



Introduction
When chronic kidney disease (CKD) progresses to end-stage renal disease (ESRD), the risk of cardiovascular mortality increases exponentially [1] and occur at a much younger age [2]. This suggests that CKD could be used as a clinical model to study premature vascular ageing [3,4]. Premature arteriosclerosis, i.e. medial vascular calcification (VC), is a distinct feature in CKD that predicts poor outcome [5]. Osteogenic factors, such as matrix Gla protein (MGP) and runt-related transcription factor 2 (RUNX2), are part of the calcification process. MGP is an important endogenous inhibitor of extracellular calcification expressed by vascular smooth muscle cells (VSMCs) [6,7], while RUNX2 has emerged as a key factor in the osteogenic transformation of VSMC in response to high phosphate and oxidative stress [8,9]. Uremic arteriosclerosis may in part be caused by increased vascular apoptosis [10,11].
Studies have associated single nucleotide polymorphisms on chromosome 9p21 close to the cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) with risk for cardiovascular disease (CVD) in the general population [12-16]. As the proteins derived from CDKN2A/B are functionally involved in maintaining cells in a state of growth arrest, and cellular senescence increases with age, expression of CDKN2A increase as a function of increasing cellular stress and organismal ageing [17,18]. Indeed, CDKN2A gene expression appears to be superior to telomere length as a biomarker of biological age [19,20]. Thus, elevated CDKN2A expression correlates with increased biological ageing as well as increased frequency of a frail phenotype [21].
Since the expression of the CDKN2A/B locus has not been investigated in uremic tissue we investigated arterial and muscle CDKN2A/B expression in ESRD. We tested if the expression of the CDKN2A/B complex correlated to chronological age, degree of VC (estimated by histological examination of arteries, semi-automated quantification and CT scans of the coronary arteries) and clinical CVD. We also related degree of VC to the number of p16INK4a and senescence associated β−-galactosidase (SA-β−Gal) positive cells.
Results
Demography, biochemical data and arterial expression of CDKN2A/p16INK4a, CDKN2B/p15INK4a, RUNX2 and MGP in the different groups of VC (none, mild, moderate and severe) are summarized in Table 1. No differences (p=0.15) in the number of no/former/present smokers were found in the calcification groups (no VC; 8/1/0, mild VC; 16/14/0, moderate VC; 7/7/0, severe VC; 3/1/0). Moreover, no difference (p=0.39) in physical activity (graded as regular, normal activity and disabled) was reported in the calcification groups (no VC; 2/6/1, mild VC; 18/13/1, moderate VC; 6/7/2, severe VC; 1/2/1). The arterial expression of CDKN2B/p15INK4a did not show any significant correlations with age, biomarkers of bone, inflammation and oxidative stress as well as extent of VC, coronary artery calcification (CAC) score or CVD. In contrast, the arterial CDKN2A/p16INK4a expression was significantly higher (Fig. 1) in patients with CVD compared to patients without CVD (median relative expression quantity 2.12 vs. 1.00, p=0.004), even after correction for age in multivariate analysis (p=0.01). Patients with CVD (632; IQR 79-1702) had significantly (p=0.0008) higher median CAC score than patients without clinical signs of CVD (0; IQR 0-64).
Table 1. Demographics, biochemical characteristics, arterial and muscle expression of the CDKN2A/B locus as well as the arterial expression of RUNX2 and Matrix Gla Protein.
| Demography | Total (n=61) | No VC (n=9) | Mild VC (n=32) | Moderate VC (n=16) | Severe VC (n=4) | P-value | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years | 45 (33-52) | 28 (24-30) | 44 (36-51) | 49 (40-56) | 53 (46-60) | p<0.0001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Males, n | 42 | 5 | 20 | 13 | 4 | p=0.1371 a | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cardiovascular disease, n | 5 | 0 | 1 | 2 | 2 | p=0.0464a | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vintage, years | 0.3 (0-1.1) | 0.7 (0-1.0) | 0.3 (0-1.0) | 0.6 (0-1.2) | 1.4 (0.3-2.0) | p=0.8156 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body mass index, kg/m2 | 23.8 (21.8-26.3) | 21.0 (19.8-25.7) | 23.5 (21.7-26.2) | 24.8 (22.6-26.7) | 23.2 (22.3-26.8) | p=0.5453 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Metabolism | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Creatinine, µmol/L | 739 (577-907) | 760 (519-1146) | 761 (592- 986) | 800 (598-906) | 914 (806-933) | p=0.9366 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cholesterol, mmol/L | 4.6 (3.9-5.3) | 4.4 (3.9-4.8) | 4.4 (3.8-5.1) | 5.1 (4.3-6.0) | 4.7 (3.8-7.2) | p=0.0853 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Triglyceride, mmol/L | 1.2 (0.9-1.7) | 1.0 (0.7-1.5) | 1.1 (0.9-1.5) | 1.4 (1.1-1.8) | 1.4 (1.1-2.9) | p=0.2950 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HDL-cholesterol, mmol/L | 1.4 (1.1-1.6) | 1.6 (1.3-1.9) | 1.4 (1.1-1.6) | 1.3 (1.0-1.5) | 1.2 (1.1-2.3) | p=0.4383 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IGF-1, ng/ml b | 255 (175-289) | 283 (261-326) | 251 (183-291) | 233 (175-282) | 168 (147-370) | p=0.5116 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Testosterone, mmol/L c | 12.3 (8.7-15.7) | 11.2 (6.5-28.6) | 13.1 (9.2-15.8) | 11.0 (7.5-15.5) | 10.7 (7.2-18.7) | p=0.6819 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Inflammation & oxidative | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| stress | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hsCRP, mg/L | 0.9 (0.5-2.5) | 0.6 (0.4-1.6) | 1.3 (0.5-3.0) | 0.8 (0.5-1.5) | 2.0 (0.5-9.4) | p=0.8237 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IL-6, pg/mL | 0.9 (0.5-1.8) | 1.0 (0.3-1.8) | 0.9 (0.6-2.1) | 0.6 (0.3-1.4) | 1.7 (1.4-16.5) | p=0.0480 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IL-8, pg/mL | 5.3 (3.5-7.9) | 5.9 (2.3-8.5) | 5.4 (3.6-8.3) | 4.4 (3.1-7.7) | 7.4 (4.8-11.3) | p=0.4705 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNF, pg/mL | 10.7 (9.0-13.1) | 9.7 (8.0-14.7) | 10.8 (9.3-12.9) | 10.4 (8.5-12.6) | 13.1 (12.2-16.7) | p=0.4624 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Albumin, g/L | 37 (33-39) | 32 (31-38) | 37 (35-39) | 37 (33-39) | 36 (32-42) | p=0.0754 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pentosidine, nmol/L d | 855 (609-1212) | 650 (466-1735) | 935 (691-1225) | 752 (607-1393) | 886 (696-984) | p=0.9495 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8-OHdG, ng/mL d | 0.18 (0.13-0.27) | 0.14 (0.08-0.19) | 0.21 (0.15-0.26) | 0.16 (0.10-0.27) | 0.30 (0.04-0.42) | p=0.4616 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Calcification & bone markers | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAC score, HU e | 0 (0-111) | 0 (0-0) | 0 (0-90) | 13 (1-171) | 541 (28-1702) | p=0.0154 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Media calcification, % f | 1.8 (0.5-5.5) | 0.2 (0-0.3) | 1.2 (0.5-2.5) | 4.9 (0.3-9.7) | 25.3 (2.5-32.9) | p<0.0001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Calcium, mmol/L | 2.3 (2.2-2.4) | 2.2 (2.1-2.4) | 2.4 (2.2-2.5) | 2.3 (2.2-2.4) | 2.2 (2.0-2.3) | p=0.2136 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phosphate, mmol/L | 1.8 (1.5-2.1) | 1.6 (1.4-2.1) | 1.8 (1.4-2.2) | 1.9 (1.5-2.1) | 1.4 (0.9-1.5) | p=0.2679 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnesium, mmol/L | 0.9 (0.8-1.0) | 0.8 (0.7-1.0) | 0.8 (0.8-0.9) | 0.9 (0.7-1.0) | 0.9 (0.7-1.1) | p=0.7395 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| iPTH, ng/L | 258 (190-400) | 216 (181-286) | 252 (190-384) | 371 (228-583) | 127 (85-210) | p=0.1306 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 25-OH D-vitamin, mmol/L g | 39 (27-55) | 33 (23-46) | 37 (27-48) | 51 (26-82) | 39 (38-44) | p=0.1777 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1,25-OH D-vitamin, pmol/L g | 18 (13-27) | 17 (12-19) | 18 (13-28) | 18 (12-29) | 18 (7-49) | p=0.6277 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FGF-23, pg/ml d | 3353 (1114-31809) | 5909 (1901-15150 | 3134 (453-33973) | 4461 (1651-58853) | 2184 (758-11994) | p=0.4184 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Klotho, pg/ml h | 395 (297-520) | 509 (434-625) | 349 (260-480) | 391 (307-489) | 409 (320-499) | p=0.3608 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alkaline phosphatase, U/L i | 60.6 (49.6-89.2) | 66.7 (47.2-126.1) | 60.2 (50.3-82.7) | 56.9 (47.9-78.1) | 90.3 (47.0-113.1) | p=0.5853 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Osteoprotegerin, pmol/L | 5.9 (5.1-7.3) | 5.3 (4.2-5.8) | 6.1 (4.9-7.0) | 5.8 (5.5-7.9) | 9.3 (7.4-14.9) | p=0.0049 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MID osteocalcin, ng/ml h | 83.6 (37.3-184.9) | 76.4 (27.5-148.3) | 125.4 (41.9-220.1) | 80.6 (48.6-204.3) | 24.9 (7.0-147.6) | p=0.4709 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GLA osteocalcin, ng/ml j | 42.5 (22.3-66.0) | 41.6 (26.7-62.4) | 49.1 (22.4-66.8) | 40.2 (27.8-74.7) | 12.8 (8.5-27.3) | p=0.3225 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GLU osteocalcin, ng/ml j | 28.4 (8.0-56.5) | 25.5 (15.7-77.6) | 33.2 (9.4-56.8) | 31.3 (6.9-75.4) | 2.6 (1.5-33.0) | p=0.5455 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vascular expression (RQ) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CDKN2B/p15INK4ak | 1.35 (0.81-1.87) | 1.91 (0.78-2.49) | 1.28 (0.97-1.74) | 1.58 (0.81-2.00) | 1.16 (0.36-2.26) | p=0.3982 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CDKN2A/p16INK4a | 1.04 (0.25-1.02) | 0.94 (0.45-1.20) | 1.02 (0.62-1.62) | 1.12 (0.49-1.69) | 2.60 (1.18-5.05) | p=0.0108 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RUNX2h | 1.56 (1.02-2.81) | 2.09 (1.00-3.04) | 1.12 (1.00-2.01) | 1.55 (1.31-2.34) | 6.12 (3.89-6.13) | p=0.0036 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Matrix Gla proteinf | 0.88 (0.61-1.30) | 0.70 (0.45-1.00) | 0.87 (0.58-1.25) | 0.91 (0.62-1.40) | 2.33 (0.70-3.65) | p=0.0002 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Muscular expression (RQ) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CDKN2A/p16INK4al | 0.63 (0.25-1.02) | 0.35 (0.10-0.86) | 0.45 (0.24-0.94) | 0.63 (0.29-2.22) | 0.88 (0.60-1.28) | p=0.7292 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| All continuous data are given as median (interquartile range). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HU = Hounsfield units; CDKN2 = Cyclin-dependent kinas; RQ = Relative quantity; PTH = Parathyroid hormone; hsCRP = high-sensitivity C-reactive protein; IL-6 = Interleukin-6; IL-8 = Interleukin-8; TNF = Tumour necrosis factor; HDL = High-density lipoprotein, 8-OHdG = 8-hydroxy-2’-deoxyguanosine, CAC = coronary artery calcification, RUNX2 = runt-related transcription factor 2 a; chi square, b; n=59; c; n=39, d; n=41, e; n=47, f; n=54, g; n=49, h; n=48, i;=44, j; n=52, k; n=58, l; n=50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
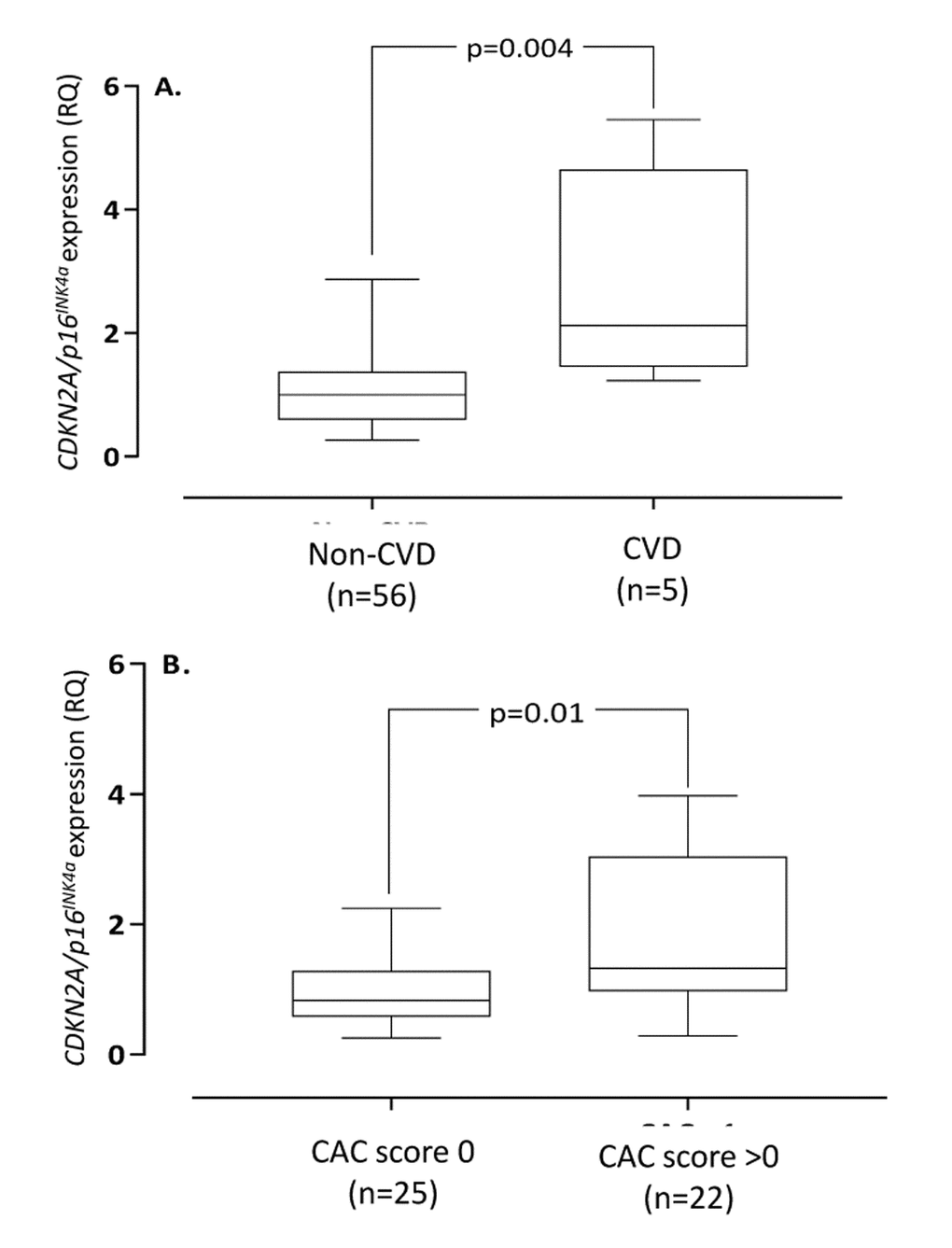
Figure 1. Arterial expression of CDKN2A/p16INK4a in end-stage renal disease patients with vs. without cardiovascular disease (A) and coronary artery calcification (CAC) score 0 vs. >0 (B). Cardiovascular disease was defined on clinical grounds. Coronary artery calcification score were obtained through CT heart (n=47). CDKN2A=cyclin-dependent kinase 2A. RQ=relative quantity.
The arterial expression of CDKN2A/p16INK4a correlated (Rho=0.34; p=0.006) with chronological age (Suppl. Fig. 2). Patients with CAC-score >0 had significantly higher (median relative expression quantity 1.32 vs. 0.83 RQ; p=0.01) arterial expression of CDKN2A/p16INK4a than patients with CAC score 0 (Fig. 1), but not after correction for age (p=0.12). The correlation between CDKN2A/p16INK4a and media calcification was borderline (Rho=0.26; p=0.06) significant in univariate analysis (Suppl. Fig. 3) but reached statistical significance (p=0.0003) after multivariate correction for chronological age. The arterial expression of CDKN2A/p16INK4a correlated with CAC score (Rho=0.41; p=0.004), IL-8 (Rho=0.31; p=0.016), IGF-1 (Rho=-0.30; p=0.02), testosterone (males only) (Rho=-0.50; p=0.001), Mid-fragments (MID)-osteocalcin (OC) (Rho=-0.52; p<0.0001), inactive GLU-type (GLU)-OC (Rho=-0.54; p<0.0001) and active GLA-type (GLA-OC) (Rho=-0.47; p=0.0004). The correlations between CDKN2A/p16INK4a and GLA-OC and CAC score are depicted in Figure 2. Median N-MID OC (27.0 vs. 115.4 ng/ml; p=0.013), GLU-OC (3.1 vs 34.4 ng/ml; p=0.02) and GLA-OC (13.3 vs. 45.5 ng/ml; p=0.014) were significantly lower in patients with CVD.
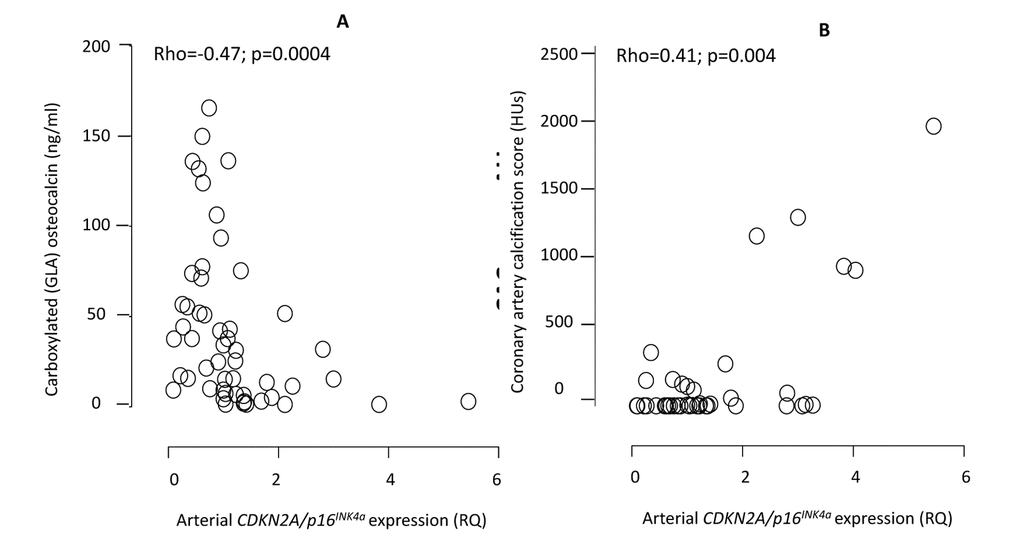
Figure 2. Correlations between the arterial expression of CDKN2A/p16INK4a and circulating levels of carboxylated (GLA) active osteocalcin (A) and coronary artery calcification by CT heart (B). CDKN2A = cyclin-dependent kinase 2A. RQ = relative quantity. HU = Hounsfield units. The exclusion of one patient on warfarin did not affect the correlation between CDKN2A/p16INK4a and GLA-OC (Rho=-0.51; p=0.001).
As shown in Figure 3, arteries with severe medial VC exhibited significantly higher vascular expression of RUNX2 (p=0.0036), MGP (p=0.0002) and CDKN2A/p16INK4a (p=0.011). The arterial expression of CDKN2A/p16INK4a was significantly correlated to the expression of MGP (Rho=0.31; p=0.02), but not RUNX2 (Rho=0.21; p=0.16). The degree of media calcification (%) correlated to total CAC score (Rho=0.59; p<0.0001) and MGP expression (Rho=0.38; p=0.008), but not to RUNX2 expression (Rho=0.14). Total CAC score correlated to arterial MGP expression (Rho=0.38; p=0.016) but not to RUNX2 expression (Rho=-0.01).
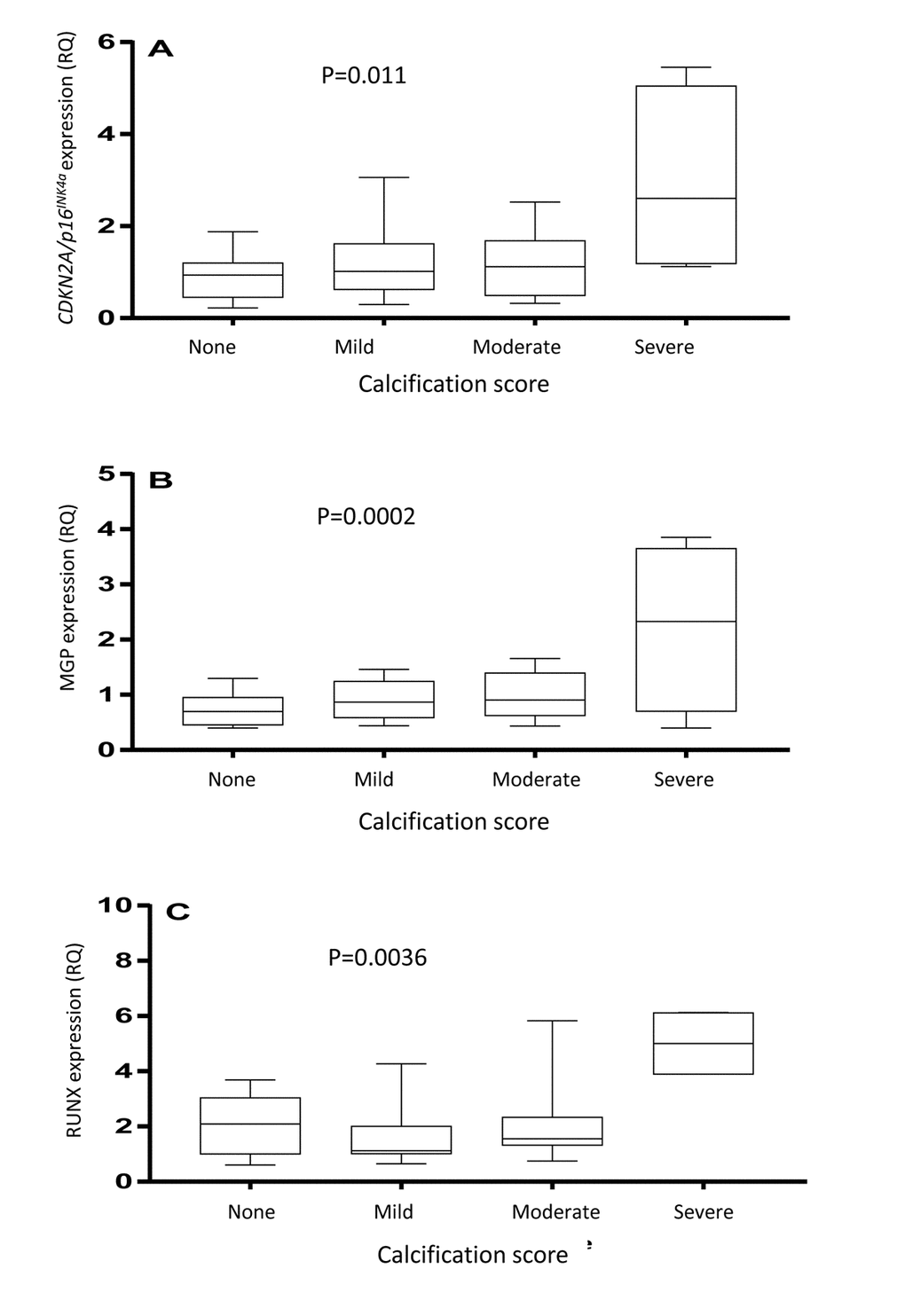
Figure 3. Arterial expression of CDKN2A/p16INK4a (A), matrix Gla protein (B) and runt-related transcription factor 2 (C) in arterial biopsies. The vascular biopsies from end-stage renal disease patients were stained with von Kossa and scored by a pathologist as having none, mild, moderate or severe media calcification. P-values represent ANOVA.
The expression of CDKN2A/p16INK4a did not differ between uremic arteries and skeletal muscle (median relative expression quantity 1.04 vs. 0.63 IU, p=0.79). No significant difference was observed in muscular CDKN2A/p16INK4a expression between patients with and without CVD (Table 1). The muscular expression of CDKN2A/p16INK4a correlated (Rho=0.30; p=0.02) with chronological age (Suppl. Fig. 2). A borderline significant (Rho=0.26; p=0.05) association was observed between the arterial and muscular expression of CDKN2A/p16INK4a (Suppl. Fig. 4). The muscular expression of CDKN2A/p16INK4a did not differ in patients with and without CVD nor between patients with CAC 0 vs. CAC >0 or the different calcification groups (Table 1).
p16INK4a protein and SA-β-Gal expression
Expression of p16INK4a in epigastric artery was absent, or low, in patients with no or mild VC, but was increased in moderate VC and further exacerbated in severe VC (Fig. 4). Cells positive for p16INK4a were localized to the vascular media and in severe VC also in the intima and were more frequent (p<0.05) in areas with extensive calcification. The cellular localization of p16INK4a was mostly nuclear. The p16INK4a protein expression pattern mirrored the expression of CDKN2A/p16INK4a. The number of SA-β-Gal positive cells was significantly (p<0.05) higher in severely calcified arteries (Fig. 4).
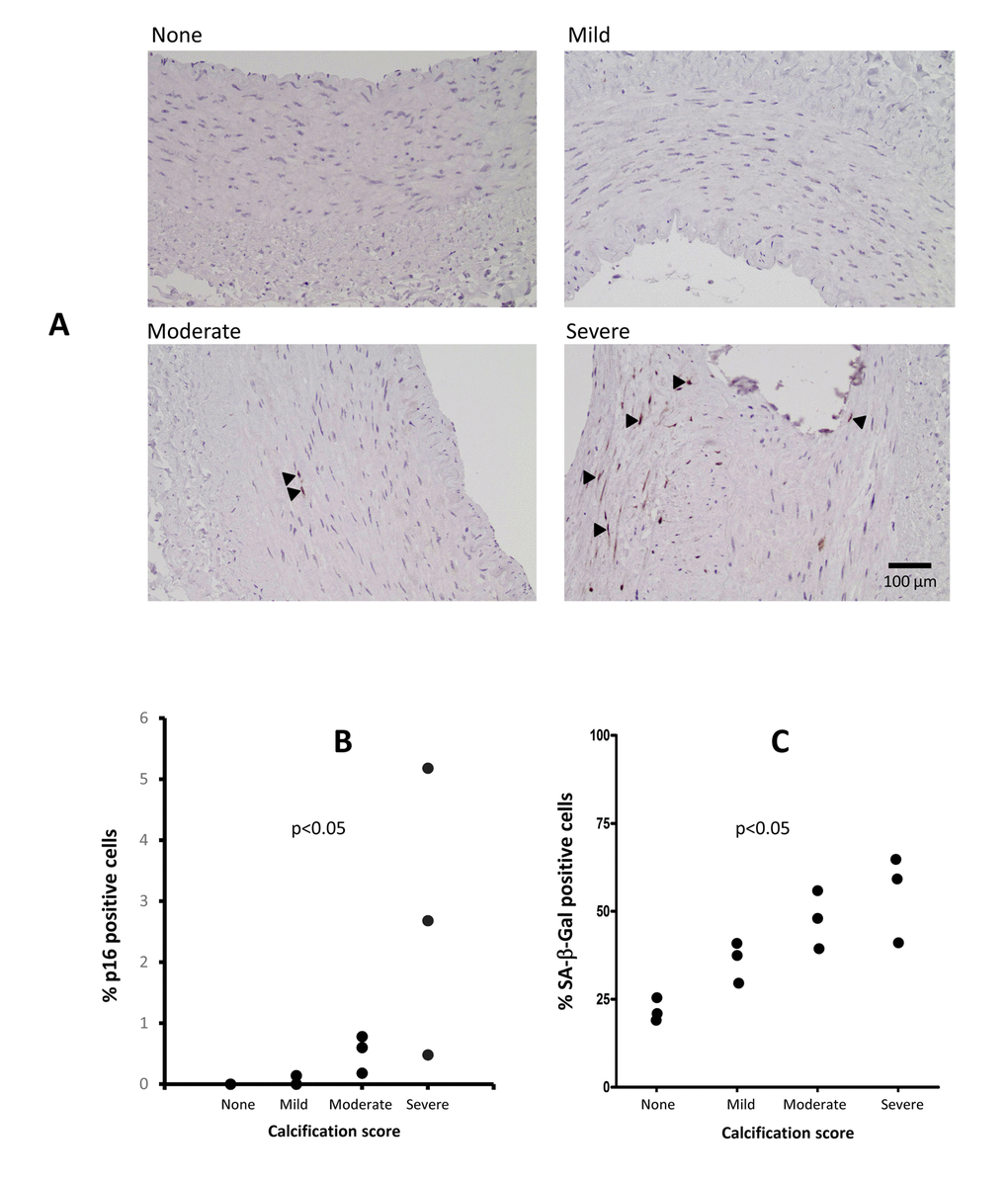
Figure 4. Immunostaining of arterial p16INK4a and SA-β-Gal in patients with varying degrees of vascular calcification (VC) (A). The p16INK4a expression was localised to the cell nucleus and involved more cells in severe VC. Staining for p16INK4a with DAB (brown) and counterstaining with hematoxylin (blue). Arrowheads indicate p16INK4a positive cells. (B) Levels of p16INK4a in epigastric arteries from patients with varying degrees of VC, expressed as the percentage of p16INK4a positive cells of total number of cells in the media and intima layers in a single arterial section (n=2 for no VC, n=3 for mild VC, n=3 for moderate VC and n=3 for severe VC). The number of positive SA-β-Gal positive cells increase with increased calcification (C); no VC; n=2, mild VC; n=3, moderate VC; n=3 and severe VC; n=3. P-values represent no vs. severe VC.
Discussion
We report that the arterial CDKN2A/p16INK4a (but not CDKN2B/p15INK4b) expression associate with CVD as well as degree of VC in ESRD. These findings are corroborated by increased number of p16INK4a and SA-β-Gal positive cells in severely calcified arteries (Fig. 4). Increased expression of p16INK4a (and the cellular senescence that follows) may be causally involved in the decline of cellular function of stem and progenitor cells [22] that are observed with organismal ageing. A recent study in mice implies a significant role of p16INK4a/β-GalpH6 positive macrophages and impaired immune system in aging [23]. Since vascular cell senescence contributes to breakdown of the blood-brain barrier [24] a consequence of increased vascular cell senescence may be higher cerebral concentrations of retained circulating cytokines, adipokines and uremic toxins. This may promote anorexia, cognitive dysfunction and depression; all common features in the uremic milieu [25,26]. The senescence-associated secretory phenotype (SASP) - a key feature of cellular senescence - results in the secretion of pro-inflammatory factors that poison the tissue in the proximity of the senescent cell. In accordance, we report a significant correlation between circulating IL-8 and the arterial expression of CDKN2A/p16INK4a and higher IL-6 and OPG in severe VC (Table 1). Since senescent cells have been considered a target for anti-aging therapies and removal of p16INK4a-positive cells in progeric mice led to an attenuation of the ageing phenotype [27], a functional involvement of p16INK4a in ageing-associated disorders is likely. The use of SASP modulators and senolytic drugs are intriguing prospect for the future treatment of uremic arteriosclerosis [28]. Indeed, a recent study in atherosclerotic mice show that chronic senolytic treatment improves established vascular dysfunction [29]. As we confirm that increased vascular senescence associates with low IGF-1 and testosterone levels [30,31], the benefit of hormone replacement therapies needs consideration in this prematurely aged patient group.
This the first report on the expression of p16INK4a in human uremic arterial tissue. Yamada et al. [32] showed increased arterial mRNA expression of Cdkn2a in rats with adenine-induced uraemia compared to control animals. Interestingly, they also demonstrated an increased arterial Runx2 mRNA expression, and both p16INK4a and Runx2 protein expression were detected in and around calcified areas of aortas from CKD rats, but not in normal rats [32]. We report higher arterial expression of RUNX2, MGP and CDKN2A/p16INK4a in ESRD patients with severe VC (Fig. 3). The higher MGP expression in arteries with severe media calcification may be regarded as an attempt to further prevent VC. Since VSMC apoptosis promote calcification [11], the relationship between the expression of the cell cycle inhibitor CDKN2A/p16INK4a and MGP is intriguing and requires further studies at the protein level.
The underlying cause(s) of the increase in CDKN2A/p16INK4a expression were beyond the scope of this study. Although it has been reported that oxidative stress may be a culprit [33] we find no associations between CDKN2A/p16INK4a expression and circulating oxidative stress biomarkers. Prelamin A accumulation promotes calcification and ageing in VSMCs – an effect which appears to be mediated, at least in part, by p16 protein. In addition, RUNX2 may be activated by prelamin A and DNA damage signalling [34,35], further linking cellular stress, p16 activity and an osteogenic potential. Another possibility for increased p16INK4a expression in ESRD is impairment of immune system function [23]. It was recently reported that the uremic toxin p-cresylsulphate induces macrophage activation, which lead to a failure in adaptive immune response [36]. Since we report on a strong inverse association between high vascular CDKN2A/p16INK4a expression and low levels of carboxylated active osteocalcin (Fig. 2) it can be hypothesized that vitamin K deficiency and decreased carboxylation of Gla proteins promote vascular senescence. Indeed, the expression of OC decreases with older age [37] and low carboxylation of OC associates not only with decreased cellular secretion of OC but also less accumulation in tissue [38]. Our data support Idelevich et al. [39] who report that OC, by promoting the osteocondrogenic differentiation in VSMC, is an active player in the VC process. Since vitamin K deficiency is a common finding in CKD [40] and promotes low carboxylation of OC our findings support a role for vitamin K deficit in premature vascular aging [41]. The protective effect of vitamin K may not only occur via carboxylation of Gla rich proteins but also via inhibition of apoptosis, which reduces the transdifferentiation of VSMC to osteoblasts [38]. It is noteworthy that vitamin K deficiency is consistently linked to features of aging, such as sarcopenia, frailty, osteoporosis and insulin resistance [42]. Since Vitamin K2 promote progenitor cell differentiation [43] its effect on vascular senescence, p16 clearance and progenitor cell mobilisation deserve further studies.
Some strengths and weaknesses of the present study deserve mentioning. An important strength is the access to highly biologically relevant methods and tissues. As opposed to being estimated by a surrogate marker of vascular stiffness the extent of VC was evaluated by three different methods. Moreover, gene expression data were obtained from the tissue directly affected by disease and we used von Kossa staining (the method of choice for detection of mineralisation). Finally, the presence of vascular senescence as estimated by arterial expression of CDKN2A/p16INK4a was mirrored by immunostaining of p16INK4a and SA-β-Gal (Fig. 4). Weak points of the study include the cross-sectional design. As many of the findings are correlational in nature, conclusions on causation are impossible to infer and we cannot conclude on the pathogenic relationship between p16INK4a overexpression and VC. However, since we observed a significant correlation between CDKN2A/p16INK4a expression and media calcification after correction for age our data imply that p16INK4a is involved in the calcification process and not only a consequence of chronological aging. It should also be appreciated that patients in the study were a selected group of younger ESRD patients with a lower prevalence of CVD than the typical Caucasian dialysis patient (where a more aggravated vascular phenotype is expected). Thus, the results may not be applicable to older and sicker dialysis patients, nor dialysis patients of other ethnicities. Another limitation is the lack of data on vascular CDKN2A/p16INK4a and p16INK4a expression in age- and gender-matched non-uremic patients with arteriosclerosis. Future studies would benefit from a control group, as it would make it possible to determine the impact of the uremic milieu on the degree of cellular senescence and CDKN2A/p16INK4a and p16INK4a expression. It should also be pointed out that VC might vary depending on arterial type and location along any specific artery. However, we reported a close relation between epigastric artery medial calcification and extent of coronary artery calcification [44]. Finally, since RNA was prepared from a homogenised arterial sample, this precludes any knowledge on how the expression varied between different cell types. Still, the multiple associations of CDKN2A/p16INK4a with different markers of VC increase the strength of our findings.
In summary, we demonstrate that in the uremic milieu, increased arterial expression of CDKN2A/p16INK4a associated with vascular progeria, independently of chronological age. These findings are supported by the increased number of p16INK4a and SA-β-Gal positive cell in arteries with severe VC. Further studies should evaluate if impaired Vitamin K mediated carboxylation contribute to premature vascular senescence.
Materials and Methods
Patient population
Adult patients undergoing living donor kidney transplantation (RTx) at the Dept. of Transplantation Surgery at the Karolinska University Hospital were invited to participate in the study. All participants provided written informed consent and the Regional Ethical Review Board in Stockholm approved the study. Basic patient characteristics are outlined in Table 1. Whereas 39 patients had undergone dialysis treatment prior to RTx (median dialysis vintage 0.3 years) 22 patients underwent pre-emptive RTx. Eighteen of the individuals that were treated with dialysis had received haemodialysis (HD), 20 had received peritoneal dialysis (PD) and one had initially received PD but switched to HD. The median age was 45 years and 69% were males. Smoking status (no, former and current smoker) and degree of physical activity (high, moderate and low) were recorded. The most common causes of CKD were chronic glomerulonephritis (n=27), polycystic kidney disease (n=10), interstitial nephritis (n=3) and other (or unknown) causes (n=21). The median systolic and diastolic blood pressures were 142 (IQR 129-152) and 86 (IQR 74-93) mmHg, respectively. The most commonly used medications were erythropoietin-stimulating agents (84%), non-calcium-based phosphate binders (75%), calcium-based phosphate binders (4%) and loop-diuretics (62%). Commonly used anti-hypertensive treatments were β-blocking agents (52%) and ACE-i/ARBs (72%); 30% of the patients were on statins. One patient was on warfarin. In this group of younger patients only five had clinical signs of cerebrovascular, cardiovascular, and/or peripheral vascular disease (grouped as CVD). Three had clinical signs of ischemic heart disease and two had peripheral ischemic atherosclerotic vascular disease.
Biopsy sampling
Within 20 minutes after skin incision, skeletal muscle and an artery (1-2 cm) were collected by sharp dissection. Muscle samples were taken from the external or internal oblique muscle or from the transverse abdominal muscle, and artery samples were obtained from the inferior epigastric artery. Samples were immediately placed in AllProtect Tissue Reagent (Qiagen, Hilden, Germany), snap-frozen in isopentane and subsequently stored at -70 °C, or fixed in 4% phosphate buffered formalin. Formalin-fixed material from the epigastric arteries was embedded in paraffin. One or two separate tissue sections (1-2 µm thick) were stained with hematoxylin and eosin and van Kossa, respectively, and were evaluated by two experienced pathologists (AW and MS). The degree of medial calcification was semi-quantified on the van Kossa stained sections and graded 0-3, where 0 indicated no calcification and 3 the highest degree of calcification [44]. Degree of calcification was also assessed as a continuous variable by semi-automated quantification as previously described [44]. The correlation (p<0.0001) between semi-quantitative histological assessment and semi-automated quantification of media calcification is presented in Suppl. Fig. 1.
Biomarker measurements
Blood samples were obtained in a fasting state in the morning the day before the surgical procedure, and stored in -80 °C. Interleukin (IL)-6, IL-8, TNF, IGF-1 and testosterone were analysed by immunometric assays on an Immulite 1000 Analyzer (Siemens Healthcare Diagnostics, Los Angeles, CA, USA) according to the instructions of the manufacturer. Plasma pentosidine was analysed by reverse-phase high performance liquid chromatography (HPLC). The following biomarkers were analysed with ELISA technique; 8-OHdG (Japan Institute for the Control of Aging, Shizuoka, Japan), osteoprotegerin (OGP) (Quidel Corporation, San Diego, CA, USA), human soluble α-Klotho (Immuno-Biological Laboratories Co., Ltd, Fujioka-shi, Japan), C-terminal FGF-23 (Immutopics Inc, San Clemente, CA, USA), Human N-MID Osteocalcin ELISA (IDS Nordic a/s, Herlev, Denmark), GLA-OC) and GLU-OC (Takara Bio Inc, Kusatsu, Japan). N-MID OC ELISA measuring total OC is more stable and reproducible than assays measuring intact OC only. Analyses of hsCRP, intact parathyroid hormone (iPTH), plasma cholesterol, triglycerides, HDL-cholesterol, creatinine, albumin, calcium, magnesium, phosphate, alkaline phosphatase (ALP), 25(OH) and 1,25(OH) D-vitamin were performed with validated routine methods at the accredited Clinical Chemical Laboratory Lab at the Karolinska University Hospital, Stockholm, Sweden.
Computed tomography
All cardiac computed tomography (CT) scans were performed using a 64-channel detector scanner (LightSpeed VCT; General Electric (GE) Healthcare, Milwaukee, WI, USA) in cine mode. Scans were ECG-gated and a standard non-contrast protocol was used with a tube voltage of 100 kV, tube current of 200 mA, 350 ms rotation time, 2.5 mm slice thickness and displayed field of 25 cm. Calcium deposits in the coronary arteries were identified by a radiologist with Level 2 competence and extensive experience of cardiac CT interpretation. Data were subsequently processed and analysed using an Advantage Workstation (GE Healthcare). Smartscore 4.0 (GE Healthcare) was used to assess CAC scores. Calcified plaques were considered to be present if values exceeded the standard threshold of 130 Hounsfield units. The CAC scores were expressed in Hounsfield units (HU) as previously described in detail [45]. Total CAC score was defined as the sum of the CAC scores in the left main artery, the left anterior descending artery, the left circumflex artery and the right coronary artery.
RNA preparation and gene expression analysis
TRIzol® Reagent (Ambion, Life Technologies, Carlsbad, USA) was used to prepare total RNA from artery and muscle biopsies according to the manufacturer’s recommendation. RNA concentration was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop products, Wilmington, USA) and RNA integrity was evaluated by an Agilent 2100 BioAnalyzer (Agilent Technologies Inc., Santa Clara, USA). For further analysis only samples with RIN>5.5 were used. Reverse transcription reaction was performed using random hexamers and SuperScriptIII® kit as per manufacturer’s instructions (Invitrogen, Life Technologies, Carlsbad, USA). QPCR was performed using individual TaqMan® gene expression assays specific to CDKN2A/p16INK4a (#Hs00923894_m1), CDKN2B/p15 (# Hs00793225_m1) and HPRT1 (# Hs02800695_m1), as a reference gene, using 7500 Fast Real Time PCR (Applied Biosystems, Life Technologies, Carlsbad, USA). Gingell-Littlejohn et al. [20] previously published the primer sequences. Expression of MGP(Hs00969490_m1) and RUNX2 (Hs00231692_m1) was analysed using a TaqMan Low Density Array on a QuantStudioTM 7 Flex Real-Time PCR system (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions and 30-1000 ng of cDNA from epigastric artery was added to the array. Amplification profiles for each sample and target were analysed individually and profiles with Ct above 35, bad passive reference signal, noise spikes and high noise were removed from further analyses. The data were analysed using ExpressionSuite®Software v1.0.4 (Applied Biosystems, Life Technologies, Carlsbad, CA, USA), and the 2-ΔΔCT formula was used to transform expression data from all genes to exponential values.
p16INK4a immunohistochemistry
Immunohistochemistry of p16INK4a was performed on paraffin-embedded sections of epigastric arteries from 11 subjects. Samples were divided according to amount of VC; no VC (n=2), minor VC (n=3), moderate VC (n=3) and severe VC (n=3). Anti-CDKN2A/p16INK4a polyclonal antibody was used (host species: rabbit, Antibody Registry ID: AB_306176, product number ab7962 Abcam, Cambridge, United Kingdom) and the analysis was performed using an established automated method at Dept. of Pathology, Karolinska University Hospital, Sweden. Staining and counterstaining were performed using diaminobenzidine (DAB) and hematoxylin, respectively. A section lacking primary antibody was used as a negative control. The score for p16INK4a positivity was calculated as the percentage of p16INK4a positive cells out of the total number of cells in the media and intima layers in one arterial section.
Senescence associated β-galactosidase (SA-β-Gal) staining
SA-β-Gal staining was performed using Senescence Cell Histochemical Staining kit (Sigma-Aldrich, UK) with sections being fixed for 1 min in room temperature, washed three times in PBS and immersed in SA-β-Gal staining solution overnight in 37°C. Tissue sections were counterstained with eosin and viewed by bright-field microscopy. The percentage of senescent cells (SA-β-Gal positive cells) and the total number of cells was counted for each section in the six independent fields. Each group consisted of three tissue specimens.
Statistical analysis
All continuous variables were expressed as median (interquartile range) and nominal variables as percentage, unless otherwise indicated. Statistical significance was set at the level of p<0.05. Comparisons between two groups were assessed with Wilcoxon rank sum test or χ2 test, as appropriate. Differences between the four groups were analysed by ANOVA. Spearman’s rank correlation (ρ) was used to determine correlations between two continuous variables. Multivariate analysis was performed using general linear model (GLM). Statistical analyses were performed using statistical software SAS 9.4 (SAS Campus Drive, Cary, NC, USA 27513).
Supplementary Materials
Acknowledgements
We thank the patients who participated in the study. We are grateful to all those who carried out the extensive laboratory work (Ann-Christin Bragfors Helin, Monica Eriksson, Björn Anderstam, Sanna Spännare and Anneli Ring).
Conflicts of Interest
The authors have no conflict of interests to declare.
Funding
Peter Stenvinkels research benefited by support from the Swedish Medical Research Council (VR), the Swedish Heart and Lung Foundation (20160384), the Strategic Research Program in Diabetes at Karolinska Institutet and Westmans Foundation.
References
- 1. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004; 351:1296–305. https://doi.org/10.1056/NEJMoa041031 [PubMed]
- 2. Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998 (Suppl 3); 32:S112–19. https://doi.org/10.1053/ajkd.1998.v32.pm9820470 [PubMed]
- 3. Kooman JP, Kotanko P, Schols AM, Shiels PG, Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol. 2014; 10:732–42. https://doi.org/10.1038/nrneph.2014.185 [PubMed]
- 4. Stenvinkel P, Larsson TE. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis. 2013; 62:339–51. https://doi.org/10.1053/j.ajkd.2012.11.051 [PubMed]
- 5. Shantouf RS, Budoff MJ, Ahmadi N, Ghaffari A, Flores F, Gopal A, Noori N, Jing J, Kovesdy CP, Kalantar-Zadeh K. Total and individual coronary artery calcium scores as independent predictors of mortality in hemodialysis patients. Am J Nephrol. 2010; 31:419–25. https://doi.org/10.1159/000294405 [PubMed]
- 6. Hale JE, Fraser JD, Price PA. The identification of matrix Gla protein in cartilage. J Biol Chem. 1988; 263:5820–24. [PubMed]
- 7. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997; 386:78–81. https://doi.org/10.1038/386078a0 [PubMed]
- 8. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008; 283:15319–27. https://doi.org/10.1074/jbc.M800021200 [PubMed]
- 9. Speer MY, Li X, Hiremath PG, Giachelli CM. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J Cell Biochem. 2010; 110:935–47. https://doi.org/10.1002/jcb.22607 [PubMed]
- 10. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011; 109:697–711. https://doi.org/10.1161/CIRCRESAHA.110.234914 [PubMed]
- 11. Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, Hiorns M, Donald AE, Deanfield J, Rees L, Shanahan CM. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008; 118:1748–57. https://doi.org/10.1161/CIRCULATIONAHA.108.783738 [PubMed]
- 12. Adams JN, Raffield LM, Freedman BI, Langefeld CD, Ng MC, Carr JJ, Cox AJ, Bowden DW. Analysis of common and coding variants with cardiovascular disease in the Diabetes Heart Study. Cardiovasc Diabetol. 2014; 13:77. https://doi.org/10.1186/1475-2840-13-77 [PubMed]
- 13. Ferguson JF, Matthews GJ, Townsend RR, Raj DS, Kanetsky PA, Budoff M, Fischer MJ, Rosas SE, Kanthety R, Rahman M, Master SR, Qasim A, Li M, et al, and CRIC Study Principal Investigators. Candidate gene association study of coronary artery calcification in chronic kidney disease: findings from the CRIC study (Chronic Renal Insufficiency Cohort). J Am Coll Cardiol. 2013; 62:789–98. https://doi.org/10.1016/j.jacc.2013.01.103 [PubMed]
- 14. Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007; 316:1491–93. https://doi.org/10.1126/science.1142842 [PubMed]
- 15. Larson MG, Atwood LD, Benjamin EJ, Cupples LA, D’Agostino RB
Sr , Fox CS, Govindaraju DR, Guo CY, Heard-Costa NL, Hwang SJ, Murabito JM, Newton-Cheh C, O’Donnell CJ, et al. Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med Genet. 2007 (Suppl 1); 8:S5. https://doi.org/10.1186/1471-2350-8-S1-S5 [PubMed] - 16. Lee JY, Lee BS, Shin DJ, Woo Park K, Shin YA, Joong Kim K, Heo L, Young Lee J, Kyoung Kim Y, Jin Kim Y, Bum Hong C, Lee SH, Yoon D, et al. A genome-wide association study of a coronary artery disease risk variant. J Hum Genet. 2013; 58:120–26. https://doi.org/10.1038/jhg.2012.124 [PubMed]
- 17. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004; 114:1299–307. https://doi.org/10.1172/JCI22475 [PubMed]
- 18. Sun LQ, Lee DW, Zhang Q, Xiao W, Raabe EH, Meeker A, Miao D, Huso DL, Arceci RJ. Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev. 2004; 18:1035–46. https://doi.org/10.1101/gad.1176104 [PubMed]
- 19. Der G, Batty GD, Benzeval M, Deary IJ, Green MJ, McGlynn L, McIntyre A, Robertson T, Shiels PG. Is telomere length a biomarker for aging: cross-sectional evidence from the west of Scotland? PLoS One. 2012; 7:e45166. https://doi.org/10.1371/journal.pone.0045166 [PubMed]
- 20. Gingell-Littlejohn M, McGuinness D, McGlynn LM, Kingsmore D, Stevenson KS, Koppelstaetter C, Clancy MJ, Shiels PG. Pre-transplant CDKN2A expression in kidney biopsies predicts renal function and is a future component of donor scoring criteria. PLoS One. 2013; 8:e68133. https://doi.org/10.1371/journal.pone.0068133 [PubMed]
- 21. Pathai S, Lawn SD, Gilbert CE, McGuinness D, McGlynn L, Weiss HA, Port J, Christ T, Barclay K, Wood R, Bekker LG, Shiels PG. Accelerated biological ageing in HIV-infected individuals in South Africa: a case-control study. AIDS. 2013; 27:2375–84. https://doi.org/10.1097/QAD.0b013e328363bf7f [PubMed]
- 22. Feng X, Xing J, Feng G, Huang D, Lu X, Liu S, Tan W, Li L, Gu Z. p16(INK4A) mediates age-related changes in mesenchymal stem cells derived from human dental pulp through the DNA damage and stress response. Mech Ageing Dev. 2014; 141-142:46–55. https://doi.org/10.1016/j.mad.2014.09.004 [PubMed]
- 23. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, Gudkov AV. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). 2016; 8:1294–315. https://doi.org/10.18632/aging.100991 [PubMed]
- 24. Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, Kanekiyo T. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016; 47:1068–77. https://doi.org/10.1161/STROKEAHA.115.010835 [PubMed]
- 25. Dong J, Pi HC, Xiong ZY, Liao JL, Hao L, Liu GL, Ren YP, Wang Q, Duan LP, Zheng ZX. Depression and cognitive impairment in peritoneal dialysis: A multicenter cross-sectional Study. Am J Kidney Dis. 2016; 67:111–18. https://doi.org/10.1053/j.ajkd.2015.06.025 [PubMed]
- 26. Carrero JJ, Qureshi AR, Axelsson J, Avesani CM, Suliman ME, Kato S, Bárány P, Snaedal-Jonsdottir S, Alvestrand A, Heimbürger O, Lindholm B, Stenvinkel P. Comparison of nutritional and inflammatory markers in dialysis patients with reduced appetite. Am J Clin Nutr. 2007; 85:695–701. [PubMed]
- 27. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 28. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015; 14:644–58. https://doi.org/10.1111/acel.12344 [PubMed]
- 29. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016; 15:973–77. https://doi.org/10.1111/acel.12458 [PubMed]
- 30. Toth P, Tarantini S, Ashpole NM, Tucsek Z, Milne GL, Valcarcel-Ares NM, Menyhart A, Farkas E, Sonntag WE, Csiszar A, Ungvari Z. IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell. 2015; 14:1034–44. https://doi.org/10.1111/acel.12372 [PubMed]
- 31. Chen YQ, Zhao J, Jin CW, Li YH, Tang MX, Wang ZH, Zhang W, Zhang Y, Li L, Zhong M. Testosterone delays vascular smooth muscle cell senescence and inhibits collagen synthesis via the Gas6/Axl signaling pathway. Age (Dordr). 2016; 38:60. https://doi.org/10.1007/s11357-016-9910-5 [PubMed]
- 32. Yamada S, Tatsumoto N, Tokumoto M, Noguchi H, Ooboshi H, Kitazono T, Tsuruya K. Phosphate binders prevent phosphate-induced cellular senescence of vascular smooth muscle cells and vascular calcification in a modified, adenine-based uremic rat model. Calcif Tissue Int. 2015; 96:347–58. https://doi.org/10.1007/s00223-014-9929-5 [PubMed]
- 33. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015; 21:1424–35. https://doi.org/10.1038/nm.4000 [PubMed]
- 34. Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013; 112:e99–109. https://doi.org/10.1161/CIRCRESAHA.111.300543 [PubMed]
- 35. Willis DM, Loewy AP, Charlton-Kachigian N, Shao JS, Ornitz DM, Towler DA. Regulation of osteocalcin gene expression by a novel Ku antigen transcription factor complex. J Biol Chem. 2002; 277:37280–91. https://doi.org/10.1074/jbc.M206482200 [PubMed]
- 36. Azevedo ML, Bonan NB, Dias G, Brehm F, Steiner TM, Souza WM, Stinghen AE, Barreto FC, Elifio-Esposito S, Pecoits-Filho R, Moreno-Amaral AN. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol Lett. 2016; 263:1–5. https://doi.org/10.1016/j.toxlet.2016.10.006 [PubMed]
- 37. Muramatsu T, Hamano H, Ogami K, Ohta K, Inoue T, Shimono M. Reduction of osteocalcin expression in aged human dental pulp. Int Endod J. 2005; 38:817–21. https://doi.org/10.1111/j.1365-2591.2005.01022.x [PubMed]
- 38. Villa JK, Diaz MA, Pizziolo VR, Martino HS. Effect of vitamin K in bone metabolism and vascular calcification: a review of mechanisms of action and evidences. Crit Rev Food Sci Nutr. 20160. https://doi.org/10.1080/10408398.2016.1211616 [PubMed]
- 39. Idelevich A, Rais Y, Monsonego-Ornan E. Bone Gla protein increases HIF-1alpha-dependent glucose metabolism and induces cartilage and vascular calcification. Arterioscler Thromb Vasc Biol. 2011; 31:e55–71. https://doi.org/10.1161/ATVBAHA.111.230904 [PubMed]
- 40. Krueger T, Westenfeld R, Ketteler M, Schurgers LJ, Floege J. Vitamin K deficiency in CKD patients: a modifiable risk factor for vascular calcification? Kidney Int. 2009; 76:18–22. https://doi.org/10.1038/ki.2009.126 [PubMed]
- 41. Brandenburg VM, Schurgers LJ, Kaesler N, Püsche K, van Gorp RH, Leftheriotis G, Reinartz S, Koos R, Krüger T. Prevention of vasculopathy by vitamin K supplementation: can we turn fiction into fact? Atherosclerosis. 2015; 240:10–16. https://doi.org/10.1016/j.atherosclerosis.2015.02.040 [PubMed]
- 42. McCann JC, Ames BN. Vitamin K, an example of triage theory: is micronutrient inadequacy linked to diseases of aging? Am J Clin Nutr. 2009; 90:889–907. https://doi.org/10.3945/ajcn.2009.27930 [PubMed]
- 43. Sada E, Abe Y, Ohba R, Tachikawa Y, Nagasawa E, Shiratsuchi M, Takayanagi R. Vitamin K2 modulates differentiation and apoptosis of both myeloid and erythroid lineages. Eur J Haematol. 2010; 85:538–48. https://doi.org/10.1111/j.1600-0609.2010.01530.x [PubMed]
- 44. Qureshi AR, Olauson H, Witasp A, Haarhaus M, Brandenburg V, Wernerson A, Lindholm B, Söderberg M, Wennberg L, Nordfors L, Ripsweden J, Barany P, Stenvinkel P. Increased circulating sclerostin levels in end-stage renal disease predict biopsy-verified vascular medial calcification and coronary artery calcification. Kidney Int. 2015; 88:1356–64. https://doi.org/10.1038/ki.2015.194 [PubMed]
- 45. Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M
Jr , Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990; 15:827–32. https://doi.org/10.1016/0735-1097(90)90282-T [PubMed]