



Introduction
Currently, mesenchymal stromal/stem cells (MSCs) derived from various sources (tissues) are often used for cell based therapies to treat a variety of diseases [1]. Such applications typically require large numbers of cells produced by in vitro expansion of cells via continuing passaging. However, as the passage number increases, the risk of genetic alterations also increases.
Indeed, high passage numbers in MSCs have been shown to contribute to the formation of chromosomal aberrations [2,3], the inability of cells to differentiate, and oncogenic transformation [4-6]. It is generally assumed that these effects are associated, through unknown mechanisms, with the process of replicative senescence, or aging, of cells [7]. However, substantial gaps in our knowledge of the genetic instability in long-term cultivated MSCs still exist. Unsolved questions include both the evaluation criteria and mechanisms of genetic instability in MSCs during cultivation, as well as the therapeutic time window, i.e. the critical number of cell passages suitable for clinical use.
Accumulation of DNA damage due to incomplete or inaccurate repair of spontaneous DNA lesions (caused by metabolic free radicals, replication and recombination errors, spontaneous chemical modifications) is the most significant contributor to genetic instability in cells that have not been exposed to external DNA damaging stimuli, such as ionizing radiation, UV, chemicals, etc [8]. Some authors consider the accumulation of DNA damage in cells as a universal cause of age-dependent changes in cells [9,10]. Among the variety of spontaneous DNA lesions, most of the interest of researchers has focused on DNA double-strand breaks (DSB). Indeed, DSBs are the most critical DNA alterations that can define the fate of cells and, if repaired incorrectly or inefficiently, can lead to serious cytogenetic abnormalities, cell death, inactivation of tumor suppressor genes or activation of oncogenes [11-14]. Moreover, in recent years, functional state of DNA DSB repair systems, as well as accumulation of DSB, have been linked to the formation of a particular phenotype inherent to aging cells [15].
An indirect method based on immunofluorescence microscopy analysis of proteins involved in DSB repair has recently gained broad use to study quantitative DSB-related changes in living cells. Complex dynamic microstructures formed during DNA DSB repair consisting of thousands of copies of proteins and visualized by immunofluorescence staining appear as bright spots of fluorescence, called DNA repair foci [16,17]. It is believed that one focus is the repair site of one single or multiple DSBs [18]. Notably, the immunofluorescence analysis of phosphorylated at serine 139 core histone Н2АX (also known as γ-Н2АХ) has been the most widely used marker of DNA DSBs [19,20]. Functioning as a binding site for the protein MDC1, γ-Н2АХ recruits key DNA repair proteins [21] and in such way, forming a vital part of the machinery that ensures genome stability. Members of the superfamily of phosphatidylinositol 3-kinase-related kinases (PIKKs), in particular Serine/Threonine protein kinases ATM (Ataxia telangiectasia mutated), ATR (ATM- and RAD3-related) and DNA-PKcs (DNA-dependent protein kinase catalytic subunit), phosphorylate H2AX in response to DSB acting as primary DSB sensor proteins [22].
The aim of our study was to investigate the pattern of change in the number of γ-Н2АХ foci during long-term (up to 22 passage) culturing of MSCs.
To reveal possible mechanisms of change in the number of γ-Н2АХ foci, we additionally performed: 1) quantitative analysis of activated (sequentially autophosphorylated at Ser1981, Ser367 and Ser1893) ATM foci in response to DSBs [23]; 2) differential quantitative analysis of γ-Н2АХ foci in actively proliferating (Ki67(+)) and resting (Ki67(-)) MSCs. The associated with ribosomal RNA transcription RNA [24] Ki67 protein present is actively proliferating (during G1, S, G2 and M phases of the cell cycle), while being absent in resting (G0 phase) cells [25].
Results
Quantitative analysis of the γH2AX and pATM foci
Quantification of γH2AX foci in MSCs at different passages is shown in Fig. 1A. It can be seen that between passages 3-16, the number of γH2AX foci did not change (r=0.66; p=0.11), whereas at passages 16-22, the number of the foci doubled. In contrast, phosphorylated ATM (pATM) foci increased gradually with the increase in the passage number (Fig. 1A). The pATM data could be best fit with a linear regression equation y=0.99 + 0.07x (r=0.83; p=0.003), where y is the number of foci per nucleus, X is the passage number. Similar pattern was observed for γH2AX foci co-localized with pATM foci: y=0.72 + 0.03x (r=0.73; p=0.017).
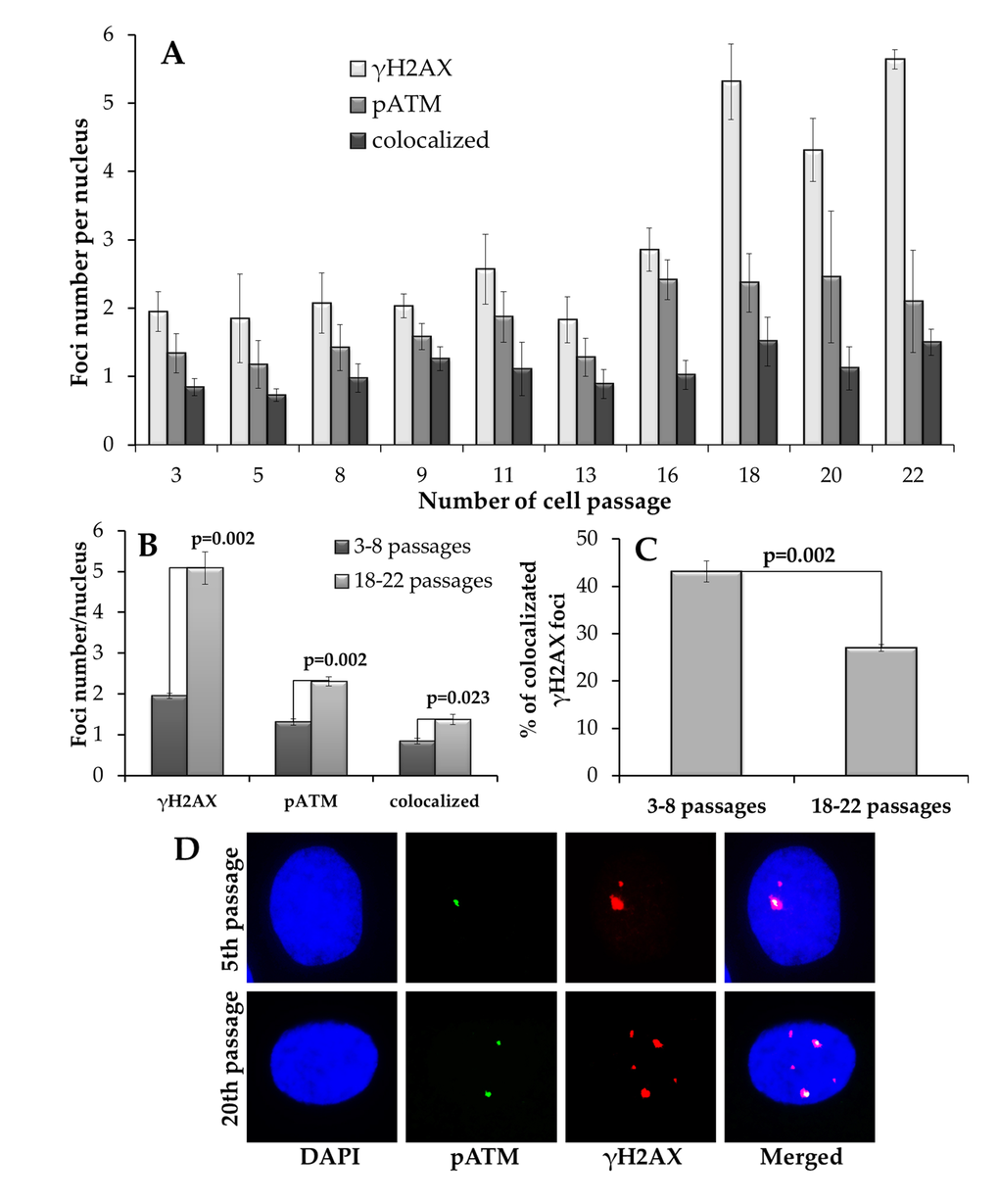
Figure 1. Immunocytochemical analysis of γH2AX and pATM foci. (A) Changes in γH2AX, pATM foci and their co-localization depending on the passage number in MSCs. (B) Comparative analysis of γH2AX, pATM foci and their co-localization in early (3-8) vs. late (18-22) passages of MSCs. (C) Fraction of γH2AX foci that co-localize with pATM at early (3-8) vs. late (18-22) passages of MSCs. (D) Representative immunofluorescent microphotographs of MSC showing γH2AX (green), pATM (red) foci and their co-localization (yellow) at passage 5 and 20. Nuclei were counterstained with DAPI.
It was interesting to compare directly foci quantities at early (3-8) and late (18-22) passages. As seen in Fig. 1B, both γH2AX and pATM foci significantly increased in late compared with early passage cells. However, whereas the number of γH2AX foci tripled, the increase for pATM foci was only 2-fold. Interestingly, comparing fractions of γH2AX foci co-localized with pATM between the early and late passages showed that the number dropped from 43±2% foci at passages 3-8 to 27±1% at passages 18-22. This data indicates that the sharp increase in γH2AX foci at late passages may not be ATM-dependent.
Differential analysis of γH2AX foci in proliferating and resting cells
Analysis of γH2AX foci in proliferating Ki67-positive (Ki67(+)) and resting Ki67-negative (Ki67(-)) is presented in Fig. 2. The number of γH2AX foci in proliferating cells was higher than that in resting cells for all examined passage numbers (Fig. 2A). No significant changes in γH2AX foci were found in proliferating cells at passages 3-16, after which the number of foci sharply increased (Fig. 2A). A different pattern was observed in resting cells: the number of γH2AX foci increased more or less gradually with the increase in passage number and, similarly to pATM foci kinetics, was well fit with a linear regression y=0.02 + 0.12x (r=0.96; p=0.00001), where y is the number of γH2AX foci and x is the passage number. Moreover, a statistically significant correlation was found between the number of γH2AX foci in resting cells and the number of pATM foci in all cells (r=0.87; p=0.001).
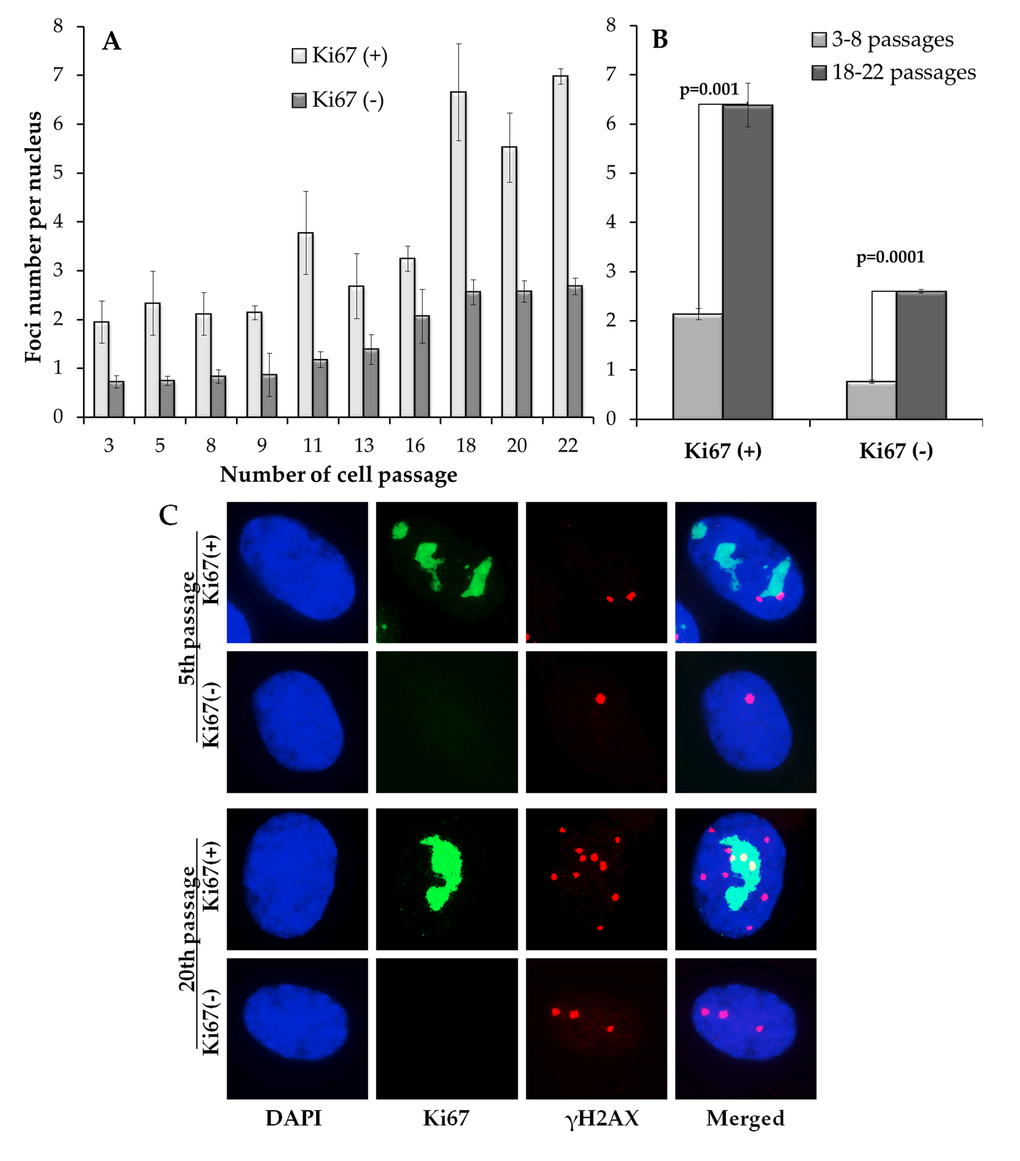
Figure 2. Differential immunocytochemical analysis of γH2AX foci in proliferating (Ki67(+)) and resting (Ki67(-)) cells. (A) Changes in the γH2AX number in Ki67(+) and Ki67(-) cells on 3-22 passages (B) Comparative analysis of γH2AX in Ki67(+) and Kib(-) cells on early (3-8) vs. late (18-22) passages; (С) Representative immunofluorescent microphotographs of MSC showing Ki67 (green), γH2AX (red) foci and their co-localization (yellow) at passage 5 and 20. Nuclei were counterstained with DAPI.
When early (3-8) and late (16-22) passages were compared directly, the number of γH2AX foci was higher in both proliferating and resting MSC (Fig. 2B). However, absolute numbers of γH2AX foci were different in proliferating vs. resting cells, 4.3 vs. 1.8 foci/nucleus, respectively. These results suggest either a higher rate of DNA DSB induction or a broader spectrum of mechanisms leading to DNA DSBs in proliferating vs. resting cells. However, diminishing DNA DSB repair mechanisms as a source of such difference between proliferating vs. resting cells cannot be ruled out as well.
Discussion
In this study we showed that long-term culture of MSCs leads to accumulation of γH2AX foci. Late passage cells were characterized by a ~3-fold increased number of foci compared with early passage cells. Apparently, two parallel processes are involved in the observed accumulation of γH2AX foci that are essentially distinct:
1. Gradual ATM-dependent accumulation of γH2AX foci in long-term cultured MSCs.
2. Step-wise ATM-independent increase of γH2AX foci numbers between passages 16 and 18.
For the first process, the ATM kinase, unlike the ATR kinase that phosphorylates H2AX upon formation of large stretches of single-stranded DNA at collapsed replication forks and nucleotide excision repair sites [26,27], phosphorylates histone H2AX in response to single or clustered DSBs [28,29]. Thus, accumulation of γH2AX co-localized with pATM suggests that these foci represent DNA DSBs in long-cultured MSCs. However, these co-localized foci had a minor contribution to an overall passage dependent increase in γH2AX foci (Fig. 1B). Accumulation of DSBs at telomeres may contribute to this process, since it was shown that repair efficiency of DNS DSBs at telomeres is low and may lead to accumulation to γH2AX foci at telomere repeats [30,31]. This process is related to cellular aging and the accumulation of γH2AX foci in senescent cells [31,32]. This is consistent with our results showing the accumulation of γH2AX foci in Ki67-negative cells, which may also represent senescent cells. Since passage related γH2AX foci in MSCs co-localized poorly with pATM foci (Fig. 1C), consistent with the results of Pospelova et al. [33], it is likely that they represent non-DNA damage related foci. Such foci have been shown to be associated with mTOR signaling pathway in senescent cells only and could be inhibited by rapamycin [33,34].
With regards to the second process, it appears to take place exclusively in proliferating cells and to be ATM-independent. This suggests an age-related increase in the rate of errors during DNA replication. For example, DNA DSBs can be formed at collapsed replication forks of late-passage cells due to increased oxidative damage associated with age-dependent dysfunction of mitochondria [35]. Phosphorylation of H2AX by a DNA-PKcs/CHK2 pathway could also contribute to this age-associated process [36].
Another explanation of the increased rates of γH2AX foci in late-passage MSCs could be the inefficient γH2AX de-phosphorylation processes, also in turn related to cellular aging [37,38]. Moreover, replicative senescence related changes in chromatin remodeling process may also represent the source of additional γH2AX foci in late-passage MSCs [38]. However, this hypothesis is inconsistent with the results of a recent study in which another marker of DNA DSBs, 53BP1 that is not relevant to H2AX, was used [39]. It is not unlikely that diminished rates of ATM-dependent phosphorylation of histone H2AX may indicate an onset of the senescence phenotype. It was shown that the concentration and the level of ATM phosphorylation, which defines its kinase activity, after exposure to ionizing radiation was lower in old compared to young mice [40]. On the contrary, ATM-mediated DNA damage response was shown to prevent further damage, induce SASP and boost protection against malignant transformation [41]. Lowered ATM activity may contribute to diminished p53-dependent responses to DNA damage, induced or spontaneous, leading to subsequent sharp increases in DNA DSBs in MSCs of high passages forming the senescence phenotype. Lastly, various DNA repair pathways were shown to be affected by age in various mouse and human tissues [42-46]. Such changes in DNA repair efficiencies may also contribute to passage-associated accumulation of γH2AX foci in MSCs found in this study. Lastly, it appears that apoptotic cells had no role in the observed accumulation of genetic instability markers as we did not observe an increase in presumptively apoptotic cells containing >25 γH2AX foci per cell, nor did we notice accumulation of nuclei with apoptotic morphology (data not shown).
Conclusions
A passage dependent accumulation of γH2AX foci in MSCs, with a sharp increase at passages 16-18, was observed in this study, indicative of genomic instability. It appears that the mechanisms of this increased rates of γH2AX foci include both a ATM-dependent, most likely representing physical DNA DSBs irrespective of cell proliferation status, slow component and a ATM-independent fast component that gets abruptly activated at passage 16-18 in proliferating cells. While precise mechanisms of the two identified components are not clear and represent obvious interest for future studies, our results provide important information with respect to the clinical applications of stem cell therapy. Indeed, understanding how passage number affects genomic instability in MSCs would allow optimizing clinical protocols for in vitro expansion of the cells to achieve higher therapeutic outcomes.
Methods
Culture and immunophenotypic characterization of MSCs
MSCs were obtained from mucosa of a 40-year old healthy male donor. Cells were cultured in low glucose DMEM (StemCell, USA) supplemented with L-glutamin, penicillin/streptomycin and 20% fetal bovine serum (StemCell, USA) at a concentration of 0.3 x 106 per flask with filter ventilated caps (25 cm2) in a humidified atmosphere of 5% CO2 and 37°C. MSCs were subcultured every 7 days up to passage 22.
For immunophenotypic characterization, cells were stained with the panels of antibodies against the following surface markers: CD3, CD13, CD14, CD19, CD25, CD29, CD31, CD34, CD38, CD44, CD45, CD69, CD73, CD90, CD105, CD106, CD166 and HLA-DR (Becton Dickinson, USA). The expression of the surface markers was then analyzed using a BD FACS Canto II (Becton Dickinson Bioscience, USA) flow cytometer. The resulting expression profiles revealed high expression levels (>60% positive cells) for CD90, CD105, CD166, CD44, CD73, medium levels (30-60%) for CD13, CD29 and CD69, and very low levels (<5%) for CD45, CD34, CD133, CD3, CD19, CD25, CD38, CD45, CD106, CD31 markers. This immunophenotype was consistent with the reported immunophenotype for MSCs [47] and did not change in the course of the experiment.
Immunofluorescence microscopy
MSCs at passages 3, 5, 8, 9, 11, 13, 16, 18, 20 and 22 were detached with 0.25% Trypsin/EDTA (StemCell Technology, USA), washed, resuspended and seeded at the density of 5 × 103 cells/cm2 in 500 μL of culture medium onto coverslips (SPL Lifesciences, South Korea) placed inside 35 mm Petri dishes (Corning, USA). To improve adhesion of cells additional volume of culture medium (1, 5 mL) was added into Petri dishes 15 minutes after seeding. Cells seeded on coverslips were incubated at 37°C and 5% CO2 for at 48 h prior to fixation.
Cells were fixed on coverslips in 4% paraformaldehyde in PBS (pH 7.4) for 15 min at room temperature followed by two rinses in PBS and permeabilization for 40 min with 0.3% Triton-X100 (in PBS, pH 7.4) supplemented with 2% bovine serum albumin (BSA) to block non-specific antibody binding. Cells were then incubated for 1 hour at room temperature with primary rabbit monoclonal antibody against γH2AX (dilution 1:200, clone EP854(2)Y, Merck-Millipore, USA) and primary mouse monoclonal antibody against phosphorylated ATM protein (dilution 1:400, clone 10H11.E12, Merck-Millipore, USA) or primary mouse monoclonal antibody against Ki67 protein (dilution 1:400, clone Ki-S5, Merck-Millipore, USA) which were diluted in PBS with 1% BSA. Following several rinses with PBS, cells were incubated for 1 hour at room temperature with secondary antibodies IgG (H+L) goat anti-mouse (Alexa Fluor 488 conjugated, dilution 1:600; Merck-Millipore, USA) and goat anti-rabbit (rhodamine conjugated, dilution 1:400; Merck-Millipore, USA) diluted in PBS (pH 7.4) with 1% BSA. Coverslips were then rinsed several times with PBS and mounted on microscope slides with ProLong Gold medium (Life Technologies, USA) with DAPI for DNA counter-staining. Cells were viewed and imaged using Nikon Eclipse Ni-U microscope (Nikon, Japan) equipped with a high definition camera ProgRes MFcool (Jenoptik AG, Germany). Filter sets used were UV-2E/C (340–380 nm excitation and 435–485 nm emission), B-2E/C (465–495 nm excitation and 515–555 nm emission) and Y-2E/C (540–580 nm excitation and 600–660 nm emission). At least 200 cells per data point were imaged. Foci were counted by manual scoring.
Statistical analyses
Statistical and mathematical analyses of the data were conducted using the Statistica 8.0 software (StatSoft). Data points in Figures are mean values obtained from three independent experiments; error bars are standard errors. Statistical significance was tested using the Student t-test at p < 0.05.
Conflicts of Interest
The authors declare no conflict of interest.
Funding
The studies were partially supported by the Russian Foundation for Basic Research grant #16-04-01810-a and with the financial support of the Ministry of Education and Science of the Russian Federation (Agreement No. 02.A03.21.0003 dated of August 28, 2013).
References
- 1. Nurkovic J, Dolicanin Z, Mustafic F, Mujanovic R, Memic M, Grbovic V, Skevin AJ, Nurkovic S. Mesenchymal stem cells in regenerative rehabilitation. J Phys Ther Sci. 2016; 28:1943–48. https://doi.org/10.1589/jpts.28.1943 [PubMed]
- 2. Capelli C, Pedrini O, Cassina G, Spinelli O, Salmoiraghi S, Golay J, Rambaldi A, Giussani U, Introna M. Frequent occurrence of non-malignant genetic alterations in clinical grade mesenchymal stromal cells expanded for cell therapy protocols. Haematologica. 2014; 99:e94–97. https://doi.org/10.3324/haematol.2014.104711 [PubMed]
- 3. Froelich K, Mickler J, Steusloff G, Technau A, Ramos Tirado M, Scherzed A, Hackenberg S, Radeloff A, Hagen R, Kleinsasser N. Chromosomal aberrations and deoxyribonucleic acid single-strand breaks in adipose-derived stem cells during long-term expansion in vitro. Cytotherapy. 2013; 15:767–81. https://doi.org/10.1016/j.jcyt.2012.12.009 [PubMed]
- 4. Moon SH, Kim JS, Park SJ, Lim JJ, Lee HJ, Lee SM, Chung HM. Effect of chromosome instability on the maintenance and differentiation of human embryonic stem cells in vitro and in vivo. Stem Cell Res (Amst). 2011; 6:50–59. https://doi.org/10.1016/j.scr.2010.08.006 [PubMed]
- 5. Casiraghi F, Remuzzi G, Abbate M, Perico N. Multipotent mesenchymal stromal cell therapy and risk of malignancies. Stem Cell Rev. 2013; 9:65–79. https://doi.org/10.1007/s12015-011-9345-4 [PubMed]
- 6. Barkholt L, Flory E, Jekerle V, Lucas-Samuel S, Ahnert P, Bisset L, Büscher D, Fibbe W, Foussat A, Kwa M, Lantz O, Mačiulaitis R, Palomäki T, et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies--bridging scientific observations and regulatory viewpoints. Cytotherapy. 2013; 15:753–59. https://doi.org/10.1016/j.jcyt.2013.03.005 [PubMed]
- 7. Goldstein S. Replicative senescence: the human fibroblast comes of age. Science. 1990; 249:1129–33. https://doi.org/10.1126/science.2204114 [PubMed]
- 8. Bautista-Niño PK, Portilla-Fernandez E, Vaughan DE, Danser AH, Roks AJ. DNA Damage: A Main Determinant of Vascular Aging. Int J Mol Sci. 2016; 17:E748. https://doi.org/10.3390/ijms17050748 [PubMed]
- 9. Busuttil RA, Garcia AM, Cabrera C, Rodriguez A, Suh Y, Kim WH, Huang TT, Vijg J. Organ-specific increase in mutation accumulation and apoptosis rate in CuZn-superoxide dismutase-deficient mice. Cancer Res. 2005; 65:11271–75. https://doi.org/10.1158/0008-5472.CAN-05-2980 [PubMed]
- 10. Gorbunova V, Seluanov A. DNA double strand break repair, aging and the chromatin connection. Mutat Res. 2016; 788:2–6. https://doi.org/10.1016/j.mrfmmm.2016.02.004 [PubMed]
- 11. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: cell cycle and proliferation-dependent regulation. Semin Cancer Biol. 2016; 37-38:51–64. https://doi.org/10.1016/j.semcancer.2016.03.003 [PubMed]
- 12. Shibata A, Jeggo PA. DNA double-strand break repair in a cellular context. Clin Oncol (R Coll Radiol). 2014; 26:243–49. https://doi.org/10.1016/j.clon.2014.02.004 [PubMed]
- 13. Osipov AN, Buleeva G, Arkhangelskaya E, Klokov D. In vivo γ-irradiation low dose threshold for suppression of DNA double strand breaks below the spontaneous level in mouse blood and spleen cells. Mutat Res. 2013; 756:141–45. https://doi.org/10.1016/j.mrgentox.2013.04.016 [PubMed]
- 14. Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst). 2014; 19:169–75. https://doi.org/10.1016/j.dnarep.2014.03.014 [PubMed]
- 15. White RR, Vijg J. Do DNA Double-Strand Breaks Drive Aging? Mol Cell. 2016; 63:729–38. https://doi.org/10.1016/j.molcel.2016.08.004 [PubMed]
- 16. Kotenko KV, Bushmanov AY, Ozerov IV, Guryev DV, Anchishkina NA, Smetanina NM, Arkhangelskaya EY, Vorobyeva NY, Osipov AN. Changes in the number of double-strand DNA breaks in Chinese hamster V79 cells exposed to γ-radiation with different dose rates. Int J Mol Sci. 2013; 14:13719–26. https://doi.org/10.3390/ijms140713719 [PubMed]
- 17. Osipov AN, Grekhova A, Pustovalova M, Ozerov IV, Eremin P, Vorobyeva N, Lazareva N, Pulin A, Zhavoronkov A, Roumiantsev S, Klokov D, Eremin I. Activation of homologous recombination DNA repair in human skin fibroblasts continuously exposed to X-ray radiation. Oncotarget. 2015; 6:26876–85. https://doi.org/10.18632/oncotarget.4946 [PubMed]
- 18. Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol. 2012; 920:613–26. https://doi.org/10.1007/978-1-61779-998-3_40 [PubMed]
- 19. Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010; 24:679–86. https://doi.org/10.1038/leu.2010.6 [PubMed]
- 20. Redon CE, Nakamura AJ, Martin OA, Parekh PR, Weyemi US, Bonner WM. Recent developments in the use of γ-H2AX as a quantitative DNA double-strand break biomarker. Aging (Albany NY). 2011; 3:168–74. https://doi.org/10.18632/aging.100284 [PubMed]
- 21. Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003; 421:961–66. https://doi.org/10.1038/nature01446 [PubMed]
- 22. Siddiqui MS, François M, Fenech MF, Leifert WR. Persistent γH2AX: A promising molecular marker of DNA damage and aging. Mutat Res Rev Mutat Res. 2015; 766:1–19. https://doi.org/10.1016/j.mrrev.2015.07.001 [PubMed]
- 23. Osipov AN, Pustovalova M, Grekhova A, Eremin P, Vorobyova N, Pulin A, Zhavoronkov A, Roumiantsev S, Klokov DY, Eremin I. Low doses of X-rays induce prolonged and ATM-independent persistence of γH2AX foci in human gingival mesenchymal stem cells. Oncotarget. 2015; 6:27275–87. https://doi.org/10.18632/oncotarget.4739 [PubMed]
- 24. Bullwinkel J, Baron-Lühr B, Lüdemann A, Wohlenberg C, Gerdes J, Scholzen T. Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J Cell Physiol. 2006; 206:624–35. https://doi.org/10.1002/jcp.20494 [PubMed]
- 25. Juríková M, Danihel Ľ, Polák Š, Varga I. Ki67, PCNA, and MCM proteins: markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016; 118:544–52. https://doi.org/10.1016/j.acthis.2016.05.002 [PubMed]
- 26. T Kulat B, Russell HM, Sarwark AE, R Zingle N, Moss ST, Mongé MC, Backer CL, and B TK. Modified TandemHeart ventricular assist device for infant and pediatric circulatory support. Ann Thorac Surg. 2014; 98:1437–41. https://doi.org/10.1016/j.athoracsur.2014.05.093 [PubMed]
- 27. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001; 276:47759–62. [PubMed]
- 28. Caron P, Choudjaye J, Clouaire T, Bugler B, Daburon V, Aguirrebengoa M, Mangeat T, Iacovoni JS, Álvarez-Quilón A, Cortés-Ledesma F, Legube G. Non-redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Reports. 2015; 13:1598–609. https://doi.org/10.1016/j.celrep.2015.10.024 [PubMed]
- 29. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001; 276:42462–67. https://doi.org/10.1074/jbc.C100466200 [PubMed]
- 30. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012; 3:708. https://doi.org/10.1038/ncomms1708 [PubMed]
- 31. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, Herbig U, Longhese MP, d’Adda di Fagagna F. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012; 14:355–65. https://doi.org/10.1038/ncb2466 [PubMed]
- 32. Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004; 6:168–70. https://doi.org/10.1038/ncb1095 [PubMed]
- 33. Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8:4112–18. https://doi.org/10.4161/cc.8.24.10215 [PubMed]
- 34. Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012; 11:2402–07. https://doi.org/10.4161/cc.20882 [PubMed]
- 35. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011; 6:e19194. https://doi.org/10.1371/journal.pone.0019194 [PubMed]
- 36. Tu WZ, Li B, Huang B, Wang Y, Liu XD, Guan H, Zhang SM, Tang Y, Rang WQ, Zhou PK. γH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013; 587:3437–43. https://doi.org/10.1016/j.febslet.2013.08.028 [PubMed]
- 37. Mamouni K, Cristini A, Guirouilh-Barbat J, Monferran S, Lemarié A, Faye JC, Lopez BS, Favre G, Sordet O. RhoB promotes γH2AX dephosphorylation and DNA double-strand break repair. Mol Cell Biol. 2014; 34:3144–55. https://doi.org/10.1128/MCB.01525-13 [PubMed]
- 38. Liu B, Yip RK, Zhou Z. Chromatin remodeling, DNA damage repair and aging. Curr Genomics. 2012; 13:533–47. https://doi.org/10.2174/138920212803251373 [PubMed]
- 39. White RR, Milholland B, de Bruin A, Curran S, Laberge RM, van Steeg H, Campisi J, Maslov AY, Vijg J. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat Commun. 2015; 6:6790. https://doi.org/10.1038/ncomms7790 [PubMed]
- 40. Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA. 2007; 104:16633–38. https://doi.org/10.1073/pnas.0708043104 [PubMed]
- 41. Menendez JA, Cufí S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L, Vazquez-Martin A. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging (Albany NY). 2011; 3:1063–77. https://doi.org/10.18632/aging.100407 [PubMed]
- 42. White RR, Sung P, Vestal CG, Benedetto G, Cornelio N, Richardson C. Double-strand break repair by interchromosomal recombination: an in vivo repair mechanism utilized by multiple somatic tissues in mammals. PLoS One. 2013; 8:e84379. https://doi.org/10.1371/journal.pone.0084379 [PubMed]
- 43. Sukup-Jackson MR, Kiraly O, Kay JE, Na L, Rowland EA, Winther KE, Chow DN, Kimoto T, Matsuguchi T, Jonnalagadda VS, Maklakova VI, Singh VR, Wadduwage DN, et al. Rosa26-GFP direct repeat (RaDR-GFP) mice reveal tissue- and age-dependence of homologous recombination in mammals in vivo. PLoS Genet. 2014; 10:e1004299. https://doi.org/10.1371/journal.pgen.1004299 [PubMed]
- 44. Vaidya A, Mao Z, Tian X, Spencer B, Seluanov A, Gorbunova V. Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS Genet. 2014; 10:e1004511. https://doi.org/10.1371/journal.pgen.1004511 [PubMed]
- 45. MacRae SL, Croken MM, Calder RB, Aliper A, Milholland B, White RR, Zhavoronkov A, Gladyshev VN, Seluanov A, Gorbunova V, Zhang ZD, Vijg J. DNA repair in species with extreme lifespan differences. Aging (Albany NY). 2015; 7:1171–84. https://doi.org/10.18632/aging.100866 [PubMed]
- 46. Hudson D, Kovalchuk I, Koturbash I, Kolb B, Martin OA, Kovalchuk O. Induction and persistence of radiation-induced DNA damage is more pronounced in young animals than in old animals. Aging (Albany NY). 2011; 3:609–20. https://doi.org/10.18632/aging.100340 [PubMed]
- 47. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006; 8:315–17. https://doi.org/10.1080/14653240600855905 [PubMed]