



Introduction
Angiomyolipoma (AML) is one of the most common benign solid tumors in the kidney [1]. Histologically, it is characterized by different proportions of blood vessels, smooth muscle, and fat [2]. However, the epithelioid AML, especially the pure epithelioid renal AML (PECOMA), could be potentially aggressive [3]. AML tends to be more common in female patients with a female to male ratio of 2:1 [4]. Abdominal pain and hematuria are the most reported symptoms. With the widespread use of CT and ultrasound, more incidental AMLs are diagnosed [5].
About 80% of renal AMLs are sporadic, and the rest are associated with hereditary tumor syndrome, such as tuberous sclerosis complex (TSC) [2]. TSC is an autosomal dominant tumor syndrome caused by the germline mutation of TSC1/2, with a birth incidence of 1 in 10000 to 1 in 6000 [6]. Patients are predisposed to types of tumors in different organs in their early ages, including the kidney, brain, skin, and lung [7]. Renal AML (RAMLs) is found in over half of TSC patients and is one of the major criteria for the clinical diagnostic criterion recommended by the International Consensus Conference in 2012 [8, 9]. TSC-related RAMLs show different phenotype from the sporadic ones, and are characterized by the early onset age (20–30 years old), bilateral and multifocal lesions, and concurrent tumors in skin, brain and lung [10]. Moreover, the TSC-related RAMLs are reported to be with a larger initial size, grow faster and have higher a risk of spontaneous hemorrhage, which can be life threatening, than the sporadic ones [11]. Thus, it is important to recognize the patients with TSC from those affected by RAMLs.
Since the clinical diagnostic criterion can only be used after the TSC-related tumors occur, clinicians now try to use the next generation sequencing (NGS) techniques to identify TSC patients at an early stage. A positive pathogenic TSC1/2 germline mutation will lead to a definite diagnosis of TSC [8]. According to the 2012 International TSC Consensus, patients with bilateral or multifocal RAMLs are recommended to take genetic testing. However, no evidence has been reported about the germline TSC1/2 gene status among patients with bilateral RAMLs. Besides, the NGS strategy is unreliable for the detection of the large deletion/amplification, which needs confirmation of the multiplex ligation-dependent probe amplification (MLPA). The standard genetic testing method is not suggested in the existing guidelines and should be chosen according to the mutation spectrum of one region. Unfortunately, The Chinese TSC1/2 mutation spectrum is still uncertain.
In this study, we evaluated the status of TSC1/2 genes among patients with bilateral RAMLs and found that about ¼ of patients carried a germline TSC1/2 variant. The result, for the first time, provided evidence for these patients to take genetic testing. Meanwhile, we described the mutation spectrum of TSC1/2 genes among Chinese TSC patients and found that almost all types of TSC1/2 variants occurred in Chinese patients. Thus NGS based techniques combined with MLPA should be recommended to be the standard genetic testing strategy for Chinese patients with TSC. These results will be helpful for the clinical-decision-making of bilateral renal AMLs and provide valuable information for genetic counseling for those Chinese patients with TSC.
Results
Clinical characteristics of patients with bilateral RAML in this study
A total of 78 individuals were enrolled for analysis, including 28 male and 50 female patients. The baseline information was demonstrated in Table 1. The onset age of RAML in our study was 32.5±14.5 years old (median 29.5, range 1 to 77), which was much earlier than the sporadic RAMLs [4]. Among all the patients, 62 patients were diagnosed before 45 years, while the other 16 ones were affected over 45. Most of the patients (73/78) had no family history. A total of 49 patients were affected by only bilateral RAMLs, while the other 29 patients were affected by bilateral RAMLs and at least one TSC-related tumors (Table 1).
Table 1. Clinical and genetic characteristics of patients with bilateral renal AMLs.
| Characteristics | No. of patients |
| Sex | |
| male | 28 |
| female | 50 |
| Onset age of AML | |
| ≤45y | 62 |
| >45y | 16 |
| Family history | |
| yes | 5 |
| no | 73 |
| Concomitant tumors | |
| yes | 29 |
| no | 49 |
| TSC1/2 gene test | |
| positive | 30 |
| negative | 48 |
Germline mutation of TSC1 or TSC2 gene in different groups of bilateral RAML patients
We carried out next-generation sequencing to detect the status of TSC1/2 genes and found 30 germline alterations (3 TSC1 and 27 TSC2). For early-onset patients who were diagnosed before 45 years old, 28.6% of them were with positive results of the TSC1/2 test. The rate decreased to 14.3% (2 out of 14) for those with onset age over 45 years old. As to the 29 patients affected by both bilateral RAMLs and other TSC-related tumors, which met the clinical criteria for TSC, 18 (62.1%) alterations in TSC1/2 were observed (Table 2).
Table 2. Germline mutation of TSC1/2 in different groups of patients with bilateral RAMLs.
| Clinical features | TSC1/2 gene test | Total | |
| + | - | ||
| Bilateral RAML | |||
| ≤45y | 10(28.6%) | 25 (71.4%) | 35 |
| >45y | 2 (14.3%) | 12 (85.7%) | 14 |
| Total | 12 (24.5%) | 37 (75.5%) | 49 |
| Bilateral RAML with other TSC-related tumors | 18(62.1%) | 11 (37.9%) | 29 |
| Total | 30 | 48 | 78 |
Among the 30 alterations, 24 were pathogenic or likely pathogenic according to the ACMG guideline, and the others were variants of uncertain significance (VUS). To illustrate the effect of the 6 VUS on protein function, we carried out the in silico prediction programs and found that 5/6 of the VUS were predicted to be harmful. The last one VUS was a synonymous mutation predicted to be benign, but according to the clinical criteria, that patient was diagnosed with TSC. We supposed this mutation might lead to TSC syndrome with a special pathogenetic mechanism. Moreover, most of the alterations were truncating, including eight frameshift mutations, eight nonsense mutations, four splicing mutations, and two deletion/duplications. The mutations in the TSC1 or TSC2 gene were compared with those in the Tuberous Sclerosis Database (http://chromium.lovd.nl/LOVD2/TSC/home), and 15 (50%) of them were first reported in this study. The detailed genotypic information and clinical manifestations were shown in Table 3.
Table 3. Genotypic and phenotypic features of patients with germline mutation of TSC1/2.
| No. | Gene | Nucleotide change | Protein change | Mutation type | Novel | Clinical manifestation | ACMG |
| 1 | TSC2/E5 | c.433G>T | p. Arg611Trp | Missense | Novel | RAML, ungual fibromas | Likely pathogenic |
| 2 | TSC2/E11 | EX11 DEL | — | Deletion | Novel | RAML, angiofibroma, forehead plaque, shagreen patch, SEGA, seizures | Likely pathogenic |
| 3 | TSC2/E11_15 | E11-15 DUP | — | Deletion | Novel | RAML, angiofibroma, | Likely pathogenic |
| 4 | TSC2/E14 | c.1372C>T | p. Arg458Ter | Nonsense | Reported | RAML | Likely pathogenic |
| 5 | TSC2/E15 | c.1513C>T | p. Arg505Ter | Nonsense | Reported | RAML, angiofibroma | Likely pathogenic |
| 6 | TSC2/E17 | c.1831C>T | p. Arg611Trp | Missense | Reported | RAML, angiofibroma, ungual fibromas | Likely pathogenic |
| 7 | TSC2/E19 | c.2083C>T | p. Gln695Ter | Nonsense | Reported | RAML | Likely pathogenic |
| 8 | TSC2/E20 | c.2102_2105 delCTGA | p. Ser701Ser fsX5 | Frameshift | Reported | RAML | Likely pathogenic |
| 9 | TSC2/E20 | c.2138T>C | p. Leu713Pro | Missense | Novel | RAML, angiofibroma | VUS |
| 10 | TSC2/E27 | c.3046delA | p. Lys1061Lys fs X14 | Frameshift | Novel | RAML, seizures | Likely pathogenic |
| 11 | TSC2/E30 | c.3412C>T | p. Arg1138Ter | Nonsense | Reported | RAML | Likely pathogenic |
| 12 | TSC2/E30 | c.3412C>T | p. Arg1138Ter | Nonsense | Reported | RAML, seizures | Likely pathogenic |
| 13 | TSC2/E30 | c.3582G>A | p. Trp1194Ter | Nonsense | Reported | RAML | Likely pathogenic |
| 14 | TSC2/E31 | c.3685C>T | p. Gln1229Ter | Nonsense | Reported | RAML, angiofibroma, ungual fibromas, Hypomelanotic macules, seizures | Likely pathogenic |
| 15 | TSC2/E31 | c.3803G>A | p. Arg1268His | Missense | Novel | RAML, angiofibroma | VUS |
| 16 | TSC2/E34 | c.4418_4419 delAG | p. Lys1473Lys fsX50 | Frameshift | Reported | RAML, angiofibroma | Likely pathogenic |
| 17 | TSC2/E34 | c.4425_4426 delAG | p. Arg1477Gly fs X46 | Frameshift | Reported | RAML, angiofibroma | Likely pathogenic |
| 18 | TSC2/E34 | c.4493_c.4493+18 delGGTGGGCCTCTTGCTTCCG | — | Frameshift | Novel | RAML, forehead plaque, | Likely pathogenic |
| 19 | TSC2/E37 | c.4737C>T | p. Gly1579Gly | Missense | Novel | RAML, angiofibroma, | VUS |
| 20 | TSC2/E37 | c.4783G>A | p. Gly1595Arg | Missense | Novel | RAML, angiofibroma | VUS |
| 21 | TSC2/E40 | c.5155G>C | p. Ala1719Pro | Missense | Novel | RAML, angiofibroma | VUS |
| 22 | TSC2/E41 | c.5175_5176del/GC | p. His1726Ser fsX2 | Frameshift | Novel | RAML, angiofibroma | Likely pathogenic |
| 23 | TSC2/E41 | c.5237_5238insC | p. His1746His fsX29 | Frameshift | Novel | RAML, angiofibroma, angual fibromas, Hypomelanotic macules, shagreen patch, seizures | Likely pathogenic |
| 24 | TSC2/IN9 | c.849-1G>A | — | Splicing | Reported | RAML | Likely pathogenic |
| 25 | TSC2/IN14 | c.1444-1G>C | — | Splicing | Novel | RAML, forehead plaque, | Likely pathogenic |
| 26 | TSC2/IN15 | c.1600-1G>C | — | Splicing | Novel | RAML, angiofibroma, | Likely pathogenic |
| 27 | TSC2/IN30 | c.3610+1G>A | — | Splicing | Reported | RAML | Likely pathogenic |
| 28 | TSC1/E6 | c.372delT | p. Thr124Thr fsX13 | Frameshift | Novel | RAML | Likely pathogenic |
| 29 | TSC1/E6 | c.433C>T | p. Gln145Ter | Nonsense | Reported | RAML | Pathogenic |
| 30 | TSC1/E15 | c.1960C>G | p. Gln654Glu | Missense | Reported | RAML | VUS |
TSC1/2 mutation spectrum in Chinese patients and the genotypic and phenotypic characteristics
To demonstrate the mutation spectrum of Chinese TSC patients, we reviewed all the reported TSC patients in China. A total of 315 alterations were involved for analysis, including 45 TSC1 and 270 TSC2 (Supplementary Table 1). TSC1 patients were more likely to be affected by nonsense mutations than TSC2 patients (51.1% vs. 20.7%, p<0.001), while the frameshift and missense mutations were more common in TSC2 patients (40% vs. 20%, 24.1% vs. 13.3%) (Figure 1A and 1B). Moreover, patients with TSC1 mutation had a significantly higher positive rate of family history compared to those with TSC2 mutation (37.8% vs. 19.6%, p=0.0067) (Table 4). The mutation spectrum of patients with TSC1 or TSC2 gene was next analyzed. For the TSC1 gene, exon 15, 8 and 18 seemed to be the hotspot mutation regions. For the TSC2 gene, we observed alterations in every exon except for exon 25. Specifically, exon 40, 33, and 29 were the most common mutation regions, which accounted for about 30% (60) of all the variants (Figure 1C and 1D).
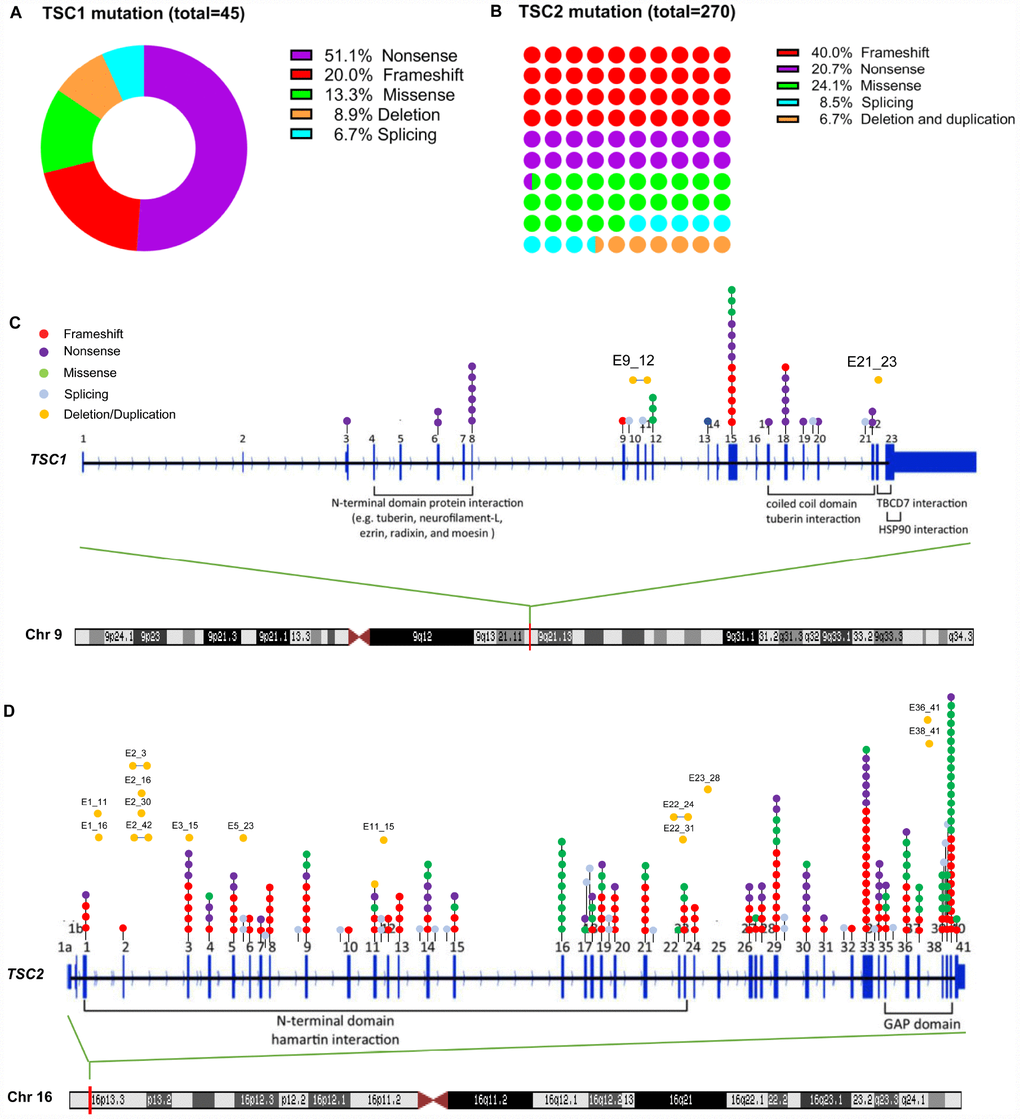
Figure 1. TSC1 and TSC2 gene mutation spectrum in Chinese patients. (A) mutation types of TSC1 gene; (B) mutation types of TSC2 gene; (C) mutation sites of TSC1 gene; (D) mutation sites of TSC2 gene.
Table 4. Differences of mutation types and family history between Chinese patients carrying TSC1 and TSC2 mutations.
| TSC1 | TSC2 | P value | |
| Mutation type | |||
| Frameshift | 9 (20%) | 108 (40%) | 0.0004 |
| Nonsense | 23 (51.1%) | 56 (20.7%) | |
| Missense | 6 (13.3%) | 65 (24.1%) | |
| Splicing | 3 (6.7%) | 23 (8.5%) | |
| Deletion and duplication | 4 (8.9%) | 18 (6.7%) | |
| Total | 45 | 270 | |
| Family history | |||
| Positive | 17 (37.8) | 53 (19.6%) | 0.0067 |
| Negative | 28 (62.2%) | 217 (80.4%) | |
| Total | 45 | 270 |
To clarify the genotypic and phenotypic differences between TSC patients from China and the western countries, we analyzed the data of sex, family history, and mutated genes in Chinses TSC patients and the TOSCA cohort. The results showed that 85.7% of Chinese TSC patients carried TSC2 alterations, which was higher than that of TOSCA (76.2%, p<0.001) (Table 5). Moreover, Chinese TSC patients were more likely to have an affected parent than those from TOSCA (22.2% vs. 13.9%, p<0.001) (Table 5). Female TSC patients were more common than male patients, but no difference was observed between the Chinese cohort and TOSCA cohort.
Table 5. Differences in mutation spectrum and phenotypic characteristics between TSC patients in China and the TOSCA project.
| China | TOSCA | P value | |
| Gene mutation | |||
| TSC1 | 45 (14.3%) | 178 (23.8%) | 0.0005 |
| TSC2 | 270(85.7%) | 571 (76.2%) | |
| Family history | |||
| positive | 70 (22.2%) | 290 (13.9%) | 0.0001 |
| negative | 245 (77.8%) | 1803 (86.1%) | |
| Sex | |||
| male | 88 (47.6%) | 1009 (48.2%) | 0.867 |
| female | 97 (52.4%) | 1084 (51.8%) |
Discussion
Renal AML is a relatively rare tumor with an overall prevalence of 0.1% to 0.44%. Only 5.2% of the patients present with multiple AMLs, and about 1.5% of them are bilateral [4]. According to the current guidelines, active surveillance is the most chosen option for most of the sporadic renal AMLs for the low growth rate and low probability of spontaneous rupture of the tumors, no matter what the initial tumor size is. And nephron-sparing surgery is the most effective treatment to reduce the rate of recurrence and secondary treatment [12]. However, for the TSC associated RAMLs, mTOR inhibitor treatment is the recommended first-line therapy for asymptomatic tumors larger than 3 cm [13, 14]. Thus, the differentiation of the TSC associated RAML and the sporadic one is of great importance for patients. Nowadays, patients with unilateral RAML are not recommended to take genetic testing if they do not develop other TSC-related tumors at the same time. But, when bilateral or multifocal RAMLs exist, gene testing is recommended according to the 2012 International TSC Consensus [8, 14]. However, the positive rate of genetic testing for patients with bilateral RAMLs remained unclear. In our study, we observed that ¼ patients with bilateral RAMLs carried TSC1/2 germline alterations. Moreover, the positive probability increased to 28% when patients were affected before 45 years old. Meanwhile, in our previous study, about 10% of patients with early-onset (<45 years old) unilateral AML were found to carry TSC1/2 germline alterations [15]. These results provide strong evidence for the utility of TSC1/2 testing for all patients with bilateral RAMLs and those with early-onset unilateral RAML.
Genotype-phenotype correlations in TSC syndrome have been studied well in the past decades. Generally, TSC2 pathogenic variants are related to a more severe phenotype than TSC1 [16]. As to renal lesions, patients carrying TSC2 mutations usually have larger initial tumor size, higher risk for a growing RAML, and higher risk for hemorrhage [17]. Meanwhile, the onset age of RAML among patients with TSC2 germline mutations are almost ten years earlier than those with TSC1 mutations [18]. Although the genotype-phenotype correlations are still challenging because of the age dependence of onset of TSC-associated manifestations, they provide valuable information for genetic counseling and give us a clue for the pathogenesis of TSC-related tumors. In this study, we observed that only a small proportion of TSC patients inherited the mutated gene from their parent, and positive family history was more common among patients with TSC1 germline mutations compared to those with TSC2 mutations. This phenotypic diversity can be partly explained by the poorer prognosis of patients carrying TSC2 mutations. In most circumstances, the severe symptoms and early onset age make the patients unable to bear a child or unwilling to give birth to an unhealthy baby when the prenatal diagnosis or preimplantation genetic diagnosis is not available. The difference in genetic anticipation between TSC1 and TSC2 families may also play a role in the phenotypic disparity. Future studies on the genetic and clinical characteristics of successive generations among TSC1 and TSC2 families are needed to confirm the hypothesis.
In line with the previous study, most of the alterations observed in our study were truncating variants, which indicates that the complete loss of function of the TSC1/2 genes is essential for the tumorigenesis of most TSC associated lesions. Interestingly, we found a silent mutation located in exon 37 of the TSC2 gene (c.4737C>T), which led to the alteration from GGC to GGT without transforming the amino acid. Silent mutations are generally considered to be normal variants and are thought to have no role in disease. However, that patient carrying this variation had been diagnosed with TSC syndrome based on the clinical criteria. In order to clarify the other atypical mutation sites in TSC1/2 genes, we screened the whole exons and the surrounding introns and found that the c.4737C>T was the only alteration of TSC1/2 genes. Although the pathogenesis of silent mutation in TSC syndrome has not been found, synonymous mutations in other genes have been reported to influence gene expression during pre-mRNA splicing and play a role in disease through the exonic splicing enhancer (ESE) and exonic splicing silencer motifs [19]. Thus, the c.4737C>T alteration may cause the skipping or retention of related exons in TSC2, leading to the dysfunction of protein. More experimental work needs to be done the reveal the detailed mechanism.
In the past several years, the NGS based methods have been recommended to screen germline or somatic alterations of TSC1/2 genes in selected individuals. However, MLPA is usually recommended in the condition of a negative result of NGS. The mutation spectrum of TSC1/2 genes in Chinese TSC patients was demonstrated in our study. Almost all kinds of mutation types were found, including frameshift mutations, nonsense mutations, small indels, large deletions/amplifications, splicing mutations, and synonymous mutations. Thus, the NGS based methods, followed by MLPA if negative, should be the routine strategies for patients suspected to be affected by TSC.
In conclusion, patients affected by merely bilateral RAMLs should receive genetic testing of TSC ½ genes, especially when they are with an early onset age. Meanwhile, the mutation spectrum of Chinese TSC patients suggests that NGS based methods and MLPA should be the recommended strategies of genetic testing. The results will be helpful to genetic counseling in clinical decision making and help to reduce the missed diagnosis rate of TSC related RAMLs and to provide accurate therapeutic strategies for patients.
Materials and Methods
Medical ethics
This project was approved by the scientific and ethics committee of the Fudan University Shanghai Cancer Center. Informed consent was obtained from patients or legal guardians.
Patient ascertainment and assessment
In this study, we recruited consecutive patients from Jan 2018 to Jan 2019, who met the following criteria: diagnosed with bilateral renal AML by image test or pathology; agreed to receive the genetic test. A total of 78 patients who met the above criteria were enrolled for analysis. Clinical characteristics including birth year, sex, onset age, and family history were collected through interviews with the probands and check of medical records. The onset age was defined as the diagnosed age of first renal AML if the patient was affected by metachronous bilateral AMLs. Positive family history was identified when one of the parents was diagnosed with TSC by the clinical criteria recommended by the International Consensus Conference in 2012, or with a pathogenic germline TSC1/2 alteration.
DNA preparation and NGS
Genomic DNA was extracted from peripheral blood of enrolled patients according to the standard procedure using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). Then the DNA was fragmented by Covaris LE220 (Massachusetts, USA) to generate a paired-end library (200–250 bp). The library was enriched by array hybridization according to previously published procedure [20], followed by elution and post-capture amplification. We next estimated the magnitude of enrichment with Agilent 2100 Bioanalyzer and ABI StepOne. All amplified libraries were subsequently sent to BGI for circularization and sequencing on the BGISEQ-500 platform. For circularization, PCR products with different barcodes were pooled together at an equimolar concentration to yield a final amount of 80 ng. Each pool was subsequently heat-denatured, and the single-strand DNA was mixed with MGIEasyTM DNA Library Prep Kit V1 (PN:85–05533-00, BGI, Shenzhen, China), containing 5 μl splint oligo, 6 μl splint Buffer, 0.6 μl ligation Enhancer, and 0.2 μl ligation (Enzyme and NF water) to form a 60 μl reaction system, which was subsequently incubated at 37°C for 30 minutes. Last, 20 μl of each single-circle-library pool was used as input to prepare the DNB. Each pool was then sequenced on 1 lane, using 100SR chemistry with BGISEQ-500RS High-throughput sequencing kit (PN: 85–05238-01, BGI). Post sequencing, the data were automatically demultiplexed by index.
Bioinformatics and variant filtering
To detect the potential variants in the family, we performed bioinformatics processing and data analysis after receiving the primary sequencing data. We used previously published filtering criteria to generate “clean reads” for further analysis [20]. The “clean reads” (with a length of 90 bp) derived from targeted sequencing and filtering were then aligned to the human genome reference (hg19) using the BWA (Burrows-Wheeler Aligner) Multi-Vision software package [21]. After alignment, the output files were used to perform sequencing coverage and depth analysis of the target region, single-nucleotide variants (SNVs) and INDEL calling. We used SOAPsnp software [22] and Samtools [23] to detect SNVs and indels. All SNVs and indels were filtered and estimated via multiple databases, including NCBI dbSNP, HapMap, 1000 human genome dataset, and database of 100 healthy Chinese adults.
To predict the effect of missense variants, we used dbNSFP [24], which contains eleven well-established in silico prediction programs. Pathogenic variants were assessed under the protocol issued by ACMG [25]. The Human Gene Mutation Database (HGMD) was used to screen mutations reported in published studies.
All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods. The PCR protocol consisted of an initial denaturation at 95°C for 3 minutes, followed by 35 cycles (95°C for 40 seconds, 55°C for 30 seconds, and 72°C for 30 seconds), with a final extension at 72°C for 10 minutes.
Statistical analysis
Chi-square test was used to compare the differences in mutation type and family history between patients with TSC1 and TSC2 gene mutation, and to compare the differences of the mutated gene, family history and sex between TSC patients from China and the TOSCA cohort. Statistical analysis was performed using SPSS20.0, and p < 0.05 was considered to be statistically significant.
Supplementary Materials
Author Contributions
SGH and YDW conceived of the project and contributed to study design; WJY performed data collection and wrote the manuscript; GG performed statistical analyses and revised the manuscript; all authors read and approved the final version of the manuscript.
Acknowledgments
The authors thank Zhao Li, a social worker, for her help to collect data.
Conflicts of Interest
The authors declare that there is no conflicts of interest.
Funding
This work was supported by the National Natural Science Foundation of China (grant number: 81902574), the Shanghai Basic Research Program (19JC1411600) and the Shanghai Sailing Program (19YF1409800).
References
- 1. Bhatt JR, Richard PO, Kim NS, Finelli A, Manickavachagam K, Legere L, Evans A, Pei Y, Sykes J, Jhaveri K, Jewett MA. Natural History of Renal Angiomyolipoma (AML): Most Patients with Large AMLs >4cm Can Be Offered Active Surveillance as an Initial Management Strategy. Eur Urol. 2016; 70:85–90. https://doi.org/10.1016/j.eururo.2016.01.048 [PubMed]
- 2. Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004; 66:924–34. https://doi.org/10.1111/j.1523-1755.2004.00838.x [PubMed]
- 3. Nese N, Martignoni G, Fletcher CD, Gupta R, Pan CC, Kim H, Ro JY, Hwang IS, Sato K, Bonetti F, Pea M, Amin MB, Hes O, et al. Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of the kidney: A clinicopathologic study of 41 cases: detailed assessment of morphology and risk stratification. Am J Surg Pathol. 2011; 35:161–76. https://doi.org/10.1097/PAS.0b013e318206f2a9 [PubMed]
- 4. Fittschen A, Wendlik I, Oeztuerk S, Kratzer W, Akinli AS, Haenle MM, Graeter T. Prevalence of sporadic renal angiomyolipoma: a retrospective analysis of 61,389 in- and out-patients. Abdom Imaging. 2014; 39:1009–13. https://doi.org/10.1007/s00261-014-0129-6 [PubMed]
- 5. Halpenny D, Snow A, McNeill G, Torreggiani WC. The radiological diagnosis and treatment of renal angiomyolipoma-current status. Clin Radiol. 2010; 65:99–108. https://doi.org/10.1016/j.crad.2009.09.014 [PubMed]
- 6. Lam HC, Siroky BJ, Henske EP. Renal disease in tuberous sclerosis complex: pathogenesis and therapy. Nat Rev Nephrol. 2018; 14:704–16. https://doi.org/10.1038/s41581-018-0059-6 [PubMed]
- 7. Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016; 2:16035. https://doi.org/10.1038/nrdp.2016.35 [PubMed]
- 8. Northrup H, Krueger DA, Northrup H, Krueger DA, Roberds S, Smith K, Sampson J, Korf B, Kwiatkowski DJ, Mowat D, Nellist M, Northrup H, Povey S, et al, and International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013; 49:243–54. https://doi.org/10.1016/j.pediatrneurol.2013.08.001 [PubMed]
- 9. Kingswood JC, Bruzzi P, Curatolo P, de Vries PJ, Fladrowski C, Hertzberg C, Jansen AC, Jozwiak S, Nabbout R, Sauter M, Touraine R, O’Callaghan F, Zonnenberg B, et al. TOSCA - first international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet J Rare Dis. 2014; 9:182. https://doi.org/10.1186/s13023-014-0182-9 [PubMed]
- 10. Seyam RM, Bissada NK, Kattan SA, Mokhtar AA, Aslam M, Fahmy WE, Mourad WA, Binmahfouz AA, Alzahrani HM, Hanash KA. Changing trends in presentation, diagnosis and management of renal angiomyolipoma: comparison of sporadic and tuberous sclerosis complex-associated forms. Urology. 2008; 72:1077–82. https://doi.org/10.1016/j.urology.2008.07.049 [PubMed]
- 11. Sheth RA, Feldman AS, Paul E, Thiele EA, Walker TG. Sporadic versus Tuberous Sclerosis Complex-Associated Angiomyolipomas: Predictors for Long-Term Outcomes following Transcatheter Embolization. J Vasc Interv Radiol. 2016; 27:1542–49. https://doi.org/10.1016/j.jvir.2016.05.029 [PubMed]
- 12. Fernández-Pello S, Hora M, Kuusk T, Tahbaz R, Dabestani S, Abu-Ghanem Y, Albiges L, Giles RH, Hofmann F, Kuczyk MA, Lam TB, Marconi L, Merseburger AS, et al. Management of Sporadic Renal Angiomyolipomas: A Systematic Review of Available Evidence to Guide Recommendations from the European Association of Urology Renal Cell Carcinoma Guidelines Panel. Eur Urol Oncol. 2019. [Epub ahead of print]. https://doi.org/10.1016/j.euo.2019.04.005 [PubMed]
- 13. Krueger DA, Northrup H, Northrup H, Krueger DA, Roberds S, Smith K, Sampson J, Korf B, Kwiatkowski DJ, Mowat D, Nellist M, Northrup H, Povey S, et al, and International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013; 49:255–65. https://doi.org/10.1016/j.pediatrneurol.2013.08.002 [PubMed]
- 14. Amin S, Kingswood JC, Bolton PF, Elmslie F, Gale DP, Harland C, Johnson SR, Parker A, Sampson JR, Smeaton M, Wright I, O'Callaghan FJ. The UK guidelines for management and surveillance of Tuberous Sclerosis Complex. QJM. 2019; 112:171–182. https://doi.org/10.1093/qjmed/hcy215 [PubMed]
- 15. Wu J, Wang H, Ricketts CJ, Yang Y, Merino MJ, Zhang H, Shi G, Gan H, Linehan WM, Zhu Y, Ye D. Germline mutations of renal cancer predisposition genes and clinical relevance in Chinese patients with sporadic, early-onset disease. Cancer. 2019; 125:1060–69. https://doi.org/10.1002/cncr.31908 [PubMed]
- 16. Peron A, Au KS, Northrup H. Genetics, genomics, and genotype-phenotype correlations of TSC: insights for clinical practice. Am J Med Genet C Semin Med Genet. 2018; 178:281–90. https://doi.org/10.1002/ajmg.c.31651 [PubMed]
- 17. Li S, Zhang Y, Wang Z, Yang Y, Gao W, Li D, Wei J. Genotype-phenotype correlation of patients with tuberous sclerosis complex-associated renal angiomyolipoma: a descriptive study. Hum Pathol. 2018; 82:61–67. https://doi.org/10.1016/j.humpath.2018.07.017 [PubMed]
- 18. Kingswood JC, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, Dahlin M, D’ Amato L, d’Augères GB, de Vries PJ, Ferreira JC, Feucht M, Fladrowski C, et al. Renal angiomyolipoma in patients with tuberous sclerosis complex: findings from the TuberOus SClerosis registry to increase disease Awareness. Nephrol Dial Transplant. 2019; 34:502–08. https://doi.org/10.1093/ndt/gfy063 [PubMed]
- 19. Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014; 156:1324–35. https://doi.org/10.1016/j.cell.2014.01.051 [PubMed]
- 20. Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T, Chen Y, Jiang H, Yang G, Zhen R, Lan Z, Qi M, Wang J, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One. 2011; 6:e29500. https://doi.org/10.1371/journal.pone.0029500 [PubMed]
- 21. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. https://doi.org/10.1093/bioinformatics/btp324 [PubMed]
- 22. Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009; 19:1124–32. https://doi.org/10.1101/gr.088013.108 [PubMed]
- 23. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–79. https://doi.org/10.1093/bioinformatics/btp352 [PubMed]
- 24. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat. 2016; 37:235–41. https://doi.org/10.1002/humu.22932 [PubMed]
- 25. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–24. https://doi.org/10.1038/gim.2015.30 [PubMed]