributor [3,4]. The relationship between old age and OA is not fully understood. Classically, it was suspected that the association was related to the “wear and tear” of articular cartilage by continuous mechanical stress. Today, we know that this model of OA is insufficient because OA involves an active response to injury comprising remodeling of articular cartilage and neighboring bone, in addition of synovial inflammation and damage to ligaments and menisci [2]. In addition, the other component of the association with old age, biological aging, has shown unsuspected complexity, including its multidimensionality, variable progression, possibility of modulation and the pivotal role played by senescent cells [5].
The many facets of biological aging have been typified in nine cellular and molecular hallmarks: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [6]. Variable progression of biological aging with dissociation between biological and chronological age is observed in progeroid syndromes. Less dramatically, it is also observed as a reflection of lifestyle with smoking, heavy drinking, obesity, stress and depression as accelerators, and exercise and caloric restriction as rejuvenators. The pivotal role of cellular senescence and of the senescence-associated secretory phenotype has been established in multiple studies, but most strikingly with the reversal of age-associated changes obtained with their removal [7]. This rejuvenation has been obtained either through genetic manipulation or with senolytic drugs in mice in spite of the eliminated senescent cells were only a minor fraction in mouse tissues [5]. All these aspects could be of relevance for OA as exemplified by the secretory phenotype that includes secretion of metalloproteases and pro-inflammatory mediators, which could be involved in OA cartilage damage [2-4]. There is already persuasive evidence of accelerated biological aging at the affected cartilage [3,4]. Many of the aging hallmarks have been described as exacerbated in OA chondrocytes and articular cartilage, including telomere length shortening, mitochondrial dysfunction, cellular senescence and genome instability [3,4]. In contrast, biological age has not been studied in any other joint tissue although a systemic component of premature aging has been suggested by accelerated telomere length shortening in blood cells of 160 hand OA subjects compared with 926 controls [8]. Telomere shortening correlated with radiographic severity of OA in the hands in this study. These findings have not yet been independently confirmed, with a small subsequent study showing no telomere attrition in blood of knee OA patients [9], and a second small study reporting telomere shortening only in knee OA patients experiencing high stress and chronic pain [10]. A systemic premature aging component in OA is an attractive hypothesis because it is congruent with some epidemiological studies that have found increased prevalence of old-age comorbidities [11-14], frailty [15], and mortality in OA patients [16-18]. The two aspects, local and systemic, of premature aging could contribute to OA by further impairing cartilage and joint function, decreasing mobility and increasing joint vulnerability.
An opportunity to explore a different aging hallmark in OA cartilage, bone and blood has become possible thanks to the recent development of biomarkers of epigenetic aging [19-24]. The available biomarkers, called DNA methylation age-measures (DmAM), combine methylation levels at CpG sites that experience methylation changes with aging. The mechanism seems to include slowly accumulating failures of methylation maintenance (epigenetic drift) that could be accelerated by somatic mutations, cell divisions and environmental stress [19,21,25,26]. Some of the changes are tissue-specific; others are shared by several tissues. This motivates a distinction between DmAM that are tissue-specific and include as few as 3 CpG sites showing strong correlation with age in blood [20,24], or in saliva [23], and biomarkers applicable to many tissues that require investigating more CpG sites [19,21]. The most comprehensive is the "epigenetic clock" method by Horvath, which includes 353 CpG and is valid for multiple tissues including bone and cartilage [19,27,28]. The DmAM are useful biomarkers of biological age that show accelerated aging in several diseases of old age [19-21,29,30] and in subjects under elevated lifetime stress [26], and that correlate with cognitive and physical fitness in the elderly and with all-cause and cause-specific mortality [30-35].
Results
The cartilage samples from OA patients showed premature aging in comparison with cartilage from controls (Figure 1A). The difference in the estimated mean age obtained with Horwath’s DmAM was of 3.7 years (Table 1). This result was obtained with the whole set of samples that included cartilage from the tibial plateau and from the femoral head. A significant premature aging was also observed with the subgroup of tibial plateau samples, with a mean difference of 5.3 years (95% CI = 2.4 to 8.2). Cartilages from the femoral heads were too few for meaningful analysis. All the comparisons were adjusted for age and sex as covariates.
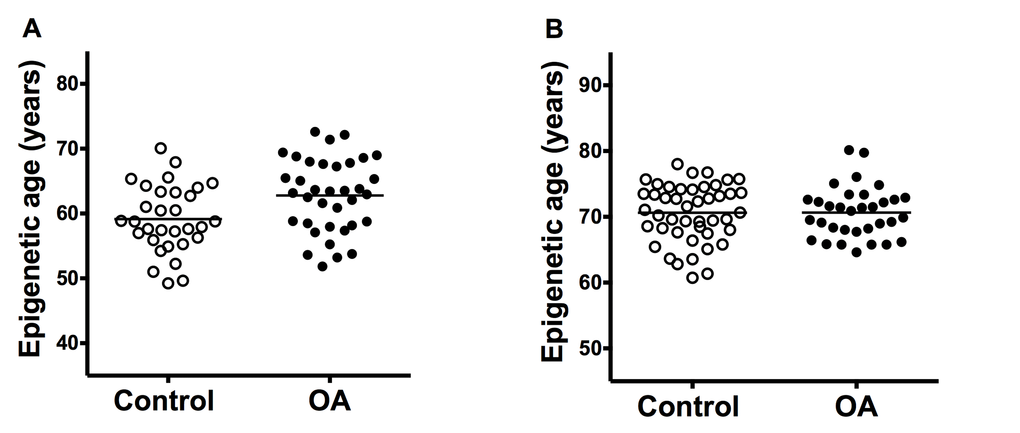
Figure 1. Comparison of epigenetic age in joint tissues from controls and patients with OA. (A) Accelerated aging in OA cartilage samples (n = 31) in comparison with control cartilage (n = 36) with ΔDmAM = 3.7 years (P = 0.008); and (B) no difference (ΔDmAM = 0.04 years, P = 0.3) in bone samples between OA patients (n = 33) and controls (n = 45). Epigenetic ages are represented as age- and sex-adjusted values with horizontal lines for the mean of each group.
Table 1. Specific premature epigenetic aging in OA cartilage compared with control cartilage. ΔDmAM = (age- and sex-adjusted mean DmAM in OA patients) – (age- and sex-adjusted mean DmAM in controls); CI = confidence interval.
| Tissue | OA set | ΔDmAMa (95% CI) | P-value |
| Cartilage | Knee/hip | 3.7 (1.1 to 6.3) | 0.008 |
| | | |
| Bone | Hip | 0.04 (-1.8 to 1.9) | 0.3 |
| | | |
| Blood | Hand | 0.01 (-1.1 to 1.1) | 0.98 |
| Knee | 0.04 (-0.9 to 1.0) | 0.9 |
| Hip | -0.7 (-1.7 to 0.3) | 0.11 |
In contrast with the cartilage results, there were no differences in epigenetic aging of bone (Figure 1B). The mean estimated age obtained from DNA methylation data was very similar in patients with hip OA and in controls (Table 1). The lack of difference was validated in a sub-analysis including only the fracture controls (ΔDmAM = 0.5 years, 95% CI = -1.50 to 2.54, P = 0.6). Bone samples from cadaver controls were too few for meaningful analysis. All the comparisons were adjusted for age and sex as covariates.
The study of epigenetic aging in blood required de novo analyzes of methylation levels at the 8 CpG sites. The MS-SNuPE assays showed a 93.0% call rate, and between-plate CV of 3.2%. Age of the 182 controls without OA was accurately predicted with the 8CpG DmAM (Figure 2), as shown by the good fit of the mean age estimate (mean difference age – DmAM = -0.1 years, SD = 8.7 years). Comparison of the epigenetic ages obtained in this way did not show differences between OA patients and controls (Figure 2). The epigenetic ages for each of the joint-specific OA subgroups were very similar to the epigenetic age for the control subjects, as shown for the hand OA patients (Figure 2A), knee OA patients (Figure 2B) and hip OA patients (Figure 2C). This similarity in blood cells was clearly shown by the near zero year ΔDmAM (Table 1). The largest difference in blood was observed between patients with hip OA and controls, but it was not significant and with direction opposed to premature aging in the OA subjects. All the comparisons were adjusted for age and sex as covariates.
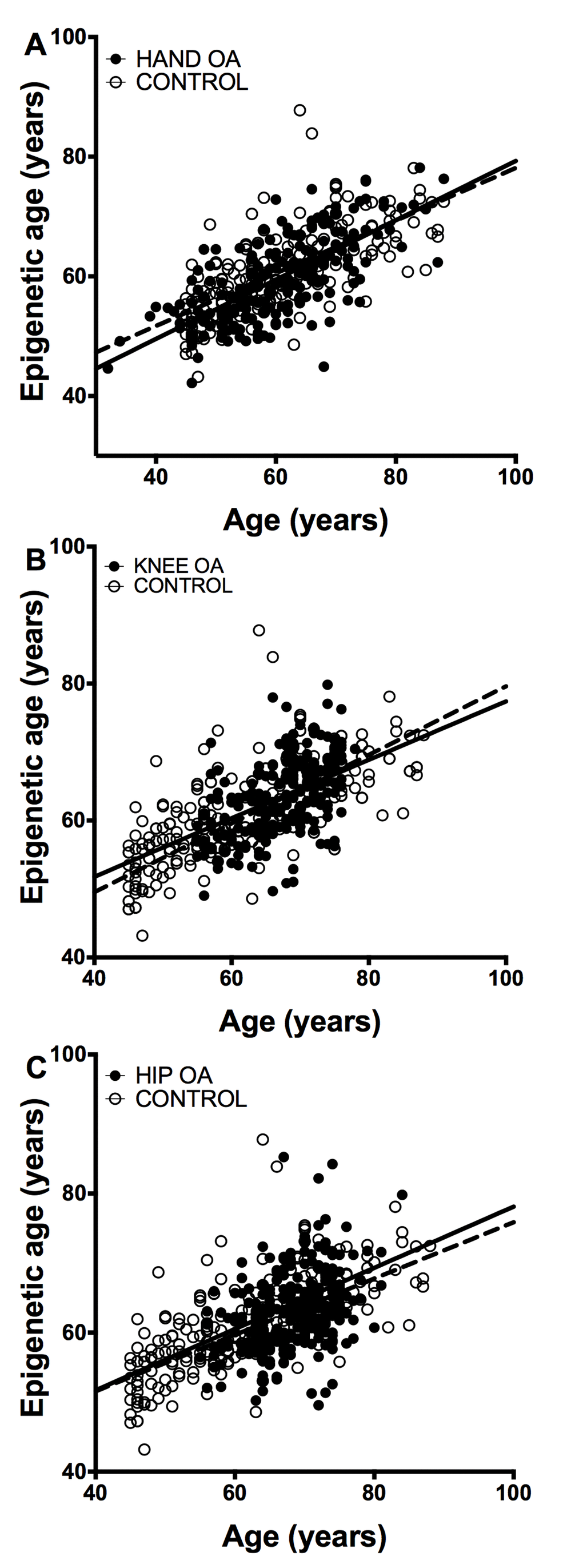
Figure 2. Lack of accelerated epigenetic aging in blood cells of OA patients. The scatterplots represent age in the horizontal axis against epigenetic age in the vertical axis from the controls without OA (empty circles, n =182) together with (A) the hand OA (n = 206), (B) the knee OA (n = 229), and (C) the hip OA (n = 273) patients (filled circles). Straight lines represent least squares regression fit to the data.
Discussion
Our results showed for the first time premature epigenetic aging as detected with DmAM in cartilage of the OA affected joint, but not in bone nearby the OA affected joint, or in blood cells of OA patients irrespective of the joint affected. These results add epigenetic aging to the list of hallmarks of aging showing accelerated changes in OA cartilage. Each of these hallmarks provide complementary and non-redundant evidence of the different facets of the premature biological aging taking place in chondrocytes and the extracellular matrix of the OA affected cartilage. In addition, our results could be interpreted as questioning systemic premature aging in OA, or even a local component of premature aging in nearby bone, but exploration of other aging hallmarks would be required to exclude them.
Previously, several hallmarks of biological aging have been found exacerbated in chondrocytes and cartilage from OA patients [3,4]. Our work adds epigenetic aging to the list of hallmarks that show premature biological aging in this tissue. This is a significant addition because the different aging hallmarks, although extensively interconnected, show tissue and disease specificity and the involvement of each of them cannot be assumed from the presence of other hallmarks [5,6,35]. They need to be tested in the specific tissue or situation under study. This necessity is exemplified by the lack of correlation between epigenetic age and telomere length observed in the elderly population [35]. In addition, diseases of abnormal telomere attrition are different from diseases in which the dominant mechanism is genomic instability and both are different from normal aging. The first group includes pulmonary fibrosis, dyskeratosis congenita and aplastic anemia, whereas genome instability is the dominant aging hallmark in progeroid syndromes such as Hutchinson-Gilford progeria syndrome and Werner’s syndrome [5,6]. In addition, the DNA methylation changes that are included in the Horvath DmAM have been shown to be independent from cellular senescence and mitotic age [19]. Similar lack of redundancy is observed between the other aging hallmarks, making it necessary to study each of them to know their involvement in OA.
Epigenetic changes with age are not restricted to DNA methylation. They encompass also histone modifications regulated by sirtuins and chromatin remodeling [6]. None of the other age-associated epigenetic changes has yet been analyzed in the context of OA, but they are of interest given their potential reversibility as with histone deacetylase inhibitors or inhibitors of histone acetyltransferases as anti-aging drugs [6]. The meaning of these epigenetic changes is still poorly understood. They likely contribute to the loss of transcriptional regulation and increase of transcriptional noise observed with aging [5,6]. Changes in DNA methylation are concentrated in genes with some functional categories including cell growth and survival, organismal development and cancer [19], and in sites within glucocorticoid response elements [26], but the pattern of hypermethylation and hypomethylation has not yet being linked to specific molecular or cellular processes [19,20]. Interpretation of the changes should also include the magnitude of the change: the increase in 3.7 years in ΔDmAM observed in the OA cartilage samples of our study is a modest acceleration compared with changes observed in tumoral tissue, but similar to the reported in a recent abstract in hip OA cartilage, which provides independent confirmation of our findings [36], and in blood of HIV infected patients [37], or in blood of Down syndrome patients [29], but larger than the observed in blood from patients with Parkinson disease [38], or in blood of women after menopause [39].
Some of the previously described aging hallmarks are strongest in the damaged cartilage and less clear in cartilage of preserved areas. Hallmarks showing this pattern are mean telomere length shortening [40-42] senescence-associated heterochromatin foci [41,42], and senescence-associated β-galactosidase (SA-β-gal). These results have been interpreted as representing, at least in part, consequences of cellular stress and the senescence status of the chondrocytes in OA. However, it is possible that epigenetic aging is a biomarker of cellular vulnerability more than of damage and, therefore, a potential target for treatment. Experiments aimed to differentiate between the two mechanisms are necessary but there are already preliminary results showing similar epigenetic aging in damaged and in preserved cartilage from the same OA patient [36]. Potential treatments could include specific senolytic drugs [5] that have not yet been assayed in chondrocytes, and other approaches with capacity to delay aging in OA chondrocytes as already shown for statins [38] and sirtuin activation [37,39].
Our results are contrary to widespread premature epigenetic aging given the lack of increased ΔDmAM at the blood and bone levels. However, the only previous direct evidence of a systemic component of accelerate aging in OA was obtained with telomere length in blood cells of OA patients [8,10], and it is likely that telomere length and DmAM capture different aspects of biological aging [6,19,21,25,27]. Telomere attrition results from cell divisions, in the absence of the enzyme telomerase, and from DNA damage induced by extrinsic stress, as oxidative or inflammatory stress. The authors that found accelerated telomere length attrition in blood of OA patient interpreted it as reflecting oxidative stress and low-level chronic inflammation [8], or associated with chronic pain and high stress [10]. In contrast, epigenetic aging as measured with DmAM seems to be due to perturbations of the DNA methylation maintenance system [19,21,25]. Therefore, our results do not question systemic accelerated aging as detected with telomere shortening, but exclude the epigenetic aspect of aging.
The lack of accelerated aging in blood and in bone was not attributable to insufficient power. In effect, blood samples were enough to exclude ΔDmAM half as fast as the observed in cartilage (1-β > 0.95 to exclude a difference of 1.83 years for each of the three joints). Bone samples, in turn, were enough to detect ΔDmAM as large as the observed in cartilage (1-β = 0.90). In addition, the use of different DmAM for cartilage and bone, in one side, and for blood, in the other, does not interfere with our results because no analysis compared results across different DmAM. We also avoided biases due to differences in age or sex between the OA patients and the controls by adjusting for these two variables, as recommended [19-21]. However, limitations of our study are that the different tissues were not from the same subjects, the lack of other joint tissues, and the absence of a larger number of cartilage samples from femoral heads allowing specific analysis of epigenetic aging at this site. The meaning of these limitations seems modest because bone and cartilage are arguably the most relevant tissues in OA [2], and because epigenetic aging in hip cartilage from OA patients has been independently found [36], as already mentioned. In any case, we cannot completely exclude that other tissues or joints show a different behavior than the reporter here, or that additional insight could be gained from analyzing several tissues from the same subjects, as epigenetic age correlation between tissues.
In summary, we have found specific accelerated aging as measured with DNA methylation in cartilage from OA affected joints. Knowledge of the mechanisms of this type of premature aging will help to understand OA pathology, but already it is apparent that these particular mechanisms are not widespread. This was indicated by the results obtained with the same DNA methylation methodology in bone near the affected joint and in blood cells. They showed absence of a systemic component of premature aging. These results cannot exclude that other hallmarks of aging could be more widespread than the DNA methylation changes analyzed here.
Methods
Cartilage and bone epigenetic age
Epigenetic age was estimated with the 353 age-related CpG probes according with Horvath [19]. Methylation information of these sites has been obtained in previous studies addressing cartilage and bone samples (Table 2 and Supplementary Table 1) [43-46]. Cartilage samples were from 31 controls and 36 OA patients (Table 2). The controls were from tibial plateau of 18 cadavers with no macroscopic signs of OA [43], and the femoral head of 10 subjects with hip fracture and without macroscopic or microscopic evidence of OA [46]. In addition, 3 cartilage samples from cadavers without information of status and location were included [45]. The OA samples included 29 from the tibial plateau of severe knee OA patients [43,46], and 7 from the femoral head of severe hip OA patients [46], obtained at the time of joint replacement. Bone samples were from 45 controls and 33 hip OA patients (Table 2). The controls included femoral heads of 34 subjects with osteoporotic hip fracture (OP) and 7 cadavers [44]. They lacked OA lesions on macroscopic examination of the hip joints and the bone pieces excluded subchondral and fractured regions. Patients with fractures due to high-energy trauma or with disorders causing secondary OP or OA were not included. In addition, 4 control bone samples from cadavers that lacked detailed information of status and place of retrieval were included [45]. The bone samples of the 33 hip OA patients were obtained from femoral heads at the time of total joint replacement for primary hip OA [44]. Methylation data were obtained either with the Human Methylation 27 BeadChip (Illumina) [43,44], or with the HumanMethylation 450 Bead-Chip microarray (Illumina, San Diego, California, USA) [45,46]. These samples were obtained with informed consent of the donors and approval of the relevant ethics committees as reported in the primary publications [43-46].
Table 2. Main characteristics of the sample collections used in this study. N = number of samples, SD = standard deviation.
| Tissue | Set | N | Age
Mean ± SD (Range) | Woman % |
| Cartilage | Control knee/hipa | 31 | 64.8 ± 15.0 (40-95) | 48.4 |
| Knee/hip OA | 36 | 67.1 ± 9.3 (41-80) | 75.0 |
| | | | |
| Bone | Control hipa | 45 | 78.0 ± 11.0 (40-104) | 93.3 |
| Hip OA | 33 | 75.4 ± 6.7 (58-89) | 100.0 |
| | | | |
| Blood | Control | 182 | 60.7 ± 11.5 (45-88) | 46.7 |
| Hand OA | 206 | 60.6 ± 10.1 (32-88) | 88.4 |
| Knee OA | 229 | 67.7 ± 5.6 (55-78) | 82.1 |
| Hip OA | 273 | 68.4 ± 5.5 (55-84) | 59.7 |
| a 3 cartilage and 4 bone control samples were from undefined localization |
Analysis of epigenetic aging in blood
Epigenetic aging in blood was assessed with a 8 CpG DmAM specific for whole blood and amenable to assay in large number of samples [24]. Methylation data were obtained for this study with methylation-sensitive single-nucleotide primer extension (MS-SNuPE) following the reported procedure [47]. Genomic DNA from 890 subjects of Spanish ancestry was assayed (Table 2 and Supplementary Table 1). This collection of samples included 182 controls recruited at the time of intravenous urography. They had not OA signs at exploration including both hands or in the radiographs either at the hip or column joints, and they did not complain of OA symptoms in a systematic questionnaire. The remaining 708 subjects were suffering from primary OA as assessed by a rheumatologist. The subjects affected by knee OA, 229, or hip OA, 273, were selected from consecutive patients aged 55–75 years at the time of surgery that were undergoing total joint replacement. The patients with hand OA, 206, were selected among those attending the Rheumatology Unit fulfilling the American College of Rheumatology classification criteria for hand OA [48]. Exclusion criteria were inflammatory, infectious, traumatic or congenic joint pathology, as well as, lesions due to crystal deposition or osteonecrosis. Morbid obesity and occupational strain were not exclusion causes. All donors provided blood DNA samples for genetic studies with written informed consent according to the Declaration of Helsinki (most recently at the General Assembly on October 2008) and the approval of the Ethics Committee for Clinical Research of Galicia, as described [49].
Statistical analysis
We estimated the epigenetic age of the cartilage and bone samples with Horvath’s DmAM [19], and of the blood samples with the 8 CpG DmAM [24]. Comparisons between samples from OA patients and controls were done with analysis of variance (ANOVA) including age and sex as covariates. Mean differences in DmAM estimates (ΔDmAM) were calculated as:
ΔDmAM = (age- and sex-adjusted mean DmAM in OA patients) – (age- and sex-adjusted mean DmAM in controls)
Age- and sex-adjustment was done with the residuals from multiple linear regression of estimated age versus age and sex. All these analyses were done with Statistica 7.0 (Stat Soft, Inc.). Post-hoc power analysis was done with G*Power 3 for α = 0.05 [50].
Acknowledgements
The authors are indebted to the patients that generously have contributed the samples and time to this work. They also thank Carmen Pena for her excellent technical support.
Funding
This work was supported by the Instituto de Salud Carlos III (Spain) [grants PI14/01651, PI12/01909, RD12/009/008, and P12/615] with participation of the European Regional Development Fund of the EU (FEDER).
Conflicts of Interest
The authors declare they have no conflict of interest.
References
-
1.
Murphy L, Schwartz TA, Helmick CG, Renner JB, Tudor G, Koch G, Dragomir A, Kalsbeek WD, Luta G, Jordan JM. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Rheum. 2008; 59:1207–13. https://doi.org/10.1002/art.24021 [PubMed]
-
2.
Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012; 64:1697–707. https://doi.org/10.1002/art.34453 [PubMed]
-
3.
Loeser RF. Aging and osteoarthritis. Curr Opin Rheumatol. 2011; 23:492–96. https://doi.org/10.1097/BOR.0b013e3283494005 [PubMed]
-
4.
Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012; 51:241–48. https://doi.org/10.1016/j.bone.2012.03.023 [PubMed]
-
5.
Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, Csoka AB. Cellular Senescence as the Causal Nexus of Aging. Front Genet. 2016; 7:13. https://doi.org/10.3389/fgene.2016.00013 [PubMed]
-
6.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
-
7.
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015; 14:644–58. https://doi.org/10.1111/acel.12344 [PubMed]
-
8.
Zhai G, Aviv A, Hunter DJ, Hart DJ, Gardner JP, Kimura M, Lu X, Valdes AM, Spector TD. Reduction of leucocyte telomere length in radiographic hand osteoarthritis: a population-based study. Ann Rheum Dis. 2006; 65:1444–48. https://doi.org/10.1136/ard.2006.056903 [PubMed]
-
9.
Tamayo M, Mosquera A, Rego JI, Fernández-Sueiro JL, Blanco FJ, Fernández JL. Differing patterns of peripheral blood leukocyte telomere length in rheumatologic diseases. Mutat Res. 2010; 683:68–73. https://doi.org/10.1016/j.mrfmmm.2009.10.010 [PubMed]
-
10.
Sibille KT, Langaee T, Burkley B, Gong Y, Glover TL, King C, Riley JL3rd, Leeuwenburgh C, Staud R, Bradley LA, Fillingim RB. Chronic pain, perceived stress, and cellular aging: an exploratory study. Mol Pain. 2012; 8:12. https://doi.org/10.1186/1744-8069-8-12 [PubMed]
-
11.
Rahman MM, Kopec JA, Anis AH, Cibere J, Goldsmith CH. Risk of cardiovascular disease in patients with osteoarthritis: a prospective longitudinal study. Arthritis Care Res (Hoboken). 2013; 65:1951–58. https://doi.org/10.1002/acr.22092 [PubMed]
-
12.
Haugen IK, Ramachandran VS, Misra D, Neogi T, Niu J, Yang T, Zhang Y, Felson DT. Hand osteoarthritis in relation to mortality and incidence of cardiovascular disease: data from the Framingham heart study. Ann Rheum Dis. 2015; 74:74–81. https://doi.org/10.1136/annrheumdis-2013-203789 [PubMed]
-
13.
Louati K, Vidal C, Berenbaum F, Sellam J. Association between diabetes mellitus and osteoarthritis: systematic literature review and meta-analysis. RMD Open. 2015; 1:e000077. https://doi.org/10.1136/rmdopen-2015-000077 [PubMed]
-
14.
Huang SW, Wang WT, Chou LC, Liao CD, Liou TH, Lin HW. Osteoarthritis increases the risk of dementia: a nationwide cohort study in Taiwan. Sci Rep. 2015; 5:10145. https://doi.org/10.1038/srep10145 [PubMed]
-
15.
Castell MV, van der Pas S, Otero A, Siviero P, Dennison E, Denkinger M, Pedersen N, Sanchez-Martinez M, Queipo R, van Schoor N, Zambon S, Edwards M, Peter R, et al. Osteoarthritis and frailty in elderly individuals across six European countries: results from the European Project on OSteoArthritis (EPOSA). BMC Musculoskelet Disord. 2015; 16:359. https://doi.org/10.1186/s12891-015-0807-8 [PubMed]
-
16.
Barbour KE, Lui LY, Nevitt MC, Murphy LB, Helmick CG, Theis KA, Hochberg MC, Lane NE, Hootman JM, Cauley JA, and Study of Osteoporotic Fractures Research Group. Hip Osteoarthritis and the Risk of All-Cause and Disease-Specific Mortality in Older Women: A Population-Based Cohort Study. Arthritis Rheumatol. 2015; 67:1798–805. https://doi.org/10.1002/art.39113 [PubMed]
-
17.
Nüesch E, Dieppe P, Reichenbach S, Williams S, Iff S, Jüni P. All cause and disease specific mortality in patients with knee or hip osteoarthritis: population based cohort study. BMJ. 2011; 342:d1165. https://doi.org/10.1136/bmj.d1165 [PubMed]
-
18.
Kluzek S, Sanchez-Santos MT, Leyland KM, Judge A, Spector TD, Hart D, Cooper C, Newton J, Arden NK. Painful knee but not hand osteoarthritis is an independent predictor of mortality over 23 years follow-up of a population-based cohort of middle-aged women. Ann Rheum Dis. 2015. [PubMed]
-
19.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. https://doi.org/10.1186/gb-2013-14-10-r115 [PubMed]
-
20.
Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jöckel KH, Erbel R, Mühleisen TW, Zenke M, Brümmendorf TH, Wagner W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014; 15:R24. https://doi.org/10.1186/gb-2014-15-2-r24 [PubMed]
-
21.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013; 49:359–67. https://doi.org/10.1016/j.molcel.2012.10.016 [PubMed]
-
22.
Florath I, Butterbach K, Müller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014; 23:1186–201. https://doi.org/10.1093/hmg/ddt531 [PubMed]
-
23.
Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS One. 2011; 6:e14821. https://doi.org/10.1371/journal.pone.0014821 [PubMed]
-
24.
Vidal-Bralo L, Lopez-Golan Y, Gonzalez A. Simplified Assay for Epigenetic Age Estimation in Whole Blood of Adults. Front Genet. 2016; 7:126. https://doi.org/10.3389/fgene.2016.00126 [PubMed]
-
25.
Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013; 22:R7–15. https://doi.org/10.1093/hmg/ddt375 [PubMed]
-
26.
Zannas AS, Arloth J, Carrillo-Roa T, Iurato S, Röh S, Ressler KJ, Nemeroff CB, Smith AK, Bradley B, Heim C, Menke A, Lange JF, Brückl T, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015; 16:266. https://doi.org/10.1186/s13059-015-0828-5 [PubMed]
-
27.
Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci USA. 2014; 111:15538–43. https://doi.org/10.1073/pnas.1412759111 [PubMed]
-
28.
Horvath S, Mah V, Lu AT, Woo JS, Choi OW, Jasinska AJ, Riancho JA, Tung S, Coles NS, Braun J, Vinters HV, Coles LS. The cerebellum ages slowly according to the epigenetic clock. Aging (Albany NY). 2015; 7:294–306. https://doi.org/10.18632/aging.100742 [PubMed]
-
29.
Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015; 14:491–95. https://doi.org/10.1111/acel.12325 [PubMed]
-
30.
Perna L, Zhang Y, Mons U, Holleczek B, Saum KU, Brenner H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics. 2016; 8:64. https://doi.org/10.1186/s13148-016-0228-z [PubMed]
-
31.
Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, Corley J, Taylor A, Murphy L, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015; 44:1388–96. https://doi.org/10.1093/ije/dyu277 [PubMed]
-
32.
Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016; 15:149–54. https://doi.org/10.1111/acel.12421 [PubMed]
-
33.
Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, Mari D, Arosio B, Monti D, Passarino G, De Rango F, D’Aquila P, Giuliani C, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY). 2015; 7:1159–70. https://doi.org/10.18632/aging.100861 [PubMed]
-
34.
Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015; 16:25. https://doi.org/10.1186/s13059-015-0584-6 [PubMed]
-
35.
Breitling LP, Saum KU, Perna L, Schöttker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016; 8:21. https://doi.org/10.1186/s13148-016-0186-5 [PubMed]
-
36.
Reynard L. The aging process and epigenetics: relationship with OA. Osteoarthritis Cartilage. 2015 (Suppl 2); 23:A18–18. https://doi.org/10.1016/j.joca.2015.02.034
-
37.
Wu Y, Chen L, Wang Y, Li W, Lin Y, Yu D, Zhang L, Li F, Pan Z. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-κB mediated inflammatory responses in osteoarthritis development. Sci Rep. 2015; 5:17602. https://doi.org/10.1038/srep17602 [PubMed]
-
38.
Yudoh K, Karasawa R. Statin prevents chondrocyte aging and degeneration of articular cartilage in osteoarthritis (OA). Aging (Albany NY). 2010; 2:990–98. https://doi.org/10.18632/aging.100213 [PubMed]
-
39.
Platas J, Guillén MI, Pérez Del Caz MD, Gomar F, Castejón MA, Mirabet V, Alcaraz MJ. Paracrine effects of human adipose-derived mesenchymal stem cells in inflammatory stress-induced senescence features of osteoarthritic chondrocytes. Aging (Albany NY). 2016; 8:1703–17. [PubMed]
-
40.
Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, Clark IM. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002; 1:57–65. https://doi.org/10.1046/j.1474-9728.2002.00008.x [PubMed]
-
41.
Harbo M, Bendix L, Bay-Jensen AC, Graakjaer J, Søe K, Andersen TL, Kjaersgaard-Andersen P, Koelvraa S, Delaisse JM. The distribution pattern of critically short telomeres in human osteoarthritic knees. Arthritis Res Ther. 2012; 14:R12. https://doi.org/10.1186/ar3687 [PubMed]
-
42.
Harbo M, Delaisse JM, Kjaersgaard-Andersen P, Soerensen FB, Koelvraa S, Bendix L. The relationship between ultra-short telomeres, aging of articular cartilage and the development of human hip osteoarthritis. Mech Ageing Dev. 2013; 134:367–72. https://doi.org/10.1016/j.mad.2013.07.002 [PubMed]
-
43.
Fernández-Tajes J, Soto-Hermida A, Vázquez-Mosquera ME, Cortés-Pereira E, Mosquera A, Fernández-Moreno M, Oreiro N, Fernández-López C, Fernández JL, Rego-Pérez I, Blanco FJ. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. 2014; 73:668–77. https://doi.org/10.1136/annrheumdis-2012-202783 [PubMed]
-
44.
Delgado-Calle J, Fernández AF, Sainz J, Zarrabeitia MT, Sañudo C, García-Renedo R, Pérez-Núñez MI, García-Ibarbia C, Fraga MF, Riancho JA. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2013; 65:197–205. https://doi.org/10.1002/art.37753 [PubMed]
-
45.
Lokk K, Modhukur V, Rajashekar B, Märtens K, Mägi R, Kolde R, Koltšina M, Nilsson TK, Vilo J, Salumets A, Tõnisson N. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014; 15:r54. https://doi.org/10.1186/gb-2014-15-4-r54 [PubMed]
-
46.
Aref-Eshghi E, Zhang Y, Liu M, Harper PE, Martin G, Furey A, Green R, Sun G, Rahman P, Zhai G. Genome-wide DNA methylation study of hip and knee cartilage reveals embryonic organ and skeletal system morphogenesis as major pathways involved in osteoarthritis. BMC Musculoskelet Disord. 2015; 16:287. https://doi.org/10.1186/s12891-015-0745-5 [PubMed]
-
47.
Kaminsky ZA, Assadzadeh A, Flanagan J, Petronis A. Single nucleotide extension technology for quantitative site-specific evaluation of metC/C in GC-rich regions. Nucleic Acids Res. 2005; 33:e95. https://doi.org/10.1093/nar/gni094 [PubMed]
-
48.
Altman R, Alarcón G, Appelrouth D, Bloch D, Borenstein D, Brandt K, Brown C, Cooke TD, Daniel W, Gray R, Greenwald R, Hochberg M, Howell D, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hand. Arthritis Rheum. 1990; 33:1601–10. https://doi.org/10.1002/art.1780331101 [PubMed]
-
49.
Rodriguez-Lopez J, Pombo-Suarez M, Liz M, Gomez-Reino JJ, Gonzalez A. Lack of association of a variable number of aspartic acid residues in the asporin gene with osteoarthritis susceptibility: case-control studies in Spanish Caucasians. Arthritis Res Ther. 2006; 8:R55. https://doi.org/10.1186/ar1920 [PubMed]
-
50.
Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007; 39:175–91. https://doi.org/10.3758/BF03193146 [PubMed]