



Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and the first cause of dementia in the elderly. Alongside memory deficits, patients with AD also display several other behavioral symptoms, including social disinhibition, apathy, anxiety, agitation, and irritability, which are part of the clinical expression of neuropsychiatric symptoms associated with AD [1–3]. Unfortunately, although it is well known that AD neuropathology extends to the frontal cortex [4,5], the biological substrates of these neuropsychiatric symptoms of AD are still poorly understood. Thus, these symptoms remain very difficult to treat, often increasing an already heavy burden on both patients and caregivers [6].
In recent years, dendritic spine and neuronal dystrophies have been recognized as part of the hallmarks of AD [7–13]. These spine and dendrite abnormalities have a direct impact in synaptic function, intercellular communication and brain function [14,15], and have also been associated with several mental retardation and psychiatric-like disorders [16]. Both neuropsychiatric diseases and AD have been associated with major metabolic and structural changes of the frontal cortex in humans as well as in animal models [17–20]. We have recently demonstrated fronto-dependent social behavior impairments coinciding with abnormal synaptic function in the frontal cortex in 3xTg-AD mice, a murine model of AD-like amyloidosis and tauopathy [21,22]. Similarly, deficits in social behavior have been reported in other transgenic models of AD [23–26]. These observations support the underlying role of synaptic defects in the prefrontal cortex in the neuropsychiatric symptoms observed in AD patients [21,27,28].
Based on these etiological convergences between AD and psychiatric-like disorders, recent studies have probed common biological substrates. An appealing idea is that AD patients may share common traits at the cellular level with patients harboring developmental mental retardation and psychiatric-like disorders [29,30]. Consistent with this idea, previous reports demonstrated that PAK1 and 3, both critical regulators of actin and dendritic spine dynamics [31] known to be implicated in mental retardation [32,33], are down-regulated in AD [9,10,34]. Such decrease in PAK has thus been proposed to play a causal role in the loss of dendritic spines and in cognitive symptoms of AD [10,34,35]. Indeed, genetically induced inactivation of neocortical PAK activity in transgenic mice reduces the number of dendritic spines and yields to long-term memory deficits [34,36].
To further define the causal role of PAK defects in AD-like neuropsychiatric symptomatology, we investigated here the role of PAK in an animal model of AD, focusing on molecular endpoints, dendritic morphology and electrophysiology correlates in the frontal cortex. Firstly, we devised a strategy to repress its activity in vivo. To this end, we intercrossed a dnPAK mouse line, a transgenic model in which the catalytic activity of p21 activated kinase is inhibited [36,37], with the 3xTg-AD model [22]. To evaluate the consequence of PAK inactivation in the frontal cortex of 3xTg-AD mice, we quantified neuropathological markers of AD, downstream signalization of PAK and synaptic proteins. Single cell morphometric analysis was used to investigate dendritic and spine defects in layer II/III of the prelimbic cortex. At the behavior level, we focused on anxiety and social interactions, which are both regulated by the frontal cortex [21,38–41] and known to be impaired in AD patients [42–44] and in 3xTg-AD mice [21,34].
Results
Phosphorylated PAK expression is decreased in the frontal cortex of 3xTg-dnPAK mice
The inactivation of PAK in 18-month-old 3xTg-AD-dnPAK mice was confirmed by the reduction in activated PAK (pPAK phosphorylated at S141) assessed in homogenates from the frontal cortex (Fig. 1). The quantification of pPAK on Western blots revealed a significant decrease in pPAK/PAK ratio in the frontal cortex of 3xTg-AD-dnPAK animals compared to NonTg (Fig. 1B) and heterozygous 3xTg-AD mice (Fig. 1B).
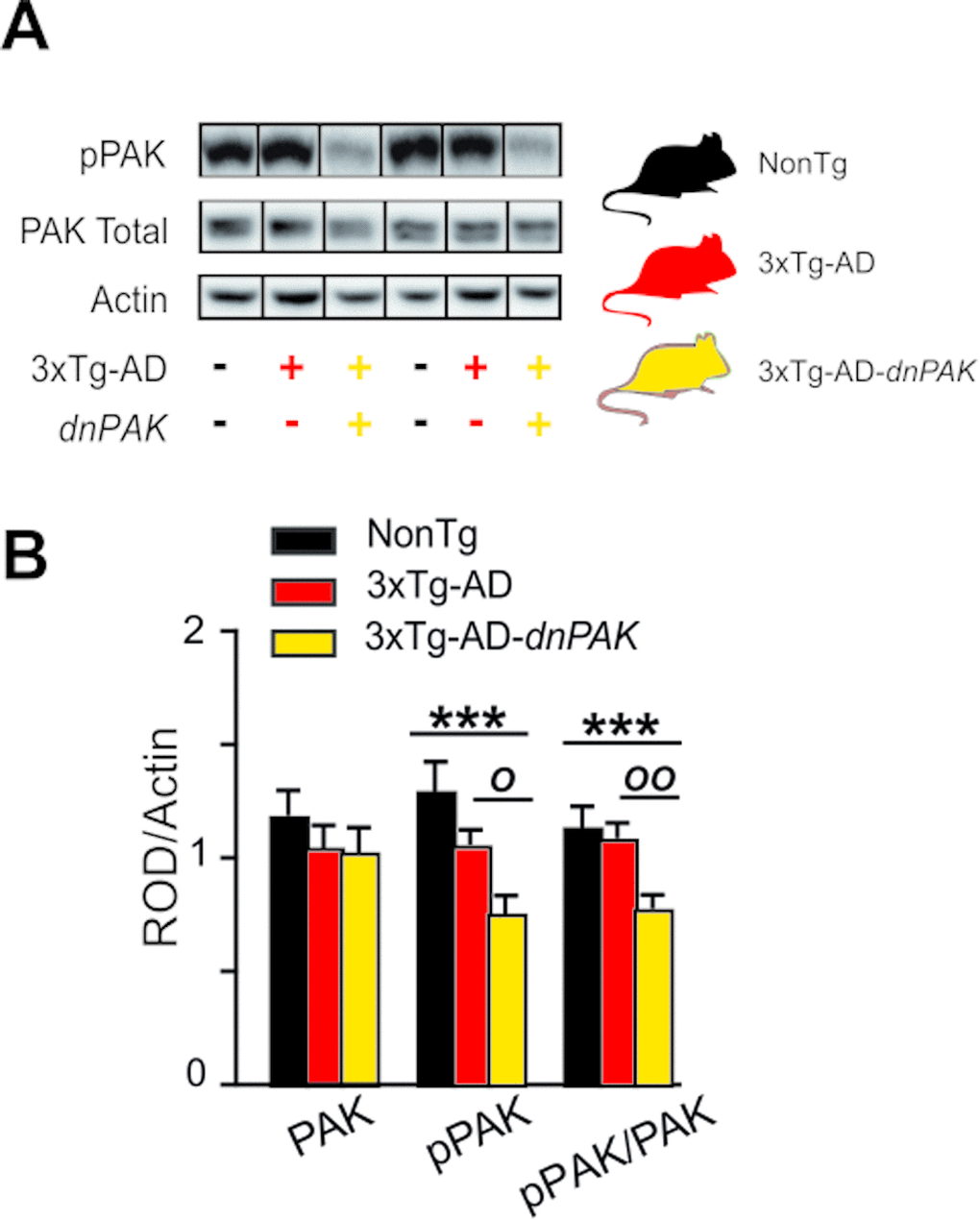
Figure 1. Experimental strategy to generate a mouse model of Alzheimer’s disease with a chronic reduction of PAK activity in the forebrain (A) dnPAK mice were crossed with 3xTg-AD or the corresponding non-transgenic mice (NonTg) yielding three genotypes: NonTg (-/-), 3xTg-AD (+/-) and 3xTg-AD-dnPAK (+/-) (B). Consistently, phosphorylation of PAK was significantly reduced in the frontal cortex of 18-month-old 3xTg-AD-dnPAK animals, confirming the inactivation of PAK. Actin served as an internal control for protein loading (N=12-13 mice for NonTg, N=19-21 mice for 3xTg-AD and N=16-17 mice for 3xTg-AD-dnPAK). Examples of Western blots were taken from the same immunoblot experiment for each primary antibody, on the same gel but run in a random order, and rearranged in the same order as the graphs (separated by black lines). O p<0.05, OO p<0.01, ***p<0.001, Tukey-Kramer post hoc test, one way ANOVA. Abbreviations: PAK: p21 activated kinase, dnPAK: dominant negative p21-activated kinase, pPAK: phospho-PAK; ROD, relative optical density.
The inhibition of PAK activity has no effect on Aβ, but increases tau phosphorylation while preventing the pathological increase of insoluble tau in the frontal cortex
We first addressed the consequences of decreased PAK activity on canonical neuropathological markers of AD. ELISA analyses did not reveal any changes in the concentrations of soluble and insoluble Aβ40 and Aβ42 in the frontal cortex of 3xTg-AD-dnPAK animals when compared with the 3xTg-AD group (Fig. 2A). On the other hand, PAK inactivation increased the proportion of tau phosphorylated at Ser202/Thr205 (CP13) and Ser396/Ser404 (AD2) in detergent-insoluble fraction but had no effect on the phosphorylation ratio at Thr181 (AT270) (Fig. 2B). Intriguingly, this rise in phosphorylated tau ratio was mostly driven by a decrease in detergent-insoluble tau as assessed with an antibody raised against human/rodent tau (tau-5) or human tau (tau-13) (Table 1).
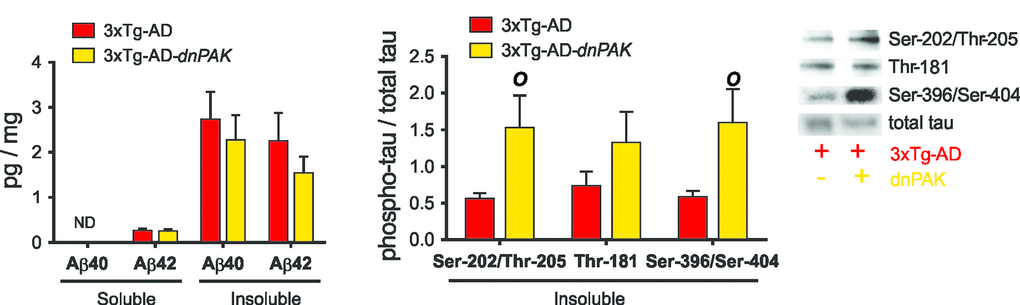
Figure 2. Consequences of the long term inhibition of PAK in 18-month-old 3xTg-AD mice on AD neuropathology markers. (A) PAK inhibition did not affect Aβ accumulation in frontal cortex, suggesting a downstream position for PAK in Aβ pathological cascade. (B) Reduced PAK activity in 3xTg-AD-dnPAK animals led to a higher proportion of tau phosphorylated at Ser202/Thr205 (CP13) and Ser396/Ser404 (AD2) in detergent-insoluble fraction but had no effect on the phospho-epitope Thr181 (AT270). Data expressed as means ± SEM over total human tau (tau-13) (p<0.05, one-way ANOVA, Tukey-Kramer post hoc, O p<0.05, student t-test).
Table 1. Statistical results.
| NonTg | 3xTg-AD | 3xTg-AD/dnPAK | Statistical | U, T or F | p value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mean+/-SEM | N | Mean+/-SEM | N | Mean+/-SEM | N | test | value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molecular quantifications (Fig. 1, 2 and 4C) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TBS-soluble fraction (cytosolic fraction; ROD/actin) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau5 | 1,19+/-0,14 | 13 | 1,02+/-0,07 | 20 | 1,07+/-0,07 | 17 | Welch ANOVA | F(2,25.94)=0.5841 | p=0.5648 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau 13 | ----- | 1,36+/-0,09 | 21 | 1,21+/-0,12 | 17 | Unpaired t-test | T(36)=1.035 | p=0.3075 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau CP13 | ----- | 0,97+/-0,08 | 21 | 1,14+/-0,11 | 17 | Unpaired t-test | T(36)=1.282 | p=0.2079 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| VGAT | 1,35+/-0,1 | 13 | 0,92+/-0,08 | 20** | 0,98+/-0,07 | 17** | One-way ANOVA | F(2,47)=7.486 | p=0.0015 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PAK total | 1,2+/-0,1 | 12 | 1,06+/-0,09 | 21 | 1,04+/-0,1 | 17 | One-way ANOVA | F(2,47)=0.6782 | p=0.5124 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pPAK (s141) | 1,31+/-0,12 | 12 | 1,07+/-0,06 | 20o | 0,77+/-0,07 | 16*** | One-way ANOVA | F(2,45)=10.03 | p=0.0002 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pPAK (s141) / PAK total | 1,15+/-0,08 | 12 | 1,1+/-0,06 | 19oo | 0,79+/-0,05 | 16*** | One-way ANOVA | F(2,44)=9.942 | p=0.0003 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GAD65 | 1,35+/-0,13 | 12 | 0,88+/-0,06 | 20*** | 0,91+/-0,07 | 16** | One-way ANOVA | F(2,45)=9.342 | p=0.0004 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DS-soluble fraction (membranous; ROD/actin) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| synaptophysin | 1,06+/-0,1 | 13 | 1,04+/-0,07 | 21 | 1,01+/-0,05 (17) | 17 | One-way ANOVA | F(2,48)=0.08795 | p=0.9160 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSD95 | 1,12+/-0,34 | 13 | 1,06+/-0,12 | 21 | 0,92+/-0,17 (17) | 17 | Welch ANOVA | F(2,24.817)=0.2841 | p=0.7551 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GABAa alpha | 0,94+/-0,11 | 13 | 1,14+/-0,14 | 21 | 1,05+/-0,15 (17) | 17 | One-way ANOVA | F(2,48)=0.4626 | p=0.6324 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| gephyrin | 1,07+/-0,11 | 13 | 1,01+/-0,05 | 21 | 0,98+/-0,09 (17) | 17 | One-way ANOVA | F(2,48)=0.2993 | p=0.7427 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| VGLUT1 | 1+/-0,08 | 13 | 1,01+/-0,06 | 21 | 1,07+/-0,07 (17) | 17 | One-way ANOVA | F(2,48)=0.2704 | p=0.7643 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Drebrin | 0,97+/-0,19 | 13 | 1,01+/-0,72 | 21 | 1,04+/-0,16 (17) | 17 | One-way ANOVA | F(2,40)=1.3420 | p=0.2728 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cofilin | 1,09+/-0,14 | 9 | 1,05+/-0,08 | 19 | 0,88+/-0,05 (18) | 18 | One-way ANOVA | F(2,43)=1.7186 | p=0.1914 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GluN2B | 0,95+/-0,08 | 13 | 1,04+/-0,08 | 21 | 1+/-0,08 (17) | 17 | One-way ANOVA | F(2,48)=0.3249 | p=0.7242 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Formic acid fraction (insoluble fraction; ROD/mg tissu) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau CP13/tau 5 | ----- | 0,5515+/-0,0845 | 14 | 1,52+/-0,4489 | 12 | Mann Whitney test | U(136,215) = 31.00 | p=0.0069 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AT270/tau5 | ----- | 0,7286+/-0,204 | 14 | 1,32+/-0,4262 | 12 | Mann Whitney test | U(168,183) = 63.00 | p=0.2917 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AD2/tau5 | ----- | 0,5782+/-0,089 | 14 | 1,59+/-0,467 | 10 | Mann Whitney test | U(139,161)=34.00 | p=0.0377 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau 5 | 0,86+/-0,17 | 10 | 1,34+/-0,12 | 16*o | 0,81+/-0,16 | 12# | One-way ANOVA | F(2,31)=3.568 | p=0.0403 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau13 | ----- | 1,23+/-0,16 | 16*o | 0,77+/-0,13 | 15# | Unpaired t-test | T(29)=2.628 | p=0.0136 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau CP13/tau 13 | ----- | 0,96+/-0,28 | 15 | 1,04+/-0,25 | 12 | Unpaired t-test | T(25)=0.2264 | p=0.8227 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abeta pathology (pg/mg tissu) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Soluble-Abeta40 | ----- | ND () | ND () | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Soluble-Abeta42 | ----- | 0,25+/-0,05 | 17 | 0,24+/-0,06 | 10 | Unpaired t-test | T(25)=0.1252 | p=0.9014 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Insoluble-Abeta40 | ----- | 2,72+/-0,62 | 17 | 2,26+/-0,57 | 10 | Unpaired t-test | T(25)=0.4989 | p=0.6222 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Insoluble-Abeta42 | ----- | 2,24+/-0,64 | 17 | 1,53+/-0,38 | 10 | Welch t-test | T(23)=0.9539 | p=0.3501 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molecular markers (ROD/g proteins) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TBS-soluble actin | 0,89+/-0,07 | 13 | 1,03+/-0,07 | 21 | 1,04+/-0,07 | 17 | One-way ANOVA | F(2,48)=1.203 | p=0.3093 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DS-soluble actin | 0,94+/-0,07 | 13 | 1+/-0,05 | 21 | 1,05+/-0,06 | 17 | One-way ANOVA | F(2,48)=0.7679 | p=0.4696 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Morphological quantifications (Fig. 3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spine density (10-60µm) | 1,3+/-0,37 | 9 | 1,43+/-0,2 | 15 | 0,7+/-0,2 | 11 | One-way ANOVA | F(2,32)=1,98 | p=0,153 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spine density (60-110µm) | 1,46+/-0,27 | 9 | 2,22+/-0,38 | 15 | 0,9+/-0,16 | 10## | One-way ANOVA | F(2,31)= 4,28 | p=0,022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spine density (110-160µm) | 1,05+/-0,2 | 8 | 2+/-0,4 | 13 | 0,4+/-0,1 | 8## | One-way ANOVA | F(2,26)= 5,64 | p=0,0092 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spine length | 1,97+/-0,024 | 892 | 2,04+/-0,01 | 2003 | 2,12+/-0,03 | 774***# | One-way ANOVA | F(2,3666)=8,1 | p=0,0003 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sholl Analysis (length-Basal dendrite) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10-29 | 50,54+/-7,55 | 9 | 48,68+/-5,78 | 15 | 44,96+/-5,58 | 11 | One-way ANOVA | F(2,33)=0.18 | p=0.83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 30-49 | 115,65+/-13,01 | 9 | 131+/-15,56 | 15 | 112,72+/-9,56 | 11 | One-way ANOVA | F(2,33)=0.54 | p=0.58 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 50-69 | 113,09+/-13,59 | 9 | 128,67+/-17,93 | 15 | 95,85+/-12,39 | 11 | One-way ANOVA | F(2.33)=1.12 | p=0.33 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 70-89 | 106,37+/-14,6 | 8 | 113,94+/-9,78 | 15 | 70+/-9,68 | 11# | One-way ANOVA | F(2,32)=1,98 | p=0.048 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 90-109 | 81,78+/-7,19 | 8 | 84,37+/-9,16 | 14 | 53,14+/-27,48 | 10*# | One-way ANOVA | F(2,30)=3.67 | p=0.037 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 110-129 | 49,91+/-7,04 | 8 | 53,63+/-7,96 | 14 | 42,27+/-7,94 | 10 | One-way ANOVA | F(2,30)=0.55 | p=0.583 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 130-149 | 22,43+/-4,05 | 7 | 22,56+/-4,34 | 11 | 20,72+/-6,53 | 8 | One-way ANOVA | F(2,24)=0.04 | p=0.96 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 150-169 | 10,8+/-2,83 | 4 | 11,3+/-10,12 | 7 | 16,16+/-7,19 | 4 | One-way ANOVA | F(2,13)=0.67 | p=0.52 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 170-189 | 8,55+/-1,5 | 2 | 10,09+/-1,61 | 5 | 8,73+/-2,03 | 2 | NA | NA | NA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 190+ | 8,3+/-NA | 1 | 6,07+/-1,3 | 3 | 7,8+/-NA ( | 1 | NA | NA | NA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Excitatory postsynaptic current (Fig. 4B) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mean frequency (Hz) | 1+/-0,39 | 6 | 1,44+/-0,39 | 5 | 0,58+/-0,18 | 6 | One-way ANOVA | F(2,15)=1,4 | p=0,27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Amplitude (pA) | 4,44+/-0,37 | 6 | 7,89+/-1,6 | 5 **oo | 4,1+/-0,58 | 6## | One-way ANOVA | F(2,15)=4.91 | p=0,024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Behavior quantifications (Fig. 5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exploration (D1) | 29,8+/-2,6 | 13 | 28,7667+/-1,8 | 25 | 25,7+/-2,3576 | 30 | One-way ANOVA | F(2,65)=0.8572 | p=0.4291 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exploration (D2) | 22,7+/-3,2 | 13 | 15,6667+/-1 | 25 | 15,9+/-2 | 30 | Welch ANOVA | F(2,26.158)=2.1611 | p=0.1353 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exploration (D3) | 18,8+/-1,9 | 13 | 12,9+/-1.0 | 25* | 13,2+/-1,6 | 30* | One-way ANOVA | F(2,65)=3.9528 | p=0.0240 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stand up duration | 1,54+/-0,04 | 16 | 1,57+/-0,06 | 24 | 1,56+/-0,07 | 21 | One-way ANOVA | F(2,58)=0.0774 | p=0.9256 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Latency | 14+/-2,3 | 16 | 24,3+/-3,9 | 26 | 28,2+/-3,6 | 20* | One-way ANOVA | F(2,59)=3.4962 | p=0.0367 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Social interaction | ----- | 166,7+/-16 | 15o | 117+/-9 | 15# | Welch t-test | F(22)=2.674 | p=0.0139 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| #p<0.05, ##p<0.01, ###p<0.001 (relative to 3xTg-AD) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| o P<0.05, oo P<0.01 (relative to 3xTg-AD/dnPAK) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ROD: relative optical density | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ND: not detected | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Pyramidal cell morphology is altered in 3xTg-AD mice with reduced PAK activity
Previous studies report neuronal and spine abnormalities in patients with AD [7–12,45–48] and in the frontal cortex of 18-month-old hAPP mice, a transgenic mouse model of AD [46]. Structural changes in dendritic spines are also observed in dnPAK mice [36,37]. Therefore, we investigated for morphological changes of layer II/III pyramidal cells in the medial prefrontal cortex in our transgenic model (Fig. 3A). Sholl analyses revealed a reduction of basal dendrite arborization in 3xTg-AD-dnPAK mice. This effect was mainly observed at a radial distance between 70 and 110 µm (middle range) (Fig. 3B). Moreover, in middle (60-110-µm radius) and distal (110-160-µm radius) dendritic segments, spine density was significantly lower in 3xTg-AD-dnPAK neurons but not in more proximal (10-60-µm radius) dendritic segments (Fig. 3C), compared to 3xTg-AD mice. Finally, our analysis of spine length, calculated as the radial distance from the tip of the spine head to the dendritic shaft [36] revealed that spines from 3xTg-AD neurons were significantly longer when compared with NonTg spines. In addition, lengthy spines in 3xTg-AD animals were further elongated by in vivo inhibition of PAK activity (Fig. 3D).
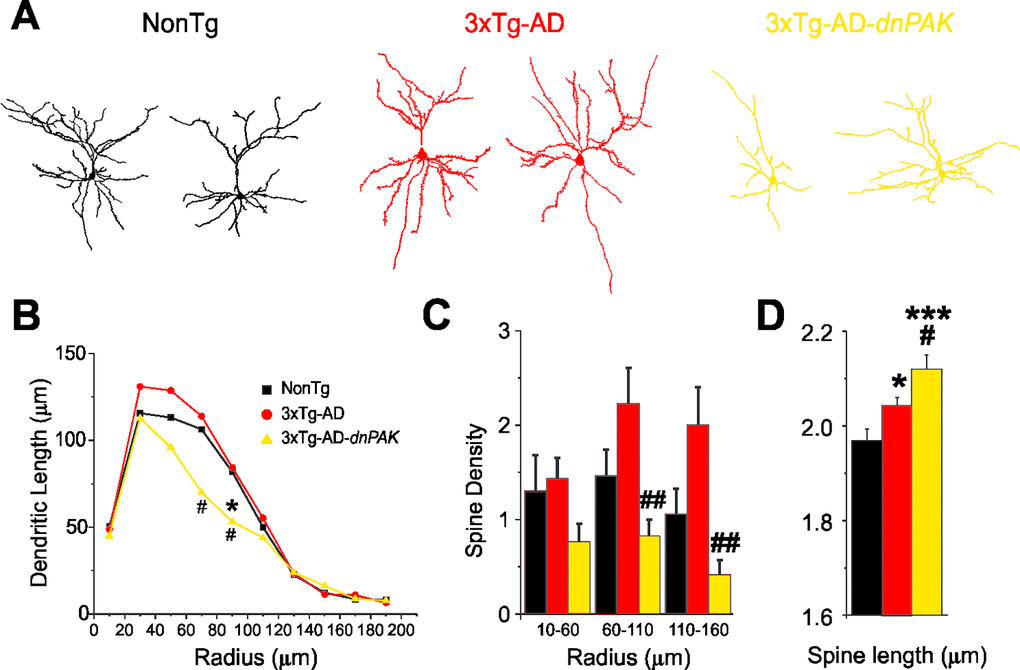
Figure 3. Abnormal dendritic and spine morphologies in 3xTg-AD-dnPAK prefrontal cortex. (A) Examples of reconstructed layer II/III prefrontal pyramidal neurons from NonTg, 3xTg-AD and 3xTg-AD-dnPAK animals. (B) Sholl analysis of the dendritic length revealed a significant reduction in dendritic arborization 3xTg-AD-dnPAK pyramidal cells. (C) Spine density in dendrites of layer II/III pyramidal neurons of the prefrontal cortex was significantly lower in 3xTg-AD-dnPAK animals. The decrease in spine density was more pronounced in intermediate (60-100 µm from cell body) and distal dendrite segments (110-160 µm), but not significantly different in proximal (10-60 µm) segments. (D) Inhibition of PAK activity potentiated the lengthening of spines observed in 3xTg-AD cells when compared with NonTg. # p<0.05 when compared to 3xTg-AD cells, # p<0.05, ## p<0.001 when compared to NonTg cells, one-way ANOVA, Fisher LSD post hoc test. Abbreviations: ROD, relative optical density.
PAK inhibition alters synaptic properties
To assess the functional consequences of the chronic inhibition of PAK activity, we performed patch clamp recordings of the synaptic activity impinging on layer II/III pyramidal neurons in the medial prefrontal cortex. Glutamate receptor-mediated mEPSC activity was recorded at a holding potential of −60 mV. The mean frequency recorded was not significantly different between groups. Our analysis revealed higher mEPSC amplitude in 3xTg-AD mice, a change not observed in 3xTg-AD-dnPAK animals compared to NonTg controls (Fig. 4A and B).
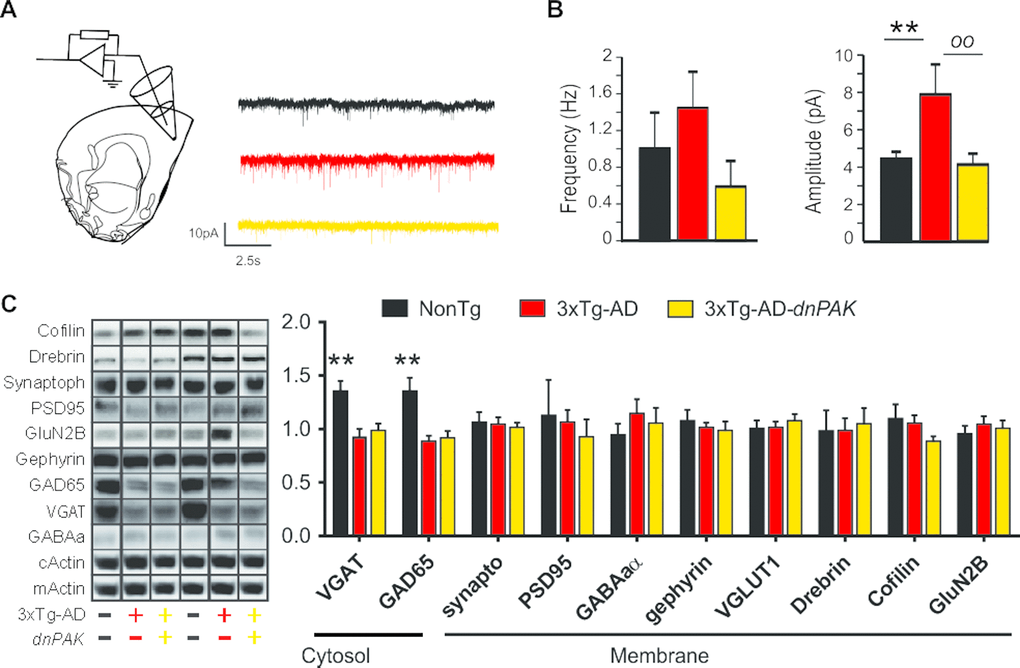
Figure 4. Reducing PAK activity counteracts the enhancement of the glutamatergic synaptic tone observed in 3xTg-AD mice. (A) Experimental design for patch clamp recordings of layer II/III pyramidal cells of the medial prefrontal cortex. (B) While the mean frequency of mEPSCs was comparable between the three genotypes, the mean amplitude of mEPSCs was larger in 3xTg-AD mice. This phenomenon was not present in 3xTg-AD-dnPAK mice. (n=5 to 6 cells per group), **p<0.01, oop<0.01, Fisher LSD post hoc test (C and D). A significant reduction in GAD65 and VGAT expression was observed in both 3xTg-AD and 3xTg-AD-dnPAK animals when compared with NonTg animals (N=12-13 mice for NonTg, N=19-21 mice for 3xTg-AD and N=16-17 mice for 3xTg-AD-dnPAK). Examples of Western blots were taken from the same immunoblot experiment for each primary antibody, on the same gel but run in a random order, and rearranged in the same order as the graphs (separated by black lines). **p<0.01, one way ANOVA Tukey-Kramer post hoc test.
Next, we investigated whether synaptic changes at the functional and structural levels were echoed at the molecular level. We did not observe any significant changes in the expression of drebrin or cofilin (Fig. 4C), two proteins regulated by PAK and known to be altered in AD [10,34,49]. However, we observed a significant reduction in the expression of GABA-related presynaptic proteins, GAD65 and VGAT, in both 3xTg-AD and 3xTg-AD-dnPAK groups when compared with NonTg mice, whereas specific markers of glutamatergic synapses (PSD95, GluN2B and Synaptophysin) and postsynaptic markers of GABAergic synapses (Gephyrin, GABAA receptor) remained unchanged (Fig. 4C). In sum, levels of postsynaptic or presynaptic proteins were not associated with functional impairments of excitatory synapses whereas the expression of AD transgenes led to a reduction of GABAergic presynaptic proteins in the frontal cortex.
PAK inhibition induces anxiety-like behavior and impairs social interaction activity
To test whether the behavioral performances of the animals were affected by the chronic autoinhibition of PAK activity, a series of evaluations were conducted. First, exploratory behavior was documented using a hole-board task for three consecutive days. Both 3xTg-AD and 3xTg-AD-dnPAK animals displayed reduced level of activity on the third day compared to NonTg animals (Fig. 5A) revealing a decreased motivation to explore their new environment. These observations were independent of changes of basic motor function, as shown by the lack of difference of mean stand-up durations between the different groups of animals (Fig. 5B). Second, we tested whether 3xTg-AD-dnPAK mice were more anxious. To this end, we exposed the mice to the light/dark box test, and we observed that 3xTg-AD-dnPAK mice displayed a higher latency to go out from the dark compartment when compared with NonTg animals (Fig. 5C). This finding suggests that PAK inhibition exacerbated anxious behavior in 3xTg-AD mice. Finally, we tested for the specific impact of PAK inhibition onto social interaction activity, known to be disrupted in 3xTg-AD mice [21,34]. When compared with 3xTg-AD animals, 3xTg-AD-dnPAK mice displayed a significant reduction in their social interaction activity (Fig. 5D), suggesting defects in fronto-dependent social behaviors caused by the inactivation of PAK.
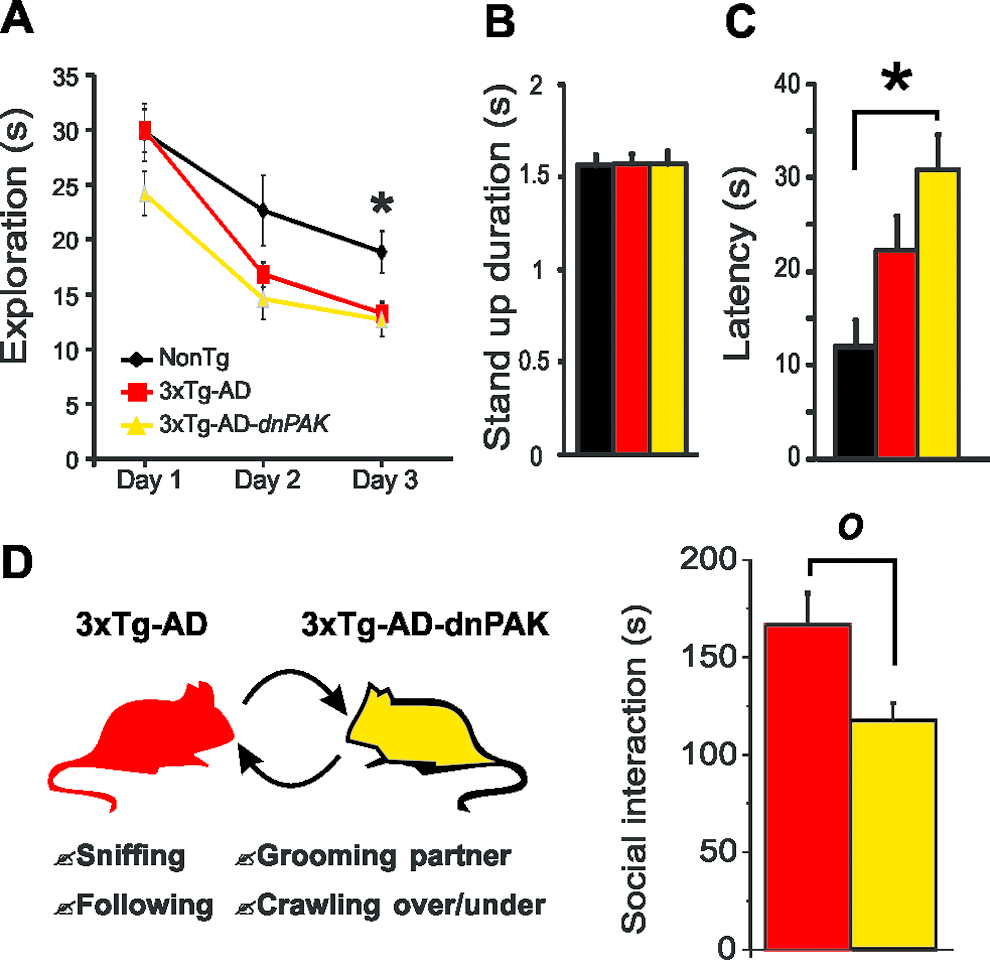
Figure 5. PAK inhibition aggravates the behavioral alterations observed in 3xTg-AD mice. (A) Both 3xTg-AD and 3xTg-AD-dnPAK displayed a reduced exploratory activity in a hole-board task. *p<0.05 when compared to 3xTg-AD and 3xTg-AD-dnPAK animals. (B) No significant difference in mean stand-up duration. (C) 3xTg-AD-dnPAK mice displayed increased anxiety in dark and light box testing as shown by the greater latency time to go out from the dark box. *p<0.05(D) Schematic representation of social interaction paradigm used to assess behavioral performance of 3xTg-AD and 3xTg-AD-dnPAK animals. Mice with genetically reduced PAK activity interacted less than 3xTg-AD animals during a social interaction test. O p<0.05, n=8 animals per group.
Discussion
Besides Aβ deposition and tangle formation, intracellular signaling pathways involved in the pathogenesis of AD are subjects of intense scrutiny as potential drug targets. More particularly, cell pathways regulating dendritic and synaptic organization attract considerable attention. As such, the PAK1-3-related cascades are of particular interest due to their potential causal role in cognitive deficits in AD, but also in mental retardation [10,29,45,50–52]. To further evaluate the role of PAK in AD, we generated a 3xTg-AD mouse with a constitutive reduction of PAK activity. Using this experimental approach, we investigated the consequences of PAK inactivation in the frontal cortex of 3xTg-AD mice at the molecular, synaptic and cellular levels, as well as on fronto-dependent behaviors reminiscent of AD neuropsychiatric symptoms.
PAK inactivation did not promote total Aβ or tau aggregation, but increased the phosphorylation ratio of insoluble tau
The 3xTg-AD mouse provides the opportunity to investigate the effect of PAK deficit on neuropathological hallmarks of AD, Aβ and tau pathologies. Firstly, the present results are consistent with a downstream position for PAK in the β amyloid cascade. Indeed, PAK inactivation had no effect on Aβ levels. In contrast, previous report emphasized a downregulation of PAK after exposure to Aβ in vitro or in the brain cortex or hippocampus of transgenic models of AD [10,34,35]. Considering the effect of PAK on tau neuropathology, our results rather suggest an upstream position for PAK1-3 in the molecular pathway controlling the phosphorylation and the accumulation of insoluble tau in 3xTg-AD-dnPAK animals. Indeed, PAK inactivation enhanced the proportion of tau phosphorylated at Ser202/Thr205 (CP13) and Ser396/Ser404 (AD2), but not at the Thr181 (AT270) phospho-epitope. Interestingly, deficits in prepulse inhibition of acoustic startle are driven by tau phosphorylation at Ser396/404 and Ser202 in insoluble protein fractions of female rTg(tauP301L)4510 mice [53]. In addition, tau phosphorylation at Thr205 may regulate the trans-synaptic transport of tau, a mechanism suspected to spread tau pathology in the brain [54]. In keeping with our present observations, elevated Ser396 phospho-tau in the frontal cortex has been found to be associated with neuropsychiatric behaviors in AD patients [55]. Since hyperphosphorylation of insoluble tau at Ser396/Ser404 is a major correlate of impaired cognition in AD [56], increased PAK activity could provide a means to improve tau phosphorylation status and AD symptoms.
Inactivation of PAK worsens dendrite atrophy and loss of spine in 3xTg-AD mice, two neurophysiological markers of AD
Our single-cell labeling analysis revealed an attrition of the basal dendritic arbor and a reduction in spine density in 3xTg-AD-dnPAK cells, whereas no major differences in these parameters were observed between hemizygous 3xTg-AD and NonTg neurons. Since a reduction of spine density was reported in the frontal cortex layer III neurons of homozygous 3xTg-AD [57], the expression degree of tau and Aß is likely to be an important factor to induce loss of synaptic spine. Our results also suggest that a repression of PAK activity precipitates the atrophy of the dendritic tree, as observed in late stage of AD [58–61]. The lack of dendritic morphological changes in 3xTg-AD mice may result from a transient increase in synaptic density or dendritic lengthening as observed in the neocortex during presymptomatic and early stages of AD [7,51,58,62–64] which may also explain the variability observed in our data. It is also known that an increase in synapse density is thought to precede morphological alterations associated with Aβ and tau-immunoreactive neuritic plaques [7,8,57,62–66]. Therefore, our results suggest that 3xTg-AD animals model an early stage of the disease [67], in which inhibiting PAK activity would accelerate synaptic decline, spine loss and dendritic atrophy, as generally observed in advanced phases of AD [8,10,29,45,49,68,69].
Spine morphometric abnormalities are aggravated by PAK inactivation in 3xTg-AD mice
We observed a lengthening of spines in 3xTg-AD-dnPAK cells, an exacerbation of a phenotype already present to a lesser degree in 3xTg-AD cells. Thinner and longer spines are generally associated with an immature state, a phenomenon suspected to underlie several neurological and psychiatric disorders [16,36,37]. Such changes in spine morphology have also been described in AD [29,70]. In the brain of rodents or in vitro, exposure to Aβ oligomers leads to a reduction in stubby and mushroom spines found in healthy neurons and is associated with the transient formation of aberrant spines with filopodial or large, branched morphologies reminiscent of structures observed in mental retardation [11,12,71]. Nevertheless, our data contrast with previous description of shorter and larger spines in the cortex of dnPAK transgenic animals when compared with non-transgenics [36,37]. This further suggests that the function of PAK might be profoundly modulated by Aβ/tau pathology in the 3xTg-AD mice, resulting in a switch in the function of PAK. The translocalization of PAK from cytosol to membrane in Tg2576 transgenic mice supports this hypothetical functional switch in an AD-like neuroenvironment [9]. Although interactions between neuropathological markers of AD and PAK function remain poorly understood, our results suggest a key role for PAK in spine morphometric abnormalities observed in AD.
PAK inactivation prevents functional impairments of glutamatergic synapses: a consequence of the effects of PAK on aggregated tau, spine density and dendrite atrophy?
An important question to investigate was whether these morphological alterations translated into functional changes at the synaptic level. Our electrophysiological recordings revealed that the synaptic activity mediated by AMPA receptors was significantly enhanced in 3xTg-AD compared to NonTg cells. The finding of a greater glutamatergic signaling in 3xTg-AD cells is consistent with the postulated redistribution of abnormally hyperphosphorylated tau to the somatodendritic compartment during AD pathogenesis, boosting excitotoxic signaling and increasing the toxic effects of Aβ [71]. In addition, the lower mEPSC amplitude in 3xTg-AD-dnPAK neurons (compared to 3xTg-AD) is in accordance with the reduction of spine density and the altered dendritic arborization in these neurons, suggesting a decrease in the number of synaptic contacts. Taken together, these results further indicate that 3xTg-AD neurons display neurophysiological defects (upregulation of glutamatergic function) seen early in the disease [62,63,67], while the combination with chronic PAK underactivity generates an animal model with synaptic features closer to advanced AD (dendrite atrophy, loss of spine density, more pronounced spine elongation and excitatory synaptic function) [72–78].
Surprisingly, the effect of 3xTg-AD transgenes expression on EPSC in frontal cortex differed with previous observations in the entorhinal cortex [34], in which a higher frequency of EPSC was recorded without any change in the mean amplitude. This contrast could be explained by a difference in the nature of the detected activity. Indeed, the previously measured synaptic activity in the entorhinal cortex was dependent of action potential (without TTX) [34] whereas here we detected pure synaptic activities (with TTX, independent of action potential) in the frontal cortex. The synaptic activity recorded in entorhinal cortex is more representative of basal brain activity since it includes activity related to neuronal activity (dependent of action potential) whereas the pure synaptic activity investigated in this study is a better parameter to focus only in synaptic function. In other words, both approaches dependent of different factors and do not provide the same information. Consequently, both results cannot be directly compared.
Transgene expression reduced presynaptic markers of GABAergic synapses but did not influence the level of drebrin/cofilin
In the last decade, studies reported neurological deregulations potentially involved in the pathophysiology or symptomatology of AD. The GABA system is one of them. The downregulation of key proteins related to the GABAergic system in the frontal, temporal and parietal cortices of AD patients is consistent with the view that GABAergic function plays a role in AD [79,80]. In addition, the memory/learning deficits in mice model of AD correlated with astrocytic GABA dysfunction and pharmacological correction of this alteration improves memory capability [81]. In agreement with this GABAergic hypothesis, we found lower levels of two GABAergic presynaptic markers (GAD65 and VGAT) in the frontal cortex of 3xTg-AD mice. Inactivation of PAK activity in transgenic mice did not influence these two molecular impairments, suggesting that PAK is not involved in GABAergic changes in frontal cortex of 3xTg-AD mice. In parallel, it has been proposed that PAK dysfunction leads to cognitive impairment through drebrin displacement and development of cofilin-positive Hirano bodies, both disrupting the actin scaffold [9,10,34]. This hypothesis stems from repeated demonstration of a drebrin loss in the brain of AD patients [7,49,82]. Our quantification did not reveal significant changes in the levels of drebrin and cofilin in the frontal cortex with transgene expression, suggesting that the effect of PAK might pertain more to activity, subcellular localization or protein interactions of drebrin and cofilin, rather than their total concentrations, at least in the frontal cortex.
Conclusion
Overall, our results indicate that repressing PAK activation aggravates anxious behavior and impairs social behavior in the 3xTg-AD mouse. These behavioral abnormalities were not coupled to changes in Aβ or excitatory synaptic markers in the frontal cortex. On the other hand, PAK inactivation induced dendritic reduction, aggravated the AD-related spine elongation and increased the relative phosphorylation of insoluble tau at Ser202/Thr205 and Ser396/Ser404 in the frontal cortex, which may be involved in the behavioral impairments observed. As such, PAK inactivation in the 3xTg-AD mouse generated or exacerbated de novo key AD-like signs and symptoms, i.e. dendritic spine defects, anxiety and social interaction deficits. Therefore, preventing PAK inactivation in the frontal cortex may prove to be a valid approach to treat neuropsychiatric-like symptoms of AD.
Materials and Methods
Animals
Animals were produced and maintained in the animal facilities of the CHUL Research Center at 22 ± 1°C under a 12-h light/dark cycle regime. Water and food were available ad libitum. All experiments were approved by the Laval University Animal Care and Use Committee in accordance with the standards of the Canadian Council on Animal Care.
Transgenic mice
Original breeders couples of homozygous 3xTg-AD mice, non-transgenic littermates and hemizygous dnPAK transgenic mice were kindly provided by Dr. Laferla [22,94] and Dr. Tonegawa [36], respectively, to start our own colonies. The 3xTg-AD mouse line was produced from the cointegration of the APP and tau transgenes in the same genetic locus, into single-cell embryos from homozygous PS1-knockin mice, generating mice with the same genetic background. Non-transgenic (NonTg) mice used here are littermates from the original PS1-knockin mice and are on the same background as 3xTg-AD mice (C57BL6/129SvJ) [22]. In the dnPAK mouse line, the mouse Camk2a promoter directs postnatal forebrain expression of the mouse dominant negative autoinhibitory domain (AID) of the p21 protein (Cdc42/Rac)-activated kinase (Pak) gene. The dnPAK transgene was purified and microinjected into C57BL6 zygotes to generate transgenic mice, as described [36,37]. The dnPAK line was intercrossed in our facilities with 3xTg-AD and their corresponding NonTg control animals. Three groups were generated: a non-transgenic group (NonTg), a hemizygous triple transgenic model of AD group (3xTg-AD) and, finally, a hemizygous triple transgenic mouse model of AD with reduced PAK activity (3xTg-AD-dnPAK) (Fig. 1A), all on the same genetic background [34]. Over one hundred mice were produced and the 3 groups were constituted according to genotype. All experiments were performed on animals between 18 and 20 months of age (18.1 ± 1 months, mean ± S.D), with an equivalent number of males (n=51) and females (n=59) per group.
ELISA and Western immunoblotting
To collect molecular endpoints, animals were perfused with 10 mM phosphate buffered saline (PBS) containing a cocktail of protease inhibitors (SIGMAFAST™, Sigma–Aldrich, St. Louis, MO) along with phosphatase inhibitors (50 mM sodium fluoride and 1 mM sodium pyrophosphate). Frozen extracts of the frontal cortex were dissected and kept at –80°C. Homogenates from cytosol (TBS-soluble), membrane (detergent-soluble) and detergent-insoluble (formic acid–soluble) fractions were generated for ELISAs and Western immunoblotting analyses as described (Arsenault et al., 2011). Insoluble and soluble Aβ40 and Aβ42 were measured using Human β-Amyloid (1-42) and (1-40) ELISA kit High Sensitive (WAKO, Osaka, Japan) as described [56,95]. Protein concentrations in samples were determined using bicinchoninic acid assays (Pierce, Rockford, IL) and equal amounts of protein per sample (15 µg of total protein per lane) were added to Laemmli’s loading buffer, heated to 95°C for 5 min before loading, and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were electroblotted onto PVDF membranes (Millipore, MA) before blocking in 5% nonfat dry milk and 1% bovine serum albumin (BSA) in PBS containing 0.1% of tween-20 for 1 h. Membranes were immunoblotted with appropriate primary and secondary antibodies followed by chemiluminescence reagents (KPL, Gaithersburg, MD). Band intensities were quantified using a KODAK Image Station 4000 Digital Imaging System (Molecular Imaging Software version, KODAK, New Haven, CT).
The following primary antibodies were used in Western immunoblotting experiments: rabbit anti-vesicular GABA transporter (VGAT; Novus Biologicals, #NB110-55238), mouse anti-PSD-95 (NeuroMab, #75-028), mouse anti-synaptophysin (Millipore, #MAB332), mouse anti-actin (ABM, #Y061021), mouse anti-tau 5 (Millipore, clone tau 5, #MAB361), mouse anti-tau (Covance, clone tau-13, #MMS-520R-500), mouse anti-tau CP13 (phosphorylated at Ser202/Thr205, 1:1000, gift from Dr Peter Davies, Albert Einstein College of Medicine, New York, NY), rabbit anti-cofilin (Cell signaling technology, #3312L), mouse anti-drebrin (Progen Biotechnik GmbH, #GP254), rabbit anti-total PAK1/2/3 (Cell signaling technology, #4750S), rabbit anti-phospho PAK1/2/3: pS141 (BioSource, #44940G), rabbit anti-gephyrin (Abcam, #ab25784), rabbit anti-GAD65 (Millipore, #ABN101), mouse anti GABAA receptor subunit 1 (Neuromab, 1:250, #75-136) and mouse anti-GluN2B (Covance, clone n59/36, #MMS-5148-100).
Morphometric analyses
Freshly sliced and fixed 250-µm-thick slices taken from 4% PFA were used to inject neurons with Lucifer Yellow (LY) (Lucifer Yellow-Lithium Salt, Invitrogen, L12926) as described previously [96]. Briefly, pyramidal neurons in layer II/III of the prelimbic area of the frontal cortex were impaled with a micropipette containing 1% LY in phosphate buffer (PB) 0.2 M and injected at 0.5-2 nA for approximately 5-10 min to fill the dendritic tree until spines on apical tuft dendrites became clearly visible. For revelation, slices were rinsed in PB 0.1 M three times for 10 min and incubated overnight at 4°C in a solution of PB 0.1 M, sucrose 5% and Triton 0.1%. On the second day three washes of 10 min in PB 0.1 M and 0.1% Triton were performed before incubating in a quenching solution (PB 0.1 M, 0.1% Triton, 0.3% H2O2) 2 h at room temperature (RT°). Slices were washed three times during 10 min in PB 0.1 M and 0.1% Triton. After 2 h at RT° in a preblocking solution (PB 0.1 M, 0.1% Triton, 10% normal goat serum, NGS), slices were incubated overnight at 4°C with a rabbit anti-LY primary antibody (ThermoFisher) # A-5750) in PB 0.1 M, 0.1% Triton, 5% NGS. On the third day, slices were incubated in a goat anti-rabbit biotinylated secondary antibody (Vector Laboratories, BA-1000) for 2 h at RT°, in PB 0.1 M, after three washes of 10 min each in PB 0.1 M. Slices were finally rinsed with three other 10-min washes in PB 0.1 M. The reaction solution was made from an ABC kit (Vectastain by Vector Laboratories, PK-6100). Sections were washed again three times for 10 min in PB 0.1 M. A 3,3´-diaminodbenzidine (DAB, Sigma-Aldrich #D5905)-Nickel 0.025% solution was filtered and applied to slices for 15 min. Then, 0.006% H2O2 was added to the well and slices were removed from the solution few seconds after and washed several times in PB 0.1 M to stop the reaction. We thus obtained long-term photostable labeling of dendritic spines and fine neuronal processes. Neurons were reconstructed and spines marked using an Olympus microscope equipped with a 63x objective (1.3 NA), a motorized stage, video camera system, and Neurolucida morphometry software (MBF Bioscience) by an experimenter blind to the experimental conditions. Only clear spines with visible neck and head were marked. Dendritic length and Sholl analysis were obtained for each neuron with NeuroExplorer software (MBF Bioscience). Only neurons with a clear pyramidal shape were kept for further analyses. Additionally, analyses and results presented were restricted to basal dendrites, because several pyramidal cells exhibited various shapes of apical dendrites (single, bitufted) displaying unclear terminal points (real vs. cut during slicing).
Electrophysiology
Subjects
None of the animals used in this study presented any evidence of tumors. A total of 27 mice were used for electrophysiological recordings. 3xTg-AD mice and NonTg were dispatched into three groups according to genotype.
Slice preparation and electrophysiology
Mice were deeply anaesthetized with ketamine and xylazine and then decapitated. The brain was removed quickly and placed in ice-cold solution containing (in mM) 210 sucrose, 3.0 KCl, 0.75 CaCl2, 3.0 MgSO4, 1.0 NaH2PO4, 26 NaHCO3, and 10 glucose, saturated with 95% O2 and 5% CO2. Coronal slices of frontal cortex including the prelimbic area were 250 µm thick and kept in artificial cerebral spinal fluid (ACSF) containing (in mM) 124 NaCl, 3.0 KCl, 1.5 CaCl2, 1.3 MgSO4, 1.0 NaH2PO4, 26 NaHCO3, and 20 glucose, gassed with 95% O2-5% CO2 at RT°. Slices were allowed to recover for at least 1 h before recording. A slice was then transferred to a chamber exposed to ACSF flowing at a rate of 2-3 ml/min. Recordings were performed between 32°C-34°C.
Whole-cell patch clamp recording
Patch pipettes (6-8 MΩ) were pulled from borosilicate glass capillaries (World Precision Instruments) and filled with an intracellular solution (pH 7.2; 275–280 mOsm) composed of (in mM): 100 Cs methyl sulfonate, 5 CsCl, 10 HEPES, 2 MgCl2, 1 CaCl2, 11 BAPTA, 4 ATP, 0.4 GTP, and 0.1% Neurobiotin Tracer (Vector Laboratories, SP-1120), 0.1% Lucifer Yellow. The junction potential of the pipette was corrected by subtracting 12 mV from recorded membrane voltages. A Multiclamp 700B amplifier (Axon Instruments) was used for the recording. The access resistance was monitored throughout each experiment and only recordings with stable access were used. Experiments were conducted using the Clampex program (pClamp 9.2, Axon Instruments), data collected were 3-kHz filtered and digitized at 10 kHz. The extracellular concentration of potassium was raised to 5 mM to increase the frequency of postsynaptic currents (PSCs) [96]. Tetrodotoxin (TTX; 1 μM, Alomone Labs, T-500) was added to the ACSF to block voltage-gated sodium channels and isolate action potential-independent miniature postsynaptic currents (mPSCs). Whole-cell patch clamp recordings were performed in voltage-clamp mode while maintaining the membrane potential at the reversal potential for GABAA receptor-mediated PSCs (-60 mV) to isolate miniature excitatory postsynaptic currents (mEPSCs). To avoid any long-term effect of the application of TTX to the whole bath, only one cell per slice was recorded. For purpose of analysis, data were filtered at 1 kHz. The Clampfit 9.2 and Origin 8 (OriginLab) software were used for analyses.
Hole-board exploration test
Exploratory behavior of mice was assessed in a rectangular arena (40 cm X 22 cm) with holes in each corner. Hole-board exploration is a behavioral paradigm that is used frequently in mice and requires no training [34]. The time spent in active exploration of the four holes was measured during a single 5 min daily session for three consecutive days.
Dark-light box test
Anxiety-like behavior was assessed for 10 min using a two-compartment box (dark and light box). Animals were positioned in the dark compartment and the latency to enter in the light compartment was measured. Additionally, the number of standings and their average duration was measured as a control for locomotor integrity [34,97,98].
Statistical analysis
All experiments and analyses were carried out blindly with respect to genotypes until the codes were broken. All results are expressed as mean ± SEM. Significance was assessed with a Mann-Whitney non-parametric U test. Statistical analysis of biochemical measurements including two groups were performed using unpaired t-test or Mann-Whitney non-parametric U test or Welch’s t-test (unequal variance). ANOVA (equal variance) followed by Tukey-Kramer or Newman-Keuls post hoc tests or with Welch’s ANOVA (unequal variance) followed by a Dunnett’s post hoc test were used to compare three groups. Significance of electrophysiological and morphological results was assessed with a Mann-Whitney non-parametric U test or one-way ANOVA followed by a protected Fisher post hoc test. All statistical analyses were performed using Origin 8.0 analysis software (OriginLab), JMP (version 9, SAS) or prism (version 4.0, GraphPad Software Inc.) and are presented in Table 1.
Abbreviations
3xTg-AD: triple transgenic mouse model of Alzheimer’s disease; Aβ: abeta peptide; AD: Alzheimer disease; AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; Cdk5: Cyclin-dependent kinase 5; dnPAK: dominant negative PAK; EPSCs: excitatory postsynaptic currents; GABA: gamma-aminobutyric acid; GAD65: glutamic acid decarboxylase 65 KDa; GluN2B: NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit; GSK3: glycogen synthase kinase 3; mEPSCs: miniature excitatory postsynaptic currents; mPSCs: miniature postsynaptic currents; NonTg: non-transgenic; PAK: p21-activated kinase; pPAK: phosphorylated PAK; PSCs: postsynaptic currents; PSD95: postsynaptic density protein 95; Ser: serine; Thr: threonine; TTX: tetradotoxin; VGAT: vesicular GABA transporter.
Author Contributions
CB performed electrophysiological recordings and data analyses and wrote the first versions of the manuscript. DA planned the study, worked on the generation of mice, tissue processing, behavioural tests and most Western blots and generated final versions of the manuscript. CT performed ELISA (Aβ) assays and some Western blots. YDK contributed to the planning of the study and provided electrophysiology equipment. FC planned the study, obtained funding and wrote the manuscript.
Acknowledgements
We thank Dr. Vincent Emond for editing of the manuscript.
Conflicts of Interest
The authors have declared that no competing interests exist.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (FC - MOP102532), the Alzheimer Society Canada (FC – ASC 0516) and the Canada Foundation for Innovation (10307). D. Arsenault held studentships from the CIHR.
References
- 1. Perri R, Monaco M, Fadda L, Caltagirone C, Carlesimo GA. Neuropsychological correlates of behavioral symptoms in Alzheimer’s disease, frontal variant of frontotemporal, subcortical vascular, and lewy body dementias: a comparative study. J Alzheimers Dis. 2014; 39:669–77. https://doi.org/10.3233/JAD-131337 [PubMed]
- 2. Brodaty H, Connors MH, Xu J, Woodward M, Ames D, and PRIME study group. The course of neuropsychiatric symptoms in dementia: a 3-year longitudinal study. J Am Med Dir Assoc. 2015; 16:380–87. https://doi.org/10.1016/j.jamda.2014.12.018 [PubMed]
- 3. Teng E, Lu PH, Cummings JL. Neuropsychiatric symptoms are associated with progression from mild cognitive impairment to Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007; 24:253–59. https://doi.org/10.1159/000107100 [PubMed]
- 4. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011; 1:a006189. https://doi.org/10.1101/cshperspect.a006189 [PubMed]
- 5. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015; 7:1198–211. https://doi.org/10.18632/aging.100864 [PubMed]
- 6. Mizrahi R, Starkstein SE. Epidemiology and management of apathy in patients with Alzheimer’s disease. Drugs Aging. 2007; 24:547–54. https://doi.org/10.2165/00002512-200724070-00003 [PubMed]
- 7. Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006; 65:592–601. https://doi.org/10.1097/00005072-200606000-00007 [PubMed]
- 8. Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009; 118:167–79. https://doi.org/10.1007/s00401-009-0536-x [PubMed]
- 9. Ma QL, Yang F, Calon F, Ubeda OJ, Hansen JE, Weisbart RH, Beech W, Frautschy SA, Cole GM. P21-activated kinase aberrant activation and translocation in Alzheimer’s disease pathogenesis. J Biol Chem. 2008; 283:14132–43. https://doi.org/10.1074/jbc.M708034200 [PubMed]
- 10. Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006; 9:234–42. https://doi.org/10.1038/nn1630 [PubMed]
- 11. Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007; 27:796–807. https://doi.org/10.1523/JNEUROSCI.3501-06.2007 [PubMed]
- 12. Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004; 24:10191–200. https://doi.org/10.1523/JNEUROSCI.3432-04.2004 [PubMed]
- 13. Herms J, Dorostkar MM. Dendritic Spine Pathology in Neurodegenerative Diseases. Annu Rev Pathol. 2016; 11:221–50. https://doi.org/10.1146/annurev-pathol-012615-044216 [PubMed]
- 14. Fainzilber M, Budnik V, Segal RA, Kreutz MR. From synapse to nucleus and back again--communication over distance within neurons. J Neurosci. 2011; 31:16045–48. https://doi.org/10.1523/JNEUROSCI.4006-11.2011 [PubMed]
- 15. Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008; 24:183–209. https://doi.org/10.1146/annurev.cellbio.24.110707.175235 [PubMed]
- 16. Calabrese B, Wilson MS, Halpain S. Development and regulation of dendritic spine synapses. Physiology (Bethesda). 2006; 21:38–47. https://doi.org/10.1152/physiol.00042.2005 [PubMed]
- 17. Rosenberg PB, Nowrangi MA, Lyketsos CG. Neuropsychiatric symptoms in Alzheimer’s disease: what might be associated brain circuits? Mol Aspects Med. 2015; 43-44:25–37. https://doi.org/10.1016/j.mam.2015.05.005 [PubMed]
- 18. Boublay N, Schott AM, Krolak-Salmon P. Neuroimaging correlates of neuropsychiatric symptoms in Alzheimer’s disease: a review of 20 years of research. Eur J Neurol. 2016; 23:1500–09. https://doi.org/10.1111/ene.13076 [PubMed]
- 19. Drevets WC. Orbitofrontal cortex function and structure in depression. Ann N Y Acad Sci. 2007; 1121:499–527. https://doi.org/10.1196/annals.1401.029 [PubMed]
- 20. Covington HE
3rd , Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, Neve RL, Deisseroth K, Nestler EJ. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010; 30:16082–90. https://doi.org/10.1523/JNEUROSCI.1731-10.2010 [PubMed] - 21. Bories C, Guitton MJ, Julien C, Tremblay C, Vandal M, Msaid M, De Koninck Y, Calon F. Sex-dependent alterations in social behaviour and cortical synaptic activity coincide at different ages in a model of Alzheimer’s disease. PLoS One. 2012; 7:e46111. https://doi.org/10.1371/journal.pone.0046111 [PubMed]
- 22. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003; 39:409–21. https://doi.org/10.1016/S0896-6273(03)00434-3 [PubMed]
- 23. Filali M, Lalonde R, Rivest S. Anomalies in social behaviors and exploratory activities in an APPswe/PS1 mouse model of Alzheimer’s disease. Physiol Behav. 2011; 104:880–85. https://doi.org/10.1016/j.physbeh.2011.05.023 [PubMed]
- 24. Deacon RM, Koros E, Bornemann KD, Rawlins JN. Aged Tg2576 mice are impaired on social memory and open field habituation tests. Behav Brain Res. 2009; 197:466–68. https://doi.org/10.1016/j.bbr.2008.09.042 [PubMed]
- 25. Lin CW, Chen CY, Cheng SJ, Hu HT, Hsueh YP. Sarm1 deficiency impairs synaptic function and leads to behavioral deficits, which can be ameliorated by an mGluR allosteric modulator. Front Cell Neurosci. 2014; 8:87. https://doi.org/10.3389/fncel.2014.00087 [PubMed]
- 26. Cheng D, Logge W, Low JK, Garner B, Karl T. Novel behavioural characteristics of the APP(Swe)/PS1ΔE9 transgenic mouse model of Alzheimer’s disease. Behav Brain Res. 2013; 245:120–27. https://doi.org/10.1016/j.bbr.2013.02.008 [PubMed]
- 27. Lopez OL, Zivkovic S, Smith G, Becker JT, Meltzer CC, DeKosky ST. Psychiatric symptoms associated with cortical-subcortical dysfunction in Alzheimer’s disease. J Neuropsychiatry Clin Neurosci. 2001; 13:56–60. https://doi.org/10.1176/jnp.13.1.56 [PubMed]
- 28. Sultzer DL, Mahler ME, Mandelkern MA, Cummings JL, Van Gorp WG, Hinkin CH, Berisford MA. The relationship between psychiatric symptoms and regional cortical metabolism in Alzheimer’s disease. J Neuropsychiatry Clin Neurosci. 1995; 7:476–84. https://doi.org/10.1176/jnp.7.4.476 [PubMed]
- 29. Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011; 14:285–93. https://doi.org/10.1038/nn.2741 [PubMed]
- 30. Shim KS, Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer’s disease and Down syndrome. Neurosci Lett. 2002; 324:209–12. https://doi.org/10.1016/S0304-3940(02)00210-0 [PubMed]
- 31. Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003; 72:743–81. https://doi.org/10.1146/annurev.biochem.72.121801.161742 [PubMed]
- 32. Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998; 20:25–30. https://doi.org/10.1038/1675 [PubMed]
- 33. Bienvenu T, des Portes V, McDonell N, Carrié A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walsh CA, Boué J, et al. Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet. 2000; 93:294–98. https://doi.org/10.1002/1096-8628(20000814)93:4<294::AID-AJMG8>3.0.CO;2-F [PubMed]
- 34. Arsenault D, Dal-Pan A, Tremblay C, Bennett DA, Guitton MJ, De Koninck Y, Tonegawa S, Calon F. PAK inactivation impairs social recognition in 3xTg-AD Mice without increasing brain deposition of tau and Aβ. J Neurosci. 2013; 33:10729–40. https://doi.org/10.1523/JNEUROSCI.1501-13.2013 [PubMed]
- 35. Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010; 31:1516–31. https://doi.org/10.1016/j.neurobiolaging.2008.08.022 [PubMed]
- 36. Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron. 2004; 42:773–87. https://doi.org/10.1016/j.neuron.2004.05.003 [PubMed]
- 37. Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA. 2007; 104:11489–94. https://doi.org/10.1073/pnas.0705003104 [PubMed]
- 38. Bi LL, Wang J, Luo ZY, Chen SP, Geng F, Chen YH, Li SJ, Yuan CH, Lin S, Gao TM. Enhanced excitability in the infralimbic cortex produces anxiety-like behaviors. Neuropharmacology. 2013; 72:148–56. https://doi.org/10.1016/j.neuropharm.2013.04.048 [PubMed]
- 39. Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009; 16:279–88. https://doi.org/10.1101/lm.1041309 [PubMed]
- 40. Varela JA, Wang J, Christianson JP, Maier SF, Cooper DC. Control over stress, but not stress per se increases prefrontal cortical pyramidal neuron excitability. J Neurosci. 2012; 32:12848–53. https://doi.org/10.1523/JNEUROSCI.2669-12.2012 [PubMed]
- 41. Freitas RL, Salgado-Rohner CJ, Hallak JE, Crippa JA, Coimbra NC. Involvement of prelimbic medial prefrontal cortex in panic-like elaborated defensive behaviour and innate fear-induced antinociception elicited by GABAA receptor blockade in the dorsomedial and ventromedial hypothalamic nuclei: role of the endocannabinoid CB1 receptor. Int J Neuropsychopharmacol. 2013; 16:1781–98. https://doi.org/10.1017/S1461145713000163 [PubMed]
- 42. Bediou B, Ryff I, Mercier B, Milliery M, Hénaff MA, D’Amato T, Bonnefoy M, Vighetto A, Krolak-Salmon P. Impaired social cognition in mild Alzheimer disease. J Geriatr Psychiatry Neurol. 2009; 22:130–40. https://doi.org/10.1177/0891988709332939 [PubMed]
- 43. Seeley WW, Allman JM, Carlin DA, Crawford RK, Macedo MN, Greicius MD, Dearmond SJ, Miller BL. Divergent social functioning in behavioral variant frontotemporal dementia and Alzheimer disease: reciprocal networks and neuronal evolution. Alzheimer Dis Assoc Disord. 2007; 21:S50–57. https://doi.org/10.1097/WAD.0b013e31815c0f14 [PubMed]
- 44. Ramakers IH, Verhey FR, Scheltens P, Hampel H, Soininen H, Aalten P, Rikkert MO, Verbeek MM, Spiru L, Blennow K, Trojanowski JQ, Shaw LM, Visser PJ, and Alzheimer’s Disease Neuroimaging Initiative and DESCRIPA Investigators. Anxiety is related to Alzheimer cerebrospinal fluid markers in subjects with mild cognitive impairment. Psychol Med. 2013; 43:911–20. https://doi.org/10.1017/S0033291712001870 [PubMed]
- 45. Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010; 13:812–18. https://doi.org/10.1038/nn.2583 [PubMed]
- 46. Bussière T, Bard F, Barbour R, Grajeda H, Guido T, Khan K, Schenk D, Games D, Seubert P, Buttini M. Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol. 2004; 165:987–95. https://doi.org/10.1016/S0002-9440(10)63360-3 [PubMed]
- 47. Terry RD, Katzman R. Life span and synapses: will there be a primary senile dementia? Neurobiol Aging. 2001; 22:347–48. https://doi.org/10.1016/S0197-4580(00)00250-5 [PubMed]
- 48. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991; 30:572–80. https://doi.org/10.1002/ana.410300410 [PubMed]
- 49. Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N
Jr , Ashe KH, Frautschy SA, Cole GM. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004; 43:633–45. https://doi.org/10.1016/j.neuron.2004.08.013 [PubMed] - 50. Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, Bredesen DE, Masliah E, Mucke L. Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer’s disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci. 2010; 30:372–81. https://doi.org/10.1523/JNEUROSCI.5341-09.2010 [PubMed]
- 51. Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006; 443:768–73. https://doi.org/10.1038/nature05289 [PubMed]
- 52. Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007; 55:697–711. https://doi.org/10.1016/j.neuron.2007.07.025 [PubMed]
- 53. Koppel J, Jimenez H, Azose M, D’Abramo C, Acker C, Buthorn J, Greenwald BS, Lewis J, Lesser M, Liu Z, Davies P. Pathogenic tau species drive a psychosis-like phenotype in a mouse model of Alzheimer’s disease. Behav Brain Res. 2014; 275:27–33. https://doi.org/10.1016/j.bbr.2014.08.030 [PubMed]
- 54. Dujardin S, Lécolle K, Caillierez R, Bégard S, Zommer N, Lachaud C, Carrier S, Dufour N, Aurégan G, Winderickx J, Hantraye P, Déglon N, Colin M, Buée L. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun. 2014; 2:14. https://doi.org/10.1186/2051-5960-2-14 [PubMed]
- 55. Guadagna S, Esiri MM, Williams RJ, Francis PT. Tau phosphorylation in human brain: relationship to behavioral disturbance in dementia. Neurobiol Aging. 2012; 33:2798–806. https://doi.org/10.1016/j.neurobiolaging.2012.01.015 [PubMed]
- 56. Tremblay C, Pilote M, Phivilay A, Emond V, Bennett DA, Calon F. Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2007; 12:377–90. https://doi.org/10.3233/JAD-2007-12411 [PubMed]
- 57. Bittner T, Fuhrmann M, Burgold S, Ochs SM, Hoffmann N, Mitteregger G, Kretzschmar H, LaFerla FM, Herms J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer’s disease mice. PLoS One. 2010; 5:e15477. https://doi.org/10.1371/journal.pone.0015477 [PubMed]
- 58. Luebke JI, Weaver CM, Rocher AB, Rodriguez A, Crimins JL, Dickstein DL, Wearne SL, Hof PR. Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct Funct. 2010; 214:181–99. https://doi.org/10.1007/s00429-010-0244-2 [PubMed]
- 59. Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008; 115:5–38. https://doi.org/10.1007/s00401-007-0312-8 [PubMed]
- 60. Anderton BH. Changes in the ageing brain in health and disease. Philos Trans R Soc Lond B Biol Sci. 1997; 352:1781–92. https://doi.org/10.1098/rstb.1997.0162 [PubMed]
- 61. Anderton BH, Callahan L, Coleman P, Davies P, Flood D, Jicha GA, Ohm T, Weaver C. Dendritic changes in Alzheimer’s disease and factors that may underlie these changes. Prog Neurobiol. 1998; 55:595–609. https://doi.org/10.1016/S0301-0082(98)00022-7 [PubMed]
- 62. Bell KF, Bennett DA, Cuello AC. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci. 2007; 27:10810–17. https://doi.org/10.1523/JNEUROSCI.3269-07.2007 [PubMed]
- 63. Bell KF, de Kort GJ, Steggerda S, Shigemoto R, Ribeiro-da-Silva A, Cuello AC. Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci Lett. 2003; 353:143–47. https://doi.org/10.1016/j.neulet.2003.09.027 [PubMed]
- 64. Bossers K, Wirz KT, Meerhoff GF, Essing AH, van Dongen JW, Houba P, Kruse CG, Verhaagen J, Swaab DF. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain. 2010; 133:3699–723. https://doi.org/10.1093/brain/awq258 [PubMed]
- 65. Leuba G, Savioz A, Vernay A, Carnal B, Kraftsik R, Tardif E, Riederer I, Riederer BM. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis. 2008; 15:139–51. https://doi.org/10.3233/JAD-2008-15112 [PubMed]
- 66. Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, Gertz HJ, Xuereb JH, Hills R, Brayne C, Huppert FA, Paykel ES, McGee M, Jakes R, Honer WG, Harrington CR, Wischik CM. Staging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol. 2000; 157:623–36. https://doi.org/10.1016/S0002-9440(10)64573-7 [PubMed]
- 67. Zahs KR, Ashe KH. ‘Too much good news’ - are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci. 2010; 33:381–89. https://doi.org/10.1016/j.tins.2010.05.004 [PubMed]
- 68. Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol Neurobiol. 2008; 37:73–82. https://doi.org/10.1007/s12035-008-8018-z [PubMed]
- 69. Bloss EB, Janssen WG, Ohm DT, Yuk FJ, Wadsworth S, Saardi KM, McEwen BS, Morrison JH. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci. 2011; 31:7831–39. https://doi.org/10.1523/JNEUROSCI.0839-11.2011 [PubMed]
- 70. Mehraein P, Yamada M, Tarnowska-Dziduszko E. Quantitative study on dendrites and dendritic spines in Alzheimer’s disease and senile dementia. Adv Neurol. 1975; 12:453–58. [PubMed]
- 71. Wilcox KC, Lacor PN, Pitt J, Klein WL. Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol Neurobiol. 2011; 31:939–48. https://doi.org/10.1007/s10571-011-9691-4 [PubMed]
- 72. Ferrer I, Guionnet N, Cruz-Sánchez F, Tuñón T. Neuronal alterations in patients with dementia: a Golgi study on biopsy samples. Neurosci Lett. 1990; 114:11–16. https://doi.org/10.1016/0304-3940(90)90420-E [PubMed]
- 73. Francis PT. Glutamatergic systems in Alzheimer’s disease. Int J Geriatr Psychiatry. 2003 (Suppl 1); 18:S15–21. https://doi.org/10.1002/gps.934 [PubMed]
- 74. DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990; 27:457–64. https://doi.org/10.1002/ana.410270502 [PubMed]
- 75. Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990; 11:29–37. https://doi.org/10.1016/0197-4580(90)90059-9 [PubMed]
- 76. Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003; 24:1029–46. https://doi.org/10.1016/j.neurobiolaging.2003.08.002 [PubMed]
- 77. Scheff SW, Price DA. Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006 (Suppl ); 9:101–15. https://doi.org/10.3233/JAD-2006-9S312 [PubMed]
- 78. Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006; 27:1372–84. https://doi.org/10.1016/j.neurobiolaging.2005.09.012 [PubMed]
- 79. Lanctôt KL, Herrmann N, Mazzotta P, Khan LR, Ingber N. GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can J Psychiatry. 2004; 49:439–53. [PubMed]
- 80. Limon A, Reyes-Ruiz JM, Miledi R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci USA. 2012; 109:10071–76. https://doi.org/10.1073/pnas.1204606109 [PubMed]
- 81. Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014; 20:886–96. https://doi.org/10.1038/nm.3639 [PubMed]
- 82. Harigaya Y, Shoji M, Shirao T, Hirai S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J Neurosci Res. 1996; 43:87–92. https://doi.org/10.1002/jnr.490430111 [PubMed]
- 83. Craig D, Mirakhur A, Hart DJ, McIlroy SP, Passmore AP. A cross-sectional study of neuropsychiatric symptoms in 435 patients with Alzheimer’s disease. Am J Geriatr Psychiatry. 2005; 13:460–68. https://doi.org/10.1097/00019442-200506000-00004 [PubMed]
- 84. Mega MS, Cummings JL, Fiorello T, Gornbein J. The spectrum of behavioral changes in Alzheimer’s disease. Neurology. 1996; 46:130–35. https://doi.org/10.1212/WNL.46.1.130 [PubMed]
- 85. Filali M, Lalonde R, Theriault P, Julien C, Calon F, Planel E. Cognitive and non-cognitive behaviors in the triple transgenic mouse model of Alzheimer’s disease expressing mutated APP, PS1, and Mapt (3xTg-AD). Behav Brain Res. 2012; 234:334–42. https://doi.org/10.1016/j.bbr.2012.07.004 [PubMed]
- 86. Filali M, Lalonde R, Rivest S. Cognitive and non-cognitive behaviors in an APPswe/PS1 bigenic model of Alzheimer’s disease. Genes Brain Behav. 2009; 8:143–48. https://doi.org/10.1111/j.1601-183X.2008.00453.x [PubMed]
- 87. Pietropaolo S, Delage P, Lebreton F, Crusio WE, Cho YH. Early development of social deficits in APP and APP-PS1 mice. Neurobiol Aging. 2012; 33:1002.e17–27. https://doi.org/10.1016/j.neurobiolaging.2011.09.012 [PubMed]
- 88. Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, Fernández-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer’s disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007; 31:125–47. https://doi.org/10.1016/j.neubiorev.2006.07.007 [PubMed]
- 89. Sterniczuk R, Antle MC, Laferla FM, Dyck RH. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 2. Behavioral and cognitive changes. Brain Res. 2010; 1348:149–55. https://doi.org/10.1016/j.brainres.2010.06.011 [PubMed]
- 90. Baeg EH, Kim YB, Jang J, Kim HT, Mook-Jung I, Jung MW. Fast spiking and regular spiking neural correlates of fear conditioning in the medial prefrontal cortex of the rat. Cereb Cortex. 2001; 11:441–51. https://doi.org/10.1093/cercor/11.5.441 [PubMed]
- 91. Kim YB, Jang J, Chung Y, Baeg EH, Kim HT, Mook-Jung I, Kim SU, Jung MW, Chung YK. Haloperidol and clozapine increase neural activity in the rat prefrontal cortex. Neurosci Lett. 2001; 298:217–21. https://doi.org/10.1016/S0304-3940(00)01765-1 [PubMed]
- 92. Prater KE, Hosanagar A, Klumpp H, Angstadt M, Phan KL. Aberrant amygdala-frontal cortex connectivity during perception of fearful faces and at rest in generalized social anxiety disorder. Depress Anxiety. 2013; 30:234–41. https://doi.org/10.1002/da.22014 [PubMed]
- 93. Behr GA, da Motta LL, de Oliveira MR, Oliveira MW, Hoff ML, Silvestrin RB, Moreira JC. Decreased anxiety-like behavior and locomotor/exploratory activity, and modulation in hypothalamus, hippocampus, and frontal cortex redox profile in sexually receptive female rats after short-term exposure to male chemical cues. Behav Brain Res. 2009; 199:263–70. https://doi.org/10.1016/j.bbr.2008.11.047 [PubMed]
- 94. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003; 24:1063–70. https://doi.org/10.1016/j.neurobiolaging.2003.08.012 [PubMed]
- 95. Arsenault D, Julien C, Tremblay C, Calon F. DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS One. 2011; 6:e17397. https://doi.org/10.1371/journal.pone.0017397 [PubMed]
- 96. Bories C, Husson Z, Guitton MJ, De Koninck Y. Differential balance of prefrontal synaptic activity in successful versus unsuccessful cognitive aging. J Neurosci. 2013; 33:1344–56. https://doi.org/10.1523/JNEUROSCI.3258-12.2013 [PubMed]
- 97. Guitton MJ, Dudai Y. Anxiety-like state associates with taste to produce conditioned taste aversion. Biol Psychiatry. 2004; 56:901–04. https://doi.org/10.1016/j.biopsych.2004.08.024 [PubMed]
- 98. St-Amour I, Paré I, Tremblay C, Coulombe K, Bazin R, Calon F. IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Aβ pathology. J Neuroinflammation. 2014; 11:54. https://doi.org/10.1186/1742-2094-11-54 [PubMed]
- 99. Salchner P, Lubec G, Singewald N. Decreased social interaction in aged rats may not reflect changes in anxiety-related behaviour. Behav Brain Res. 2004; 151:1–8. https://doi.org/10.1016/j.bbr.2003.07.002 [PubMed]
Social interaction paradigm
Animals were submitted to a social interaction test performed in a transparent plastic arena (40 cm x 22 cm x 18 cm). Pairs of age-matched animals, unfamiliar with each other, were placed in the unfamiliar test arena for an observation period of 20 min. Their social behavior and interaction time was then scored as follows: number of social events (sniffing, following, grooming the partner, crawling over or under). The interaction time was defined as the time spent in active social interactions (sniffing, following, grooming the partner, wrestling, crawling over or under) [21,97,99]. The interaction time represents the total time spent in active behavior, regardless of the number of individual events. After each trial, animals were returned to their home cages and the arena was changed to a new and clean one, in order to avoid any odor cues from one pair to the next one.