



Introduction
Neurodegenerative diseases comprise one of the major public health concerns worldwide, with Alzheimer’s disease (AD), Frontotemporal disease (FTD), and Parkinson’s disease (PD) being the most common types. Although these diseases are considered distinct entities, each with its distinct etiological mechanisms, affected brain regions and clinical characteristics, they share some common features. For instance, some behavioral disturbances can characterize the initial phases of AD patients, while some FTD patients can manifest deficit in the episodic memory domain [1]. From a neuropathological point of view, accumulation of intracellular tau protein is seen in both AD and FTD [2]; moreover, TAR-DNA binding protein (TDP)-43 deposition has been reported in AD, in a subtype of FTD and, in some rare PD cases, associated with a leucine-rich repeat kinase 2 (LRRK2) mutation [3,4]. Evidence also point to potential genetic overlap among these disorders [5]. The MAPT gene, extensively investigated in FTD [2], has been also implicated in AD and PD [6,7]. In addition, evidences suggest that genetic variants within the HLA locus contribute to the development of these disorders [8–10]. Taken together, such overlapping features may reflect some common underlying etiological factors.
The impairment of mitochondrial functioning is now emerging as an upstream event in the chain of pathological events leading to neuronal degeneration and a shared feature of these neurodegenerative diseases [11–15]. What is more, mitochondrial dysfunction, oxidative stress and abnormal accumulation of misfolded and aggregated proteins seem to be interdependent phenomena that work in concert, reinforcing each other to drive these pathologies, as summarized in two recent reviews [16,17]. An inherent part of mitochondrial physiology which could impact on neuronal functioning is the uncoupling of respiration from oxidative phosphorylation, a process mediated by three out of five mitochondrial uncoupling proteins (UCP2,4,5) which affects energy production, oxidative stress and intracellular calcium homeostasis [18,19].
We recently found that a non-coding variant (rs9472817) in the last intron of UCP4/SLC25A27 gene affects the risk of developing sporadic and familial late onset Alzheimer's disease (LOAD) and strongly modulates the effect of APOE-ε4 on the disease risk [20]. UCP4, which is prominently expressed in neurons from hippocampus, cortex, Substantia nigra pars compacta, striatum and cerebellum [21,22] and, at lower levels, in astrocytes [23], appears to have a critical role in helping neurons to cope with conditions of metabolic and oxidative stress [22]. Thus, it is reasonable to hypothesize that UCP4 genotypes may foster the development of multiple neurodegenerative conditions.
In order to test this hypothesis, we investigated whether the rs9472817 variation of UCP4 gene also influences the risk of developing FTD and PD and whether this eventual risk is modulated by the APOE-ε4 genotypic variability.
Results
Descriptive information about participants is presented in Table 1. It is of note that there was no difference in age and gender between patients and healthy controls. Table S1 reports the allele and genotype frequencies for the UCP4-rs9472817 in patients and controls. Genotype distributions were in agreement with the HW equilibrium (P-value > 0.05). We did not detect any significant effect of the rs9472817 polymorphism on the risk of sporadic and familial PD and familial FTD (Figure 1). On the contrary, our results suggest that UCP4 may be a susceptibility gene for sporadic FTD. The data from this subgroup of patients showed, as previously reported for LOAD [20], that individuals with one or more copies of the C allele for rs9472817 are at increased risk of disease compared to those with the GG genotype, with a per-allele increased risk of 1.513 (95% CI 1.091-2.098, P-value= 0.013). After adjusting for possible confounders, this risk resulted almost unchanged (OR=1.506, 95% CI 1.085-2.090, P-value=0.014). We also evaluated the potential association of the UCP4 rs9472817-C allele with the disease progression and severity, as measured by age of disease’s onset and MMSE values We did not find statistically significant difference between carriers and non-carriers of the C allele according to age of disease’s onset and MMSE scores. We found that the presence of the APOE-ε4 allele was associated with an increased risk of FTD, both in familial (OR= 6.341, 95%CI: 3.747-10.731, P-value=5.97*10-12) and sporadic cases (OR= 3.621, 95%CI: 2.027-6.470, P-value=1.4*10-5), as well as in the familial (OR=4.764, 95%CI: 2.205-10.294, P-value=7.2*10-5) and in the sporadic form of PD (OR= 2.552, 95%CI: 1.171-5.562, P-value=0.018) (Table 2, model 1). However, the reported effect of UCP4-rs9472817 variation on sporadic FTD remained statistically significant also after adjustment for APOE-ε4 status (OR=1.599, 95%CI: 1.135-2.252, P-value=0.007, model 2). In other words, in sporadic FTD, the UCP4-rs9472817 variation affects the risk of dementia independently of the presence of APOE-ε4 allele. Furthermore, interaction analysis enabled us to verify that in these models, the interaction terms were not significantly different from 0 (model 3).
Table 1. Characteristics of the analyzed sample in cases and controls.
| FTD | Parkinson | Controls (N=442) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Familial (N=113) | Sporadic (N=100) | Familial (N=41) | Sporadic (N=55) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (mean ± SD) | 73.9 ± 6.8 | 75.2 ± 6.9 | 74.9 ± 6.8 | 74.8 ± 6.9 | 73.7 ± 8.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Males [n (%)] | 49 (43.4) | 43 (43.0) | 23 (56.1) | 29 (52.7) | 225 (50.9) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age onset (mean ± SD) | 72.9 ± 5.3 | 72.5 ± 5.7 | 66.7 ± 6.4 | 67.0 ± 8.2 | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMSE1 (mean ± SD) | 11.2 ± 8.1 | 13.1 ± 7.5 | 24.1 ± 5.2 | 23.8 ± 5.4 | 23.5 ± 4.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| APOE-ε4 carriers [n (%)] | 42 (37.2) | 25 (25.3) | 12 (30.8) | 10 (19.2) | 32 (8.5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1MMSE scores were adjusted for educational level and age at inclusion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
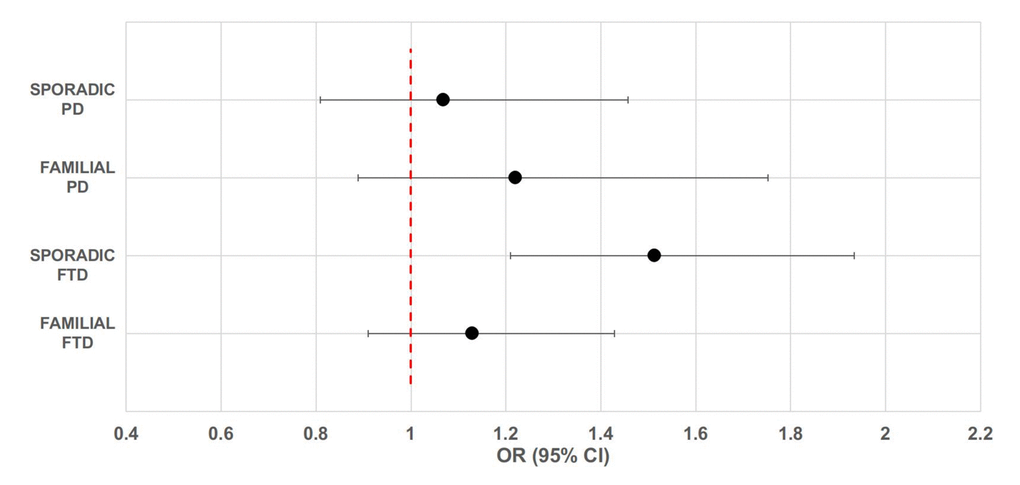
Figure 1. Forest plot of overall analysis for the association between rs9472817 in UCP4 and risk of familial and sporadic FTD and PD. The circle and horizontal lines represent odds ratio (OR) and 95% confidence interval (CI).
Table 2. Results of the logistic regression models for FTD and PD genetic risk.
| Familial | Sporadic | |||||||
| Group | Model | OR | 95% CI | P-value | OR | 95% CI | P-value | |
| FTD | Model 1 | APOE-ε4 | 6.341 | 3.747-10.731 | 5.97*10-12 | 3.621 | 2.027-6.470 | 1.4*10-5 |
| Model 2 | APOE-ε4 | 6.598 | 3.776-11.530 | 3.45*10-11 | 3.557 | 1.917-6.559 | 5.7*10-5 | |
| UCP4-rs9472817 | 1.293 | 0.916-1.825 | 0.144 | 1.599 | 1.135-2.252 | 0.007 | ||
| Model 3 | APOE-ε4 | 4.888 | 1.827-13.078 | 0.002 | 3.189 | 1.067-9.535 | 0.038 | |
| UCP4-rs9472817 | 1.207 | 0.817-1.783 | 0.346 | 1.567 | 1.071-2.292 | 0.021 | ||
| APOE-ε4*UCP4-rs9472817 | 1.357 | 0.592-3.112 | 0.471 | 1.111 | 0.465-2.657 | 0.813 | ||
| PD | Familial | Sporadic | ||||||
| Model | OR | 95% CI | P-value | OR | 95% CI | P-value | ||
| Model 1 | APOE-ε4 | 4.764 | 2.205-10.294 | 7.2*10-5 | 2.552 | 1.171-5.562 | 0.018 | |
| Model 2 | APOE-ε4 | 5.255 | 2.376-11.624 | 4.2*10-5 | 2.082 | 0.889-4.880 | 0.091 | |
| UCP4-rs9472817 | 1.314 | 0.795-2.172 | 0.288 | 1.162 | 0.756-1.786 | 0.494 | ||
| Model 3 | APOE-ε4 | 3.572 | 0.859-14.848 | 0.080 | 2.138 | 0.537-8.511 | 0.281 | |
| UCP4-rs9472817 | 1.182 | 0.655-2.134 | 0.579 | 1.167 | 0.731-1.863 | 0.517 | ||
| APOE ε4*UCP4-rs9472817 | 1.447 | 0.473-4.427 | 0.518 | 0.972 | 0.295-3.200 | 0.962 | ||
Discussion
FTD, PD and LOAD are clinically distinct conditions although they share similar dysfunctional phenotypes, such as protein aggregation, neuronal cell death, and cognitive decline. Yet, the extent of the genetic overlap between these disorders is still not fully known, despite considerable research to date. Assessing genetic overlap between complex traits is based on the notion that genes or genetic variants may play a role in related phenotypes in the context of different genetic backgrounds and under different environmental conditions because of their pleiotropic functions [24].
Building on our prior work implicating the involvement of the rs9472817 in UCP4 gene in LOAD [20], in the current study we assessed the potential pleiotropic effect of this variant on late-onset familial and sporadic FTD and PD. Our data suggest that this SNP is a marker also for sporadic FTD. The lack of association with familial FTD suggests that this variant probably confers a low risk of disease, which could be covered by major risk factors involved in the familial form of the disease. On the other hand, association studies indicate that sporadic FTD is a polygenic trait, arising from the influences of multiple pleiotropic loci with small individual effects [5], likely including UCP4 gene.
As we stated previously, FTD and LOAD are characterized by impairment in different cognitive domains. However, there are levels of overlap between these disorders, i.e. some behavioral disturbances can characterize the initial phases of AD patients, while some FTD patients can manifest deficit in the episodic memory domain, that likely reflect the fact that FTD and AD are associated with progressive impairment of similar brain circuits [1]. This is suggestive of a potential pathological overlap and of the existence of common genetic mechanisms that drive the pathological events leading to their onset. Thus, our data may provide new insights into the underlying shared pathogenic mechanisms between FTD and LOAD.
No association was detected between rs9472817 and PD, indicating that this genetic factor is likely not implicated in the development of this type of dementia. However, given the reduced number of PD patients evaluated in the present study, this could be also due to a type II error and thus, a role of rs9472817 on disease risk, even if small, cannot be rule out.
We assessed the functional consequences of the rs9472817 on gene expression by querying three different brain-specific expression quantitative trait loci (eQTLs) datasets (see Supplementary Table S2 and Figure S1). Data are consistent regarding an increased expression of UCP4 in different brain regions associated to the presence of the risk C allele of rs9472817, possibly indicating that higher levels of UCP4 might be the mechanism by which the association at this locus is mediated. In terms of biological plausibility this would seem to be not in accordance with the effect we found on the risk of FTD, since experimental evidences document that increased levels of UCP4 allow to preserve the physiological functions of neurons by maintaining energy and redox balance and by decreasing mitochondrial calcium accumulation [21,25,26]. On the other hand, the increased expression of UCP4 associated to the C allele is in accordance with our previous study showing that this is a longevity allele [27]. The reasons for these contrasting results are not clear, however it can be hypothesized that a phenomenon of genetic epistasis (the phenotypic effect of the allele depends on the specific alleles at another locus) is present or that the phenotypic effect of the allele vary depending on cell’s biological context. As for the latter case, for example, evidence indicate that UCP4 acts as a buffering mechanism that finely tunes the entry of cytosolic Ca2+ into the mitochondria, avoiding the calcium overload in the organelle. In other conditions, such as in neurodegenerative diseases where increased levels of cytosolic Ca2+ have been documented [28–30], an increased expression of UCP4, reducing the uptake of Ca2+ in mitochondria might worsen the cytosolic overload, further accruing the pathological accumulation of this element.
It must however be pointed out that, according to eQTL data, the risk C allele is also significantly associated with decreased TDRD6 expression in brain tissues. This gene encodes a tudor domain-containing protein, a male germ line-specific protein involved in spermiogenesis, chromatoid body formation, regulation of miRNA expression and the nonsense mediated decay pathway [31,32]. To our knowledge, there is no evidence to support a link between TDRD6 and FTD or other neurodegenerative diseases. Therefore, although a role cannot be ruled out, it seems unlikely that alterations in the levels of this gene might be the molecular basis of the observed associations, and this deserves further attention.
With respect to the involvement of APOE in FTD and PD, results in literature are inconsistent regarding the effect of the ε4 allele: some studies report an increased risk associated to this allele whereas others report no association [33–36]. In particular, studies focusing on the role of APOE in PD remain largely inconclusive, with some studies reporting APOE-ε2 allele associated with higher prevalence of sporadic PD [37]. Here we found that in all subgroups analyzed, carriers of APOE-ε4 allele are at higher risk of disease. In our previous study on LOAD, we observed a strong interaction between rs9472817 genotype and APOE-ε4 in determining the disease risk [20]. Here, we did not observe any significant interactions. However, subjects carrying at least one APOE-ε4 allele combined with the rs9472817-CC genotype had a significantly higher risk of sporadic FTD. The lack of interaction may be a consequence of an insufficient sample size, and hence the power to unveil a small interaction effect is limited, or it may be attributed to the lower risk conferred by the APOE-ε4 to FTD [38]. Alternatively, and more simply, it may be due to the fact that in FTD there is no interaction between these variants.
There are some limitations to our study. First, the relatively small size of our sample makes it necessary to replicate the current result with a larger sample and, possibly, in other countries. It should be noted however, that this study was designed to replicate our previous data implicating the involvement of UCP4-rs9472817 with LOAD. Furthermore, we did not provide experimental evidence in support of the hypothesis that the detrimental effect of the C allele may depend on the increased expression of UCP4, so functional studies in this sense should be carried out. Although our study does not allow definitive conclusions, data here presented can be a source of inspiration for future studies, to better clarify the role of mitochondrial uncoupling in the pathogenesis of neurodegenerative diseases and the possible causative mechanisms at the origin of these disorders.
Methods
Subject selection
Unrelated patients with late-onset FTD (N=213) and PD (N=96) were recruited at the Regional Neurogenetic Centre (Calabria, southern Italy). Patients without other affected members in the family were classified as sporadic case (100 subjects with FTD and 55 subjects with PD), while patients with a positive family history of disease were classified as familial case (113 subjects with FTD and 41 subjects with PD) (Table 1).
A control group of 442 unrelated healthy subjects matched for age, sex and ethnicity was recruited in the same population.
The study was approved by the local ethics committee and conducted in accordance with the provisions of the Helsinki Declaration and a written informed consent was obtained from all individuals involved in the study; for disabled patients consent was given by their legal tutors.
Clinical assessment
Diagnosis of FTD was assessed by using multiple operational criteria and was based on specific clinical neuropsychological features and neuro-radiological profiles according to the Neary criteria [39] or the revised criteria for behavioural FTD [40]. No known pathological mutations of MAPT (microtubuleassociated protein tau gene), GRN (progranulin) and C9orf72 (chromosome 9 open reading frame 72) were detected in FTD patients. Diagnosis of Parkinson's disease was ascertained according to Gelb’s diagnostic criteria for PD [41].
The same complete set of clinical-laboratory procedures and neurological assessment of cognitive status used for patients were also performed in the control group.
Genotyping and statistical analysis
Genotyping and statistical analysis of the data were performed as previously described [20]. Briefly, the genotype of the UCP4-rs9472817 SNP was determined by Sequenom iPLEX Gold platform. Genotyping of the two SNPs, rs439358 and rs7412, used to determine APOE genotype was conducted according to the protocol described in [42]. Allele and genotype frequencies were estimated by gene counting from the observed genotypes. Hardy–Weinberg (HW) equilibrium was tested by Fisher’s exact test. The association between the analyzed genetic variants and the disease phenotype was assessed by fitting logistic regression models. In these models genotypes at the UCP4-rs9472817 polymorphism were coded in an additive fashion (number of copies of the minor allele), while those at APOE locus were coded in a dominant fashion (carrier/non carrier of ε4 allele). The same model was also used for evaluating possible gene-gene interactions on the disease susceptibility. In such models, age, gender, MMSE score and age of disease’s onset were also used as confounder factors.
Supplementary Materials
Acknowledgements
The work has been made possible by the collaboration with Gruppo Baffa (Sadel Spa, Sadel San Teodoro srl, Sadel CS srl, Casa di Cura Madonna dello Scoglio, AGI srl, Casa di Cura Villa del Rosario srl, Savelli Hospital srl, Casa di Cura Villa Ermelinda).
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This research was funded by grants from the Italian Ministry of University and Research (PRIN: Progetti di Ricerca di rilevante Interesse Nazionale – 2015, Prot. 20157ATSLF) to GR.
References
- 1. Silveri MC. Frontotemporal dementia to Alzheimer’s disease. Dialogues Clin Neurosci. 2007; 9:153–60. [PubMed]
- 2. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013; 12:609–22. https://doi.org/10.1016/S1474-4422(13)70090-5 [PubMed]
- 3. Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016; 131:571–85. https://doi.org/10.1007/s00401-016-1537-1 [PubMed]
- 4. Ling H, Kara E, Bandopadhyay R, Hardy J, Holton J, Xiromerisiou G, Lees A, Houlden H, Revesz T. TDP-43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol Aging. 2013; 34:2889.e5–9. https://doi.org/10.1016/j.neurobiolaging.2013.04.011 [PubMed]
- 5. Ferrari R, Wang Y, Vandrovcova J, Guelfi S, Witeolar A, Karch CM, Schork AJ, Fan CC, Brewer JB, Momeni P, Schellenberg GD, Dillon WP, Sugrue LP, et al, and International FTD-Genomics Consortium (IFGC), and International Parkinson’s Disease Genomics Consortium (IPDGC), and International Genomics of Alzheimer’s Project (IGAP). Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry. 2017; 88:152–64. https://doi.org/10.1136/jnnp-2016-314411 [PubMed]
- 6. Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK, Holland D, Brewer JB, Chen CH, Thompson WK, Harold D, Williams J, Owen MJ, et al, and ADNI, ADGC, GERAD, CHARGE and IPDGC Investigators. Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol Psychiatry. 2015; 20:1588–95. https://doi.org/10.1038/mp.2015.6 [PubMed]
- 7. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009; 41:1303–07. https://doi.org/10.1038/ng.485 [PubMed]
- 8. Yokoyama JS, Wang Y, Schork AJ, Thompson WK, Karch CM, Cruchaga C, McEvoy LK, Witoelar A, Chen CH, Holland D, Brewer JB, Franke A, Dillon WP, et al, and Alzheimer’s Disease Neuroimaging Initiative. Association Between Genetic Traits for Immune-Mediated Diseases and Alzheimer Disease. JAMA Neurol. 2016; 73:691–97. https://doi.org/10.1001/jamaneurol.2016.0150 [PubMed]
- 9. Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB, Dobson-Stone C, Brooks WS, Schofield PR, Halliday GM, Hodges JR, Piguet O, Bartley L, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014; 13:686–99. https://doi.org/10.1016/S1474-4422(14)70065-1 [PubMed]
- 10. Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010; 42:781–85. https://doi.org/10.1038/ng.642 [PubMed]
- 11. Hroudová J, Singh N, Fišar Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. BioMed Res Int. 2014; 2014:175062. https://doi.org/10.1155/2014/175062 [PubMed]
- 12. Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, Verschueren A, Rouzier C, Le Ber I, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014; 137:2329–45. https://doi.org/10.1093/brain/awu138 [PubMed]
- 13. Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016 (Suppl 1); 139:216–31. https://doi.org/10.1111/jnc.13731 [PubMed]
- 14. Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017; 143:418–31. https://doi.org/10.1111/jnc.14037 [PubMed]
- 15. Franco-Iborra S, Vila M, Perier C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front Neurosci. 2018; 12:342. https://doi.org/10.3389/fnins.2018.00342 [PubMed]
- 16. Ganguly G, Chakrabarti S, Chatterjee U, Saso L. Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des Devel Ther. 2017; 11:797–810. https://doi.org/10.2147/DDDT.S130514 [PubMed]
- 17. Kawamata H, Manfredi G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J Cell Biol. 2017; 216:3917–29. https://doi.org/10.1083/jcb.201709172 [PubMed]
- 18. Cioffi F, Senese R, de Lange P, Goglia F, Lanni A, Lombardi A. Uncoupling proteins: a complex journey to function discovery. Biofactors. 2009; 35:417–28. https://doi.org/10.1002/biof.54 [PubMed]
- 19. Cardoso S, Correia S, Carvalho C, Candeias E, Plácido AI, Duarte AI, Seiça RM, Moreira PI. Perspectives on mitochondrial uncoupling proteins-mediated neuroprotection. J Bioenerg Biomembr. 2015; 47:119–31. https://doi.org/10.1007/s10863-014-9580-x [PubMed]
- 20. Montesanto A, Crocco P, Anfossi M, Smirne N, Puccio G, Colao R, Maletta R, Passarino G, Bruni AC, Rose G. The Genetic Variability of UCP4 Affects the Individual Susceptibility to Late-Onset Alzheimer’s Disease and Modifies the Disease’s Risk in APOE-ɛ4 Carriers. J Alzheimers Dis. 2016; 51:1265–74. https://doi.org/10.3233/JAD-150993 [PubMed]
- 21. Liu D, Chan SL, de Souza-Pinto NC, Slevin JR, Wersto RP, Zhan M, Mustafa K, de Cabo R, Mattson MP. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 2006; 8:389–414. https://doi.org/10.1385/NMM:8:3:389 [PubMed]
- 22. Ramsden DB, Ho PW, Ho JW, Liu HF, So DH, Tse HM, Chan KH, Ho SL. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012; 2:468–78. https://doi.org/10.1002/brb3.55 [PubMed]
- 23. Perreten Lambert H, Zenger M, Azarias G, Chatton JY, Magistretti PJ, Lengacher S. Control of mitochondrial pH by uncoupling protein 4 in astrocytes promotes neuronal survival. J Biol Chem. 2014; 289:31014–28. https://doi.org/10.1074/jbc.M114.570879 [PubMed]
- 24. Gratten J, Visscher PM. Genetic pleiotropy in complex traits and diseases: implications for genomic medicine. Genome Med. 2016; 8:78. https://doi.org/10.1186/s13073-016-0332-x [PubMed]
- 25. Ho PW, Ho JW, Tse HM, So DH, Yiu DC, Liu HF, Chan KH, Kung MH, Ramsden DB, Ho SL. Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial Complex II in neuroblastoma cells. PLoS One. 2012; 7:e32810. https://doi.org/10.1371/journal.pone.0032810 [PubMed]
- 26. Chan SL, Liu D, Kyriazis GA, Bagsiyao P, Ouyang X, Mattson MP. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J Biol Chem. 2006; 281:37391–403. https://doi.org/10.1074/jbc.M605552200 [PubMed]
- 27. Rose G, Crocco P, De Rango F, Montesanto A, Passarino G. Further support to the uncoupling-to-survive theory: the genetic variation of human UCP genes is associated with longevity. PLoS One. 2011; 6:e29650. https://doi.org/10.1371/journal.pone.0029650 [PubMed]
- 28. Zündorf G, Reiser G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal. 2011; 14:1275–88. https://doi.org/10.1089/ars.2010.3359 [PubMed]
- 29. Berridge MJ. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion. 2013; 7:2–13. https://doi.org/10.4161/pri.21767 [PubMed]
- 30. Palluzzi F, Ferrari R, Graziano F, Novelli V, Rossi G, Galimberti D, Rainero I, Benussi L, Nacmias B, Bruni AC, Cusi D, Salvi E, Borroni B, Grassi M. A novel network analysis approach reveals DNA damage, oxidative stress and calcium/cAMP homeostasis-associated biomarkers in frontotemporal dementia. PLoS One. 2017; 12:e0185797. https://doi.org/10.1371/journal.pone.0185797 [PubMed]
- 31. Vasileva A, Tiedau D, Firooznia A, Müller-Reichert T, Jessberger R. Tdrd6 is required for spermiogenesis, chromatoid body architecture, and regulation of miRNA expression. Curr Biol. 2009; 19:630–39. https://doi.org/10.1016/j.cub.2009.02.047 [PubMed]
- 32. Fanourgakis G, Lesche M, Akpinar M, Dahl A, Jessberger R. Chromatoid Body Protein TDRD6 Supports Long 3′ UTR Triggered Nonsense Mediated mRNA Decay. PLoS Genet. 2016; 12:e1005857. https://doi.org/10.1371/journal.pgen.1005857 [PubMed]
- 33. Verpillat P, Camuzat A, Hannequin D, Thomas-Anterion C, Puel M, Belliard S, Dubois B, Didic M, Lacomblez L, Moreaud O, Golfier V, Campion D, Brice A, Clerget-Darpoux F. Apolipoprotein E gene in frontotemporal dementia: an association study and meta-analysis. Eur J Hum Genet. 2002; 10:399–405. https://doi.org/10.1038/sj.ejhg.5200820 [PubMed]
- 34. Ji Y, Liu M, Huo YR, Liu S, Shi Z, Liu S, Wisniewski T, Wang J. Apolipoprotein Ε ε4 frequency is increased among Chinese patients with frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2013; 36:163–70. https://doi.org/10.1159/000350872 [PubMed]
- 35. Pulkes T, Papsing C, Mahasirimongkol S, Busabaratana M, Kulkantrakorn K, Tiamkao S. Association between apolipoprotein E genotypes and Parkinson’s disease. J Clin Neurosci. 2011; 18:1333–35. https://doi.org/10.1016/j.jocn.2011.01.028 [PubMed]
- 36. Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis. 2012; 46:389–92. https://doi.org/10.1016/j.nbd.2012.02.002 [PubMed]
- 37. Huang X, Chen PC, Poole C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology. 2004; 62:2198–202. https://doi.org/10.1212/01.WNL.0000130159.28215.6A [PubMed]
- 38. Maiti TK, Konar S, Bir S, Kalakoti P, Bollam P, Nanda A. Role of apolipoprotein E polymorphism as a prognostic marker in traumatic brain injury and neurodegenerative disease: a critical review. Neurosurg Focus. 2015; 39:E3. https://doi.org/10.3171/2015.8.FOCUS15329 [PubMed]
- 39. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998; 51:1546–54. https://doi.org/10.1212/WNL.51.6.1546 [PubMed]
- 40. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011; 134:2456–77. https://doi.org/10.1093/brain/awr179 [PubMed]
- 41. Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999; 56:33–39. https://doi.org/10.1001/archneur.56.1.33 [PubMed]
- 42. Carrieri G, Bonafè M, De Luca M, Rose G, Varcasia O, Bruni A, Maletta R, Nacmias B, Sorbi S, Corsonello F, Feraco E, Andreev KF, Yashin AI, et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum Genet. 2001; 108:194–98. https://doi.org/10.1007/s004390100463 [PubMed]