



Introduction
Appropriate cellular responses to oxygen levels are essential for tissue homeostasis. Hypoxia-inducible factors (HIFs) are evolutionarily-conserved proteins that govern cellular response to low oxygen tension by initiating the transcription of genes associated with angiogenesis, erythropoiesis, and metabolism. While HIFs are essential for metabolic regulation in both adults and embryos, they also play key roles in pathologic processes such as tumorigenesis, metastasis, tumor-associated angiogenesis, and congenital erythrocytosis [1]. Germline and somatic mutations in genes involved in the hypoxia-sensing pathway, including Von Hippel-Lindau (VHL) and Egl-9 homolog 1 (EGLN1; also known as prolyl hydroxylase domain-containing protein 2 [PHD2]), have been described in neuroendocrine tumor syndromes associated with polycythemia [2–6]. Recently, somatic gain-of-function mutations in the oxygen-dependent domain (ODD) of HIF-2α were discovered in the context of multiple paraganglionomas, somatostatinomas, and polycythemia [7–9]. Dysregulation of the oxygen-sensing pathway occurs in many neuroendocrine tumors, but the exact mechanisms by which this dysregulation promotes oncogenesis remain largely unknown.
The liver is a primary site of hematopoiesis in embryos and extramedullary hematopoiesis in adults [10], and circulating hematopoietic stem cells may be capable of hepatocyte differentiation [11]. Polycythemia has been observed in patients with hepatocellular carcinoma (HCC) and is thought to result from the tumor’s secretion of erythropoietin [12, 13]. HCC is a relatively rare disease with an incidence between 3 and 18 per 100,000 in the United States [14, 15], but it may be as frequent as 24 per 100,000 in endemic countries [16]. Major risk factors for HCC include infection with oncogenic hepatitis B virus as well as chronic hepatitis due to hepatitis C virus infection, alcoholism, smoking, non-alcoholic fatty liver disease, and aflatoxin B1 intake [17–24]. In light of recent findings that HIF2A mutations can cause polycythemia in certain tumor syndromes, polycythemia in HCC might also involve genetic alternations in hypoxia-induced signaling pathways. In this study, we therefore sought to identify mutations in HIF2A in HCC patients presenting with polycythemia. We also examined in depth a previously undescribed germline c.G1645A (p.E549K) mutation in a critical oxygen-sensing region identified in one of the 302 HCC patients screened; this patient also exhibited polycythemia that resolved upon tumor resection.
Results
HIF2A mutational analysis in HCC patients
Three hundred and two HCC patients underwent genomic DNA sequencing to identify mutations in HIF2A. Of the 302 patients, 104 exhibited polycythemia, while the remaining 198 patients did not. A unique heterozygous c.G1645A (p.E549K) mutation in the HIF2A gene was identified in the tumor DNA of only one patient (index patient) who exhibited concurrent polycythemia.
Clinical history of index patient
The index patient was a 43-year-old male who was referred to the Division of Hepatobiliary and Pancreatic Surgery clinic at the First Affiliated Hospital of Zhejiang University in August 2012 for a suspected hepatic hemangioma identified by abdominal computed tomography (CT). The patient presented with non-radiating abdominal pain on the right side below the xyphoid process that did not worsen with eating or changing position. He also complained of mild general weakness but denied any nausea or vomiting. Physical exam revealed no jaundice. Laboratory results are presented in Table 1. A viral hepatitis panel was positive for hepatitis B surface (HBs) antigen and antibodies to hepatitis B core antigen (HBc) and envelope antigen (HBe); there were no detectable antibodies to hepatitis C virus (HCV). Serum α-fetoprotein (AFP) was elevated at 89.5 ng/mL (reference range: < 20 ng/mL). A complete blood count (CBC) revealed a hemoglobin level of 188 g/L (reference range: 131-172 g/L), a red blood cell count of 5.8 × 1012/L (reference range: 4.09-5.74 × 1012/L), and a hematocrit of 54.2% (reference range: 38.0-50.8%). Abdominal ultrasonography revealed a hypo-echoic mass in the right liver and possible cholecystitis. A subsequent non-contrast abdominal CT revealed a 5.2 cm × 3.4 cm hypodense mass at the same location (Figure 1A). Hepatectomy was performed to remove the tumor.
Table 1. Laboratory values of the index patient and family members.
| Index case | Sister (II-C) | Sister (II-E) | Niece (III-C) | Reference range | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pre-operative | 4 days post-operative | 7 weeks post-operative | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Serum α-fetoprotein (ng/ml) | 89.5 | Undetectable | Undetectable | 2.9 | ND | ND | < 20 ng/mL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Viral hepatitis panel | ND | ND | ND | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HBs Ag | Positive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HBs Ab | Negative | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HBc Ab | Positive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HBe Ag | Negative | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HBe Ab | Positive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HCV Ab | Negative | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complete blood count | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Erythrocytes × 1012/L | 5.8 | 4.1 | 5.18 | 4.38 | 4.44 | 4.16 | 4.09 – 5.74 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hematocrit (%) | 54.2 | 37.5 | 48.3 | 39.7 | 35.3 | 37.8 | 38.0 – 50.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hemoglobin (g/L) | 188 | 135 | 165 | 143 | 115 | 133 | 131 – 172 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Leukocytes × 109/L | 7.7 | 20.7 | 7.3 | 5.1 | 4.4 | 5.4 | 4 – 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Platelets × 109/L | 227 | 186 | 178 | 217 | 379 | 314 | 83 – 303 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The reference ranges presented are those used at the Massachusetts General Hospital. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
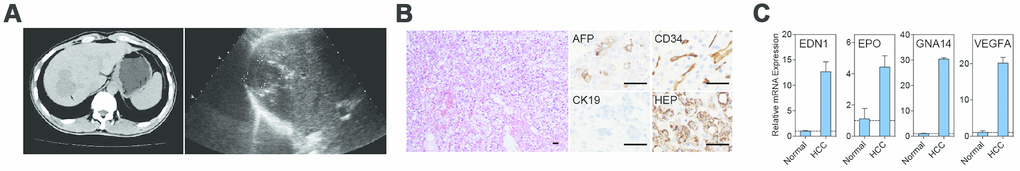
Figure 1. Imaging, histopathology, and mutant HIF2A signaling in the index patient. (A) Abdominal CT imaging and ultrasound revealed a hypodense and hypoechoic mass in the patient’s right liver. (B) Hematoxylin and eosin staining of the resected tumor revealed hepatocellular carcinoma, clear cell variant, with prominent angiogenesis. Tumor cells were positive for AFP and HEP and negative for CK19 by immunohistochemistry; endothelial cells stained positively for CD34. (C) Real-time PCR demonstrated overexpression of the HIF2A transcriptional targets EDN1, EPO, GNA14, and VEGFA within the tumor bed. Scale bars: 50 μm.
The patient was stable post-operatively and his recovery was unremarkable. Histological examination of the tumor specimen revealed tumor cells with prominent clear cytoplasm arranged in a trabecular growth pattern (Figure 1B). Tumor cells were positive for AFP and Hep par 1 (HEP) stains but negative for cytokeratin 19 (CK19). The tumor also contained numerous capillary channels positive for CD34 staining. These observations supported a diagnosis of hepatocellular carcinoma, clear cell variant. On post-operative day four, CBC revealed a hemoglobin level of 135 g/L, a red blood cell count of 4.1 × 1012/L, and a hematocrit of 37.5%.
Germline mutational analysis in DNA of index patient and family
To determine whether this was a somatic or germline mutation, additional HIF2A analyses were performed on DNA obtained from normal peritumoral tissue and blood lymphocytes of the index patient. Sanger sequencing confirmed the presence of the p.E549K mutation in both normal liver tissue and blood (Figure 2A), confirming a germline etiology.
Subsequently, additional studies were performed to identify this mutation in family members of the index patient. Blood lymphocytes from the proband’s family members revealed the p.E549K mutation was present in two sisters and one niece (Figure 2A). However, CBCs failed to demonstrate polycythemia in any family member, and none were diagnosed with HCC. These findings are illustrated in a genetic pedigree (Figure 2B).
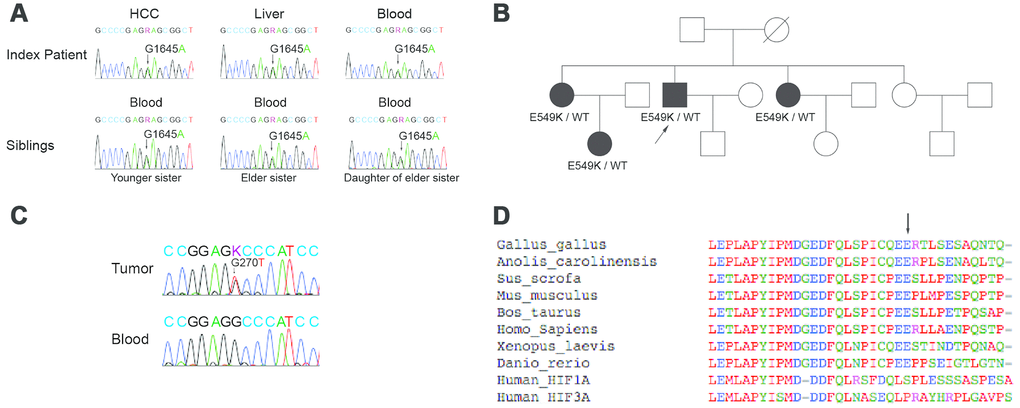
Figure 2. Sanger sequencing, genetic pedigree, and localization of germline mutation in the index patient and family members. (A) Direct Sanger sequencing results are shown for the HIF2A gene in the index patient and affected family members. The patient’s blood lymphocytes as well as the tumor and surrounding liver tissue carried a G1645A mutation in the gene coding for HIF2A. This mutation was also present in the blood lymphocytes of two of the patient’s sisters and one niece. (B) A family pedigree is shown. (C) Sanger sequencing of the p53 gene in the tumor tissue and blood lymphocytes of the index patient is shown. The p53 mutation was present only in the index patient’s tumor tissue. (D) Multiple peptide sequence alignment was performed; the E549 site was critically conserved across species (arrow). As such, a mutation at this site could potentially alter HIF-2α signaling.
P53 mutational analysis in tumor of index patient
Because none of the index patient’s family members had been diagnosed with HCC, further studies were performed to identify any additional mutations that had accumulated in the tumor. Mutations in the p53 tumor suppressor gene are common in HCC [25–27]. As such, Sanger sequencing for p53 was performed on DNA obtained from the tumor and blood lymphocytes of the index patient. A c.G270T (p.R90S) mutation was identified in the tumor DNA but not in the blood, confirming a somatic etiology (Figure 2C).
Localization of HIF2A mutation
Having established a germline origin for the p.E549K mutation, additional studies were undertaken to elucidate the function of this genetic alteration. Multiple peptide sequence alignment demonstrated that E549 of HIF-2α is located proximal to the protein’s primary hydroxylation domain and is conserved across species (arrow, Figure 2D). An amino acid substitution at this site could therefore potentially affect the conformation of the HIF-2α hydroxylation center and alter its recognition by VHL.
Stability of the mutant HIF-2α protein
To determine the effect of the c.G1645A mutation on HIF-2α protein stability, we performed an immunoprecipitation assay to analyze HIF-2α ubiquitination and von Hippel-Lindau protein (pVHL) recognition (Figure 3A). Under normoxic conditions, HIF-2α is hydroxylated and subsequently ubiquitinated by pVHL. The mutant E549K exhibited lower affinity for VHL, preventing its ubiquitination and subsequent degradation by the proteasome. A CHX assay confirmed that the stabilization of mutant HIF-2α extended the half-life of the mutant peptide (Figure 3B).
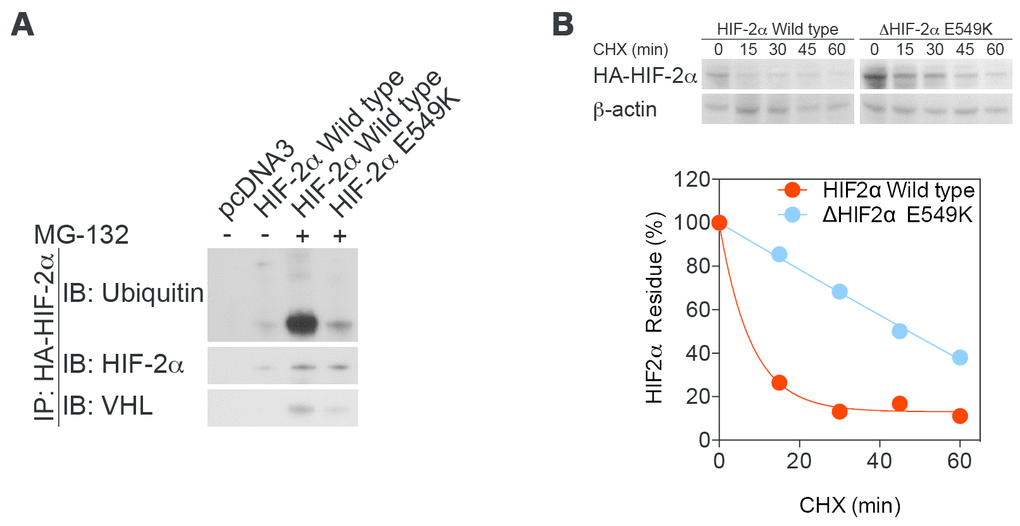
Figure 3. Stability is increased, while enzymatic function is maintained, in mutant HIF-2α protein. (A) An immunoprecipitation assay demonstrated lower affinity of mutant HIF-2α protein for binding to VHL and subsequent decreases in ubiquitination and proteasome degradation. (B) A cycloheximide assay confirmed that stabilization of mutant HIF-2α extended the half-life of the mutant protein.
Transcriptional activity of mutant HIF-2α
Transcription of the mutant HIF-2α peptide was determined using real-time PCR (Figure 4A) and a luciferase assay (Figure 4B). Transcription of mutant HIF-2α increased under normoxic conditions and was maintained during hypoxia, demonstrating that the p.E549K mutation results in a stable HIF-2α peptide without affecting its transcription.
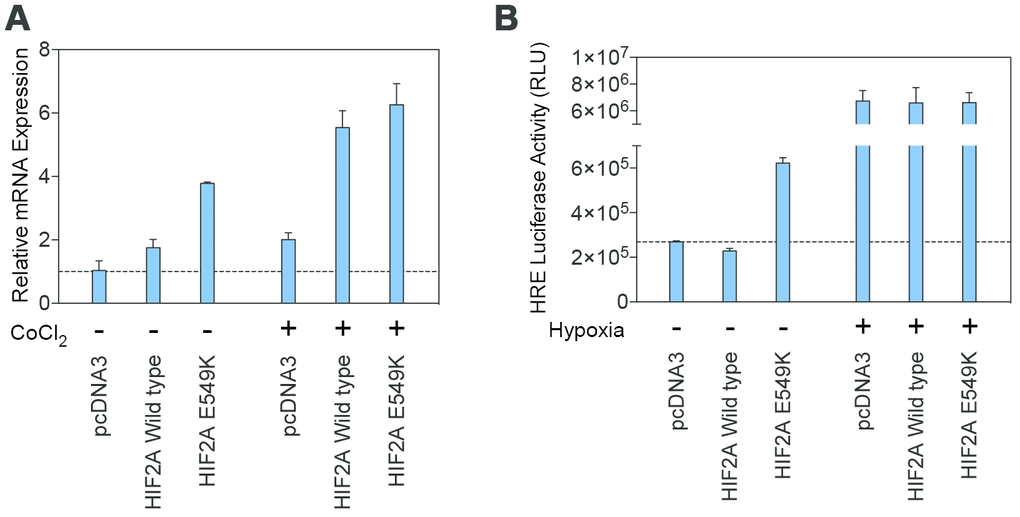
Figure 4. Transcription of the mutant HIF-2α protein is maintained under normoxic and hypoxic conditions. (A) Results from real-time PCR demonstrated similar transcription levels for the wildtype and mutant HIF2A genes under conditions of normoxia and simulated hypoxia. (B) A luciferase assay using Hep3B cells transfected with wildtype and p.E549K HIF2A confirmed these findings.
Discussion
This report documents a novel germline gain-of-function HIF2A mutation in a family in which one individual developed HCC and polycythemia. Interestingly, the germline mutation itself was not associated with erythrocytosis; none of the affected family members who did not develop HCC had a history of polycythemia or associated symptoms. Furthermore, although all cells carried the mutation, only tumor cells from the index patient exhibited upregulation of the transcriptional targets of HIF-2α. Removal of the tumor resolved his polycythemia.
Several HIF2A mutations are known to cause polycythemia. All of them result in amino acid substitutions or in-frame deletions in the primary hydroxylation domain, thereby subverting normal oxygen sensing and driving tumor formation [7–9]. The first such mutation discovered was p.G537W, which was originally reported as a germline mutation found across three generations in a single family that causes Familial Erythrocytosis Type 4 per Online Mendelian Inheritance in Man (OMIM) [28]. Since then, numerous somatic gain-of-function mutations have been identified, including p.P534L, p.M535V, p.M535I, p.G537W, p.G537R, and p.D539E, all of which are located in the ODD of HIF2A [29]. Like these mutations, the p.E549K mutation lies in the evolutionarily-conserved primary hydroxylation site of HIF-2α. This precludes oxygen-dependent HIF-2α hydroxylation by PHD enzymes, ubiquitination by the pVHL-E3 ligase complex, and subsequent proteasomal degradation, resulting in constitutive hypoxia signaling in the presence of normal oxygen tension.
More recently, HIF2A mutations have been implicated in unique clinical syndromes in patients with neuroendocrine tumors associated with polycythemia. These include the somatic mutations p.A530V [30] and p.A530E [31], somatic mosaic mutations p.L529P and p.L542P [32], and the germline mutation p.F374Y [33]. While HIF2A overexpression promotes oncogenesis in renal cell carcinoma [34], the p.E549K mutation described in the index patient here is the first HIF2A mutation to be identified in HCC. Previous studies have shown that prolonged HIF activation may accelerate the development of fibrosis, promote viral replication in chronic liver disease, and promote tumor cell growth and metastasis [35–38]. Hepatocellular carcinoma is typical of metabolically heterogeneous cancers in that tumor cells often generate energy through aerobic glycolysis rather than relying on oxidative phosphorylation in the mitochondria, even when oxygen supplies are adequate [23, 24]. This phenomenon, known as the Warburg effect, promotes tumor cell growth not only by providing an energy source for rapid cell proliferation, but also by minimizing the production of reactive oxygen species [39, 40]. The p.E549K mutation observed here stabilized the mutant peptide, which may transcriptionally up-regulate many genes encoding glycolytic enzymes [39]. Additionally, the HIF2α transcriptional targets EDN1, EPO, GNA14, and VEGFA were significantly upregulated in the tumor and might also promote tumor progression. Interestingly, despite its germline inheritance, the p.E549K mutation upregulated HIF-2α targets in the tumor but not the surrounding tissue, indicating that it selectively activated hypoxia signaling within the tumor. Moreover, the lack of obvious necrosis in the tumor suggests aberrant hypoxia signaling. Why HIF2A expression was not upregulated in the patient’s normal cells and why the other affected family members did not exhibit polycythemia remain unclear. These findings suggest that this mutation may show incomplete penetrance in causing the polycythemia phenotype. Well-known examples of gain-of-function mutations with incomplete penetrance and tumor predisposition include RET mutations in multiple endocrine neoplasia (MEN) syndromes and GNAS1 mutations in McCune-Albright syndrome. Like these syndromes, tumor development arising from a germline p.E549K mutation likely requires additional mutagenic changes, such as the p53 mutation reported in the index patient.
The mechanisms underlying potential carcinogenic interactions between the p53 tumor suppressor pathway and HIF expression are not fully understood. HIF expression is associated with increased genomic instability [41] and can promote malignant transformation through antagonism of p53-mediated tumor suppression [42, 43]. In clear-cell renal cell carcinoma, for instance, HIFs act upon the p53 pathway to mediate the DNA damage response [44], and overexpression of HIF2A suppresses Hdm2-mediated p53 activity, conferring chemotherapeutic resistance [43]. Likewise, inhibition of HIF-2α activity abrogates p53 activity, significantly sensitizing clear-cell renal carcinoma cells to radiation-induced apoptosis [45]. Therefore, further studies are needed to elucidate any potential synergy or causality between p53 mutations and gain-of-function mutations in hypoxia induced signaling pathways. In the case of the index patient, it is unclear whether selective activation of HIF2A signaling was attributable to or further propagated by the p53 mutation.
Given that the index patient’s polycythemia resolved upon total tumor resection, it is probable that the erythrocytosis in this case was due to upregulation of HIF2A targets by the novel mutant protein. However, this mutation was found in only one of 104 total HCC patients with concurrent polycythemia, suggesting that genetic alterations in HIF2A are involved in only a subset of HCC patients with erythrocytosis. There are numerous other possible etiologies for polycythemia, including mutations in PHD enzymes, VHL proteins, alterations in angiotensin II signaling, upregulation of insulin-like growth factor-1 signaling, and constitutive activation of the JAK/STAT pathway [46–50]. Additional investigations are needed to determine the molecular mechanisms and therapeutic significance of polycythemia in HCC patients.
Materials and Methods
Patients
302 patients with HCC including 104 with polycythemia were referred to the Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery at the First Affiliated Hospital of Zhejiang University and fulfilled the diagnostic criteria for hepatocellular carcinoma described by the American Association for the Study of Liver Diseases (AASLD) [51]. Patients with hematological system disorders were excluded from the study. Polycythemia was defined as an elevated hematocrit or red blood cell count that did not meet the 2007 World Health Organization (WHO) criteria for diagnosis of polycythemia vera [52].
Study oversight
This study was approved by the institutional review board of the First Affiliated Hospital, College of Medicine, Zhejiang University (Reference number: 2014-272). Pedigree investigation participants provided written informed consent for participation in this study. Written informed consent was not obtained from other screened patients because this study used only preexisting data and pathological tissues; in these cases, we informed the participants of their right to refuse enrolment via telephone interview. These informed consent and enrolment procedures are in accordance with the detailed regulations described in the guidelines.
Laboratory studies
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were probed with primary antibodies against AFP (mouse monoclonal; Zhongshan Golden Bridge Biotechnology Company Limited, Beijing, China), CD34 (mouse monoclonal; Long Island Biotechnology Company Limited, Shanghai, China), CK19 (mouse monoclonal; Zhongshan), or HEP (mouse monoclonal; Zhongshan). Slides were then stained with 3,3’-diaminobenzidine (DAB) and visualized using a Leica DM LB light (Solms, Germany) microscope at magnifications of 100× or 400× as indicated.
Mutation analysis
Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Venlo, Netherlands). HIF2A exons were amplified by polymerase chain reaction (PCR) using previously described primer sets [7]. Likewise, P53 exons were amplified by PCR using previously described primer sets [53]. Amplicon sequences were then determined by Sanger sequencing.
Plasmids and mutagenesis
A pcDNA3 plasmid (Addgene, Cambridge, MA; plasmid 18950 [7]) containing the human HIF2A coding sequence was used for wild-type HIF2A expression analysis. E549K-mutant HIF2A was cloned and inserted into an expression plasmid using a Quikchange Lightning Site-directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Sanger sequencing was used to confirm successful insertion.
Real-time PCR
Total RNA was extracted from microdissected tumor samples using an RNeasy Plus Mini Kit (Qiagen). Messenger RNA (mRNA) expression of HIF target genes was examined by real-time polymerase chain reaction (PCR) using an Eco Real-Time PCR System (Illumina, San Diego, CA). Gene-specific primer sets included endothelin-1 (EDN1; Qiagen QT00088235), erythropoietin (EPO; Origene, Rockville, MD; HP200740), guanine nucleotide binding protein alpha 14 (GNA14; Qiagen QT00099379), vascular endothelial growth factor A (VEGFA; Qiagen QT01682072), and beta-actin (ACTB; Qiagen QT00095431).
Immunoprecipitation
HIF2A-transfected HEK293 cells were treated with MG-132 (10 μM) for 6 hr. Total protein was extracted using immunoprecipitation (IP) lysis buffer supplemented with a protease inhibitor cocktail (Thermo, Waltham, MA). According to the manufacturer’s instructions, 500 μg total protein extract was incubated with 3 μg monoclonal anti-hemagglutinin (HA) antibody (Covance, Princeton, NJ) and a Dynabeads Protein G Immunoprecipitation Kit (Life Technologies, Carlsbad, CA) overnight at 4°C.
The eluent was then subjected to Western blot and probed with antibodies for ubiquitin (Abcam, Cambridge, UK), HIF-2α (Novus Biologicals, Littleton, CO), or VHL protein (Cell Signaling Technology, Danvers, MA).
Cycloheximide assay
HIF2A-transfected HEK293 cells were treated with cycloheximide (CHX) (50 μM, Sigma-Aldrich, St. Louis, MO) for 0, 15, 30, 45, or 60 min as indicated. Total protein was extracted using RIPA lysis buffer supplemented with a protease inhibitor cocktail (Thermo). HIF-2α protein levels were then determined by Western blot using an anti-HA antibody (Covance).
Luciferase assay
Hep3B cells stably expressing HRE-luciferase (Promega, Madison, WI) were transfected with HIF2A recombinants. Cells were incubated in 21% or 1% O2 for 12 hr and luciferase activity was determined using the ONE-Glo Luciferase assay system (Promega).
Author Contributions
H.C. and Z.Z., the coprincipal investigators, designed and supervised the study. Z.Z. and H.C. obtained the funding. J.Y. and X.S. assisted with recruitment, clinical management, and data collection and completed immunohistochemistry and mutation analysis. C.Y., P.D., C.L.N., and C.S.H. detected gene function and performed statistical analyses. K.P. and P.B. provided technical support. H.C., Z.Z., L.L., and C.Y. drafted the article, and all authors reviewed and approved the final version.
Acknowledgments
The authors wish to thank the family and index patient described in this study. In addition, they acknowledge Prof. Shusen Zheng, Dr. Lin Zhou, Dr. Qiyi Zhang, and Dr. Wei Ding of the First Affiliated Hospital, College of Medicine, Zhejiang University for providing the patient histories and samples.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by the National Science and Technology Major Project (No. 2012ZX10002004), the Chinese High-Tech Research and Development (863) Program (No. SS2013AA020102), and the Science Fund for Creative Research Groups of the National Natural Science Foundation of China (No. 81121002). This work was also supported by the intramural research programs at the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, both at the National Institutes of Health (NIH). Additional support was provided by the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer, Inc., The Doris Duke Charitable Foundation, The Alexandria Real Estate Equities, Inc., and Mr. and Mrs. Joel S. Marcus and the Howard Hughes Medical Institute, as well as other private donors. For a complete list, please visit the Foundation at: https://fnih.org/what-we-do/programs/mrsp.
References
- 1. Jochmanová I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst. 2013; 105:1270–83. https://doi.org/10.1093/jnci/djt201 [PubMed]
- 2. Da Silva JL, Lacombe C, Bruneval P, Casadevall N, Leporrier M, Camilleri JP, Bariety J, Tambourin P, Varet B. Tumor cells are the site of erythropoietin synthesis in human renal cancers associated with polycythemia. Blood. 1990; 75:577–82. https://doi.org/10.1182/blood.V75.3.577.577 [PubMed]
- 3. Krieg M, Marti HH, Plate KH. Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von Hippel-Lindau tumor suppressor gene loss of function. Blood. 1998; 92:3388–93. https://doi.org/10.1182/blood.V92.9.3388 [PubMed]
- 4. Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA. 2006; 103:654–59. https://doi.org/10.1073/pnas.0508423103 [PubMed]
- 5. Taïeb D, Barlier A, Yang C, Pertuit M, Tchoghandjian A, Rochette C, Zattara-Canoni H, Figarella-Branger D, Zhuang Z, Pacak K, Metellus P. Somatic gain-of-function HIF2A mutations in sporadic central nervous system hemangioblastomas. J Neurooncol. 2016; 126:473–81. https://doi.org/10.1007/s11060-015-1983-y [PubMed]
- 6. Yang Y, Zhang YJ, Zhu Y, Cao JZ, Yuan ZY, Xu LM, Wu JX, Wang W, Wu T, Lu B, Zhu SY, Qian LT, Zhang FQ, et al. Prognostic nomogram for overall survival in previously untreated patients with extranodal NK/T-cell lymphoma, nasal-type: a multicenter study. Leukemia. 2015; 29:1571–77. https://doi.org/10.1038/leu.2015.44 [PubMed]
- 7. Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012; 367:922–30. https://doi.org/10.1056/NEJMoa1205119 [PubMed]
- 8. Yang C, Sun MG, Matro J, Huynh TT, Rahimpour S, Prchal JT, Lechan R, Lonser R, Pacak K, Zhuang Z. Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood. 2013; 121:2563–66. https://doi.org/10.1182/blood-2012-10-460972 [PubMed]
- 9. Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, Prchal JT, Tischler AS, Lechan RM, Zhuang Z. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol. 2013; 31:1690–98. https://doi.org/10.1200/JCO.2012.47.1912 [PubMed]
- 10. Swain A, Inoue T, Tan KS, Nakanishi Y, Sugiyama D. Intrinsic and extrinsic regulation of mammalian hematopoiesis in the fetal liver. Histol Histopathol. 2014; 29:1077–82. https://doi.org/10.14670/HH-29.1077 [PubMed]
- 11. Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Teperman L, Henegariu O, Krause DS. Liver from bone marrow in humans. Hepatology. 2000; 32:11–16. https://doi.org/10.1053/jhep.2000.9124 [PubMed]
- 12. Kew MC, Fisher JW. Serum erythropoietin concentrations in patients with hepatocellular carcinoma. Cancer. 1986; 58:2485–88. https://doi.org/10.1002/1097-0142(19861201)58:11<2485::AID-CNCR2820581122>3.0.CO;2-N [PubMed]
- 13. Sakisaka S, Watanabe M, Tateishi H, Harada M, Shakado S, Mimura Y, Gondo K, Yoshitake M, Noguchi K, Hino T, Nohno R, Majima Y, Hirai K, et al. Erythropoietin production in hepatocellular carcinoma cells associated with polycythemia: immunohistochemical evidence. Hepatology. 1993; 18:1357–62. https://doi.org/10.1002/hep.1840180612 [PubMed]
- 14. El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003; 139:817–23. https://doi.org/10.7326/0003-4819-139-10-200311180-00009 [PubMed]
- 15. Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based study. Gastroenterology. 2004; 127:1372–80. https://doi.org/10.1053/j.gastro.2004.07.020 [PubMed]
- 16. Skolnick AA. Armed with epidemiologic research, China launches programs to prevent liver cancer. JAMA. 1996; 276:1458–59. https://doi.org/10.1001/jama.1996.03540180012005 [PubMed]
- 17. Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki H, Kawashima T. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med. 1993; 328:1797–801. https://doi.org/10.1056/NEJM199306243282501 [PubMed]
- 18. Trichopoulos D, Bamia C, Lagiou P, Fedirko V, Trepo E, Jenab M, Pischon T, Nöthlings U, Overved K, Tjønneland A, Outzen M, Clavel-Chapelon F, Kaaks R, et al. Hepatocellular carcinoma risk factors and disease burden in a European cohort: a nested case-control study. J Natl Cancer Inst. 2011; 103:1686–95. https://doi.org/10.1093/jnci/djr395 [PubMed]
- 19. El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004; 126:460–68. https://doi.org/10.1053/j.gastro.2003.10.065 [PubMed]
- 20. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010; 51:1972–78. https://doi.org/10.1002/hep.23527 [PubMed]
- 21. Yu MC, Tong MJ, Govindarajan S, Henderson BE. Nonviral risk factors for hepatocellular carcinoma in a low-risk population, the non-Asians of Los Angeles County, California. J Natl Cancer Inst. 1991; 83:1820–26. https://doi.org/10.1093/jnci/83.24.1820 [PubMed]
- 22. Chen CJ, Wang LY, Lu SN, Wu MH, You SL, Zhang YJ, Wang LW, Santella RM. Elevated aflatoxin exposure and increased risk of hepatocellular carcinoma. Hepatology. 1996; 24:38–42. https://doi.org/10.1002/hep.510240108 [PubMed]
- 23. Cassim S, Raymond VA, Dehbidi-Assadzadeh L, Lapierre P, Bilodeau M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle. 2018; 17:903–16. https://doi.org/10.1080/15384101.2018.1460023 [PubMed]
- 24. Cassim S, Raymond VA, Lacoste B, Lapierre P, Bilodeau M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget. 2018; 9:26868–83. https://doi.org/10.18632/oncotarget.25525 [PubMed]
- 25. Lunn RM, Zhang YJ, Wang LY, Chen CJ, Lee PH, Lee CS, Tsai WY, Santella RM. p53 mutations, chronic hepatitis B virus infection, and aflatoxin exposure in hepatocellular carcinoma in Taiwan. Cancer Res. 1997; 57:3471–77. [PubMed]
- 26. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, Parker JS, Li J, Powers S, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013; 58:1693–702. https://doi.org/10.1002/hep.26540 [PubMed]
- 27. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clément B, Balabaud C, Chevet E, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012; 44:694–98. https://doi.org/10.1038/ng.2256 [PubMed]
- 28. Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, Lee FS. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008; 358:162–68. https://doi.org/10.1056/NEJMoa073123 [PubMed]
- 29. Lee FS, Percy MJ. The HIF pathway and erythrocytosis. Annu Rev Pathol. 2011; 6:165–92. https://doi.org/10.1146/annurev-pathol-011110-130321 [PubMed]
- 30. Taïeb D, Yang C, Delenne B, Zhuang Z, Barlier A, Sebag F, Pacak K. First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2013; 98:E908–13. https://doi.org/10.1210/jc.2013-1217 [PubMed]
- 31. Toyoda H, Hirayama J, Sugimoto Y, Uchida K, Ohishi K, Hirayama M, Komada Y. Polycythemia and paraganglioma with a novel somatic HIF2A mutation in a male. Pediatrics. 2014; 133:e1787–91. https://doi.org/10.1542/peds.2013-2419 [PubMed]
- 32. Buffet A, Smati S, Mansuy L, Ménara M, Lebras M, Heymann MF, Simian C, Favier J, Murat A, Cariou B, Gimenez-Roqueplo AP. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2014; 99:E369–73. https://doi.org/10.1210/jc.2013-2600 [PubMed]
- 33. Lorenzo FR, Yang C, Ng Tang Fui M, Vankayalapati H, Zhuang Z, Huynh T, Grossmann M, Pacak K, Prchal JT. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl). 2013; 91:507–12. https://doi.org/10.1007/s00109-012-0967-z [PubMed]
- 34. Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007; 17:71–77. https://doi.org/10.1016/j.gde.2006.12.006 [PubMed]
- 35. Ju C, Colgan SP, Eltzschig HK. Hypoxia-inducible factors as molecular targets for liver diseases. J Mol Med (Berl). 2016; 94:613–27. https://doi.org/10.1007/s00109-016-1408-1 [PubMed]
- 36. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016; 352:175–80. https://doi.org/10.1126/science.aaf4405 [PubMed]
- 37. Chen C, Lou T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget. 2017; 8:46691–703. https://doi.org/10.18632/oncotarget.17358 [PubMed]
- 38. Chen J, Chen J, Huang J, Li Z, Gong Y, Zou B, Liu X, Ding L, Li P, Zhu Z, Zhang B, Guo H, Cai C, Li J. HIF-2α upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway. Aging (Albany NY). 2019; 11:10839–60. https://doi.org/10.18632/aging.102488 [PubMed]
- 39. Chen Z, Zuo X, Zhang Y, Han G, Zhang L, Wu J, Wang X. MiR-3662 suppresses hepatocellular carcinoma growth through inhibition of HIF-1α-mediated Warburg effect. Cell Death Dis. 2018; 9:549. https://doi.org/10.1038/s41419-018-0616-8 [PubMed]
- 40. De Matteis S, Scarpi E, Granato AM, Vespasiani-Gentilucci U, La Barba G, Foschi FG, Bandini E, Ghetti M, Marisi G, Cravero P, Gramantieri L, Cucchetti A, Ercolani G, et al. Role of SIRT-3, p-mTOR and HIF-1α in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin. Int J Mol Sci. 2019; 20:E1503. https://doi.org/10.3390/ijms20061503 [PubMed]
- 41. Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell. 2005; 17:793–803. https://doi.org/10.1016/j.molcel.2005.02.015 [PubMed]
- 42. Obacz J, Pastorekova S, Vojtesek B, Hrstka R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol Cancer. 2013; 12:93. https://doi.org/10.1186/1476-4598-12-93 [PubMed]
- 43. Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011; 2:e164. https://doi.org/10.1038/cddis.2011.48 [PubMed]
- 44. Selvarajah J, Nathawat K, Moumen A, Ashcroft M, Carroll VA. Chemotherapy-mediated p53-dependent DNA damage response in clear cell renal cell carcinoma: role of the mTORC1/2 and hypoxia-inducible factor pathways. Cell Death Dis. 2013; 4:e865. https://doi.org/10.1038/cddis.2013.395 [PubMed]
- 45. Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B, Brown EJ, Nathanson KL, Simon MC. HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci USA. 2009; 106:14391–96. https://doi.org/10.1073/pnas.0907357106 [PubMed]
- 46. Mrug M, Stopka T, Julian BA, Prchal JF, Prchal JT. Angiotensin II stimulates proliferation of normal early erythroid progenitors. J Clin Invest. 1997; 100:2310–14. https://doi.org/10.1172/JCI119769 [PubMed]
- 47. Shih LY, Huang JY, Lee CT. Insulin-like growth factor I plays a role in regulating erythropoiesis in patients with end-stage renal disease and erythrocytosis. J Am Soc Nephrol. 1999; 10:315–22. [PubMed]
- 48. Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993; 74:227–36. https://doi.org/10.1016/0092-8674(93)90414-L [PubMed]
- 49. Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002; 32:614–21. https://doi.org/10.1038/ng1019 [PubMed]
- 50. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011; 365:537–47. https://doi.org/10.1056/NEJMra1011165 [PubMed]
- 51. Bruix J, Sherman M, and American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology. 2011; 53:1020–22. https://doi.org/10.1002/hep.24199 [PubMed]
- 52. Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer. 2009; 115:3842–47. https://doi.org/10.1002/cncr.24440 [PubMed]
- 53. Hsueh C, Wang H, Gonzalez-Crussi F, Lin JN, Hung IJ, Yang CP, Jiang TH. Infrequent p53 gene mutations and lack of p53 protein expression in clear cell sarcoma of the kidney: immunohistochemical study and mutation analysis of p53 in renal tumors of unfavorable prognosis. Mod Pathol. 2002; 15:606–10. https://doi.org/10.1038/modpathol.3880573 [PubMed]