




Introduction
Population aging has resulted in a rapid increase in lung cancer cases as well as corresponding surgeries among elderly patients [1]. Indeed, the median age at diagnosis of lung cancer is 70 years old [2]. Further, lung cancer leads as a cause of cancer deaths among men ≥40 years old and women ≥60 years old [3].
Progression of lung cancer is, in part, due to accumulation of genomic instability as well as age-related declines in system integrity and function [4]. Thus, even for individuals exposed to similar levels of risk factors, lung cancer severity can vary as a function of individual differences in aging. Therefore, compared to predictive guidance for the overall population, effective predictive guidance for age-specific populations, especially elderly patients, is needed to better guide postoperative treatment and improve survival. Developing such guidance necessitates identifying exclusive prognostic indicators of lung cancer for the elderly.
Epigenetic mechanisms represent the molecular interface mediating gene–environment interactions throughout the lifecycle [5]. DNA methylation, a reversible epigenetic modification, correlates with tumor prognosis in almost all cancers including non-small cell lung cancer (NSCLC) [6–9]. DNA methylation events may potentially be cancer biomarkers as well as therapeutic targets to improve cancer treatment [10].
Alterations to DNA methylation often occur during aging [11]. One of these alterations, known as “epigenetic drift”, may further contribute to tumorigenesis in the elderly [12]. Changes in DNA methylation also can contribute to senescence [13]. However, it remains largely unclear whether alterations of methylation patterns resulting from aging, accumulating environmental exposures throughout life [14], and other events also have varied effects on cancer survival during aging. Such phenomena may further explain the increased alteration of cancer mortality risk with age and may increase the effectiveness of cancer prediction and treatment.
We hypothesized that the methylation effect on cancer survival changes during aging. Thus, identifying age-specific signatures will be critical for prognosis prediction, underpinning potential preventative strategies, and improving survival for elderly patients. However, most epigenome-wide association studies are designed to identify main effects of DNA methylation and fail to provide knowledge about changes in epigenetic effects during aging. Thus, we performed an epigenome-wide methylation–age interaction analysis to identify age-specific, prognosis-associated epigenetic biomarkers using NSCLC patients from four cohorts, along with an independent population from The Cancer Genome Atlas (TCGA) to confirm our results.
Results
After quality control (QC) procedures, 1,230 lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) patients with 311,891 CpG probes remained for subsequent association analysis. There were 613 (NLUAD = 492; NLUSC = 121) patients in the discovery phase, and 617 (NLUAD = 332; NLUSC = 285) patients in the validation phase. The average age was 66.4 and 66.5 years for patients in the discovery and validation phases, respectively. Most NSCLC patients were in stage I (77.5% in discovery; 63.7% in validation) (Supplementary Table 1).
We only observed two significant methylation–age interactions for LUAD patients in the discovery phase (Figure 1, Supplementary Figure 1, Supplementary Table 2), and none for LUSC patients. Results of the epigenome-wide association study are shown in Supplementary Materials 1 and 2. In the validation phase, only one LUAD-specific CpG probe, located in proline dehydrogenase 1 (PRODH) (Supplementary Table 3), remained significant.
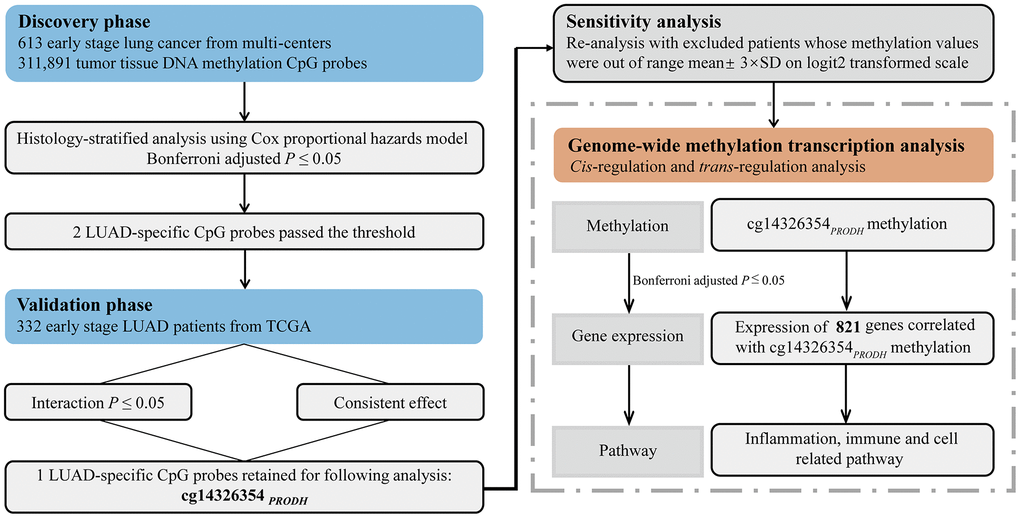
Figure 1. Flow chart of study design and statistical analyses.
Low methylation of cg14326354PRODH interacted with age to affect survival of patients (discovery phase: hazard ratio (HR)interaction = 0.982; 95% CI: 0.976–0.989; P = 1.11×10–7; validation phase: HRinteraction = 0.981; 95% CI: 0.966–0.997; P = 0.0202; combined data: HRinteraction = 0.989; 95% CI: 0.986–0.994; P = 9.18×10–7). Further, the robustly significant interaction effect was confirmed in sensitivity analysis by removing outliers in methylation data (Supplementary Table 4). When using leave-one-out method for validation, the interaction remained significant (Supplementary Figure 2). Moreover, meta-analysis also exhibited significant (HRinteraction = 0.983; 95% CI: 0.976–0.990; P = 3.95×10–6) and homogenous (PHeterogeneity = 0.97) interaction effects across five cohorts (Supplementary Figure 3). Based on stratified analysis by smoking status, sex, clinical stage, and study cohort, there was no significant heterogeneity of interaction effect between subgroups of any of these covariates (Supplementary Table 5).
With increased age, there was an increased protective effect for low methylation of cg14326354PRODH on LUAD survival (Figure 2A, Supplementary Figure 4). Thus, age was a modifier of the association between cg14326354PRODH and survival. To better understand the interaction between DNA methylation and age, patients were categorized into young and elderly groups based on the boundary of 95% CI (BoCI) of HR (<57 vs >65 years in Figure 2A) or the United Nations (UN) standard (≤65 vs >65 years). The BoCI standard provided stable results in both phases as well as combined data (Supplementary Table 6), with varied effects of cg14326354PRODH methylation across different age groups. Low methylation of cg14326354PRODH showed a risk effect on survival for young patients (HRBoCI = 1.20; 95% CI: 1.03–1.40; P = 1.97×10–2; HRUN = 1.10; 95% CI: 0.99–1.22; P = 8.71×10–2) but benefited survival of elderly LUAD patients (HRBoCI = 0.81; 95% CI: 0.75–0.88; P = 5.38×10–7; HRUN = 0.81; 95% CI: 0.75–0.88; P = 5.38×10–7) (Figure 2B). Kaplan-Meier curves also confirmed reversed effect patterns across age groups based on BoCI standard (HRyoung = 2.44; 95% CI: 1.26–4.72; P = 8.34×10-3; HRelderly = 0.58; 95% CI: 0.42–0.82; P = 1.67×10-3), with methylation groups defined by median values. There was significant heterogeneity of the low cg14326354PRODH methylation effect between young and elderly patients (I2 = 93.03%, Q = 14.35, P = 1.52×10-4) (Figure 2C). These results indicated that elderly LUAD patients had better survival with lower methylation of cg14326354PRODH.
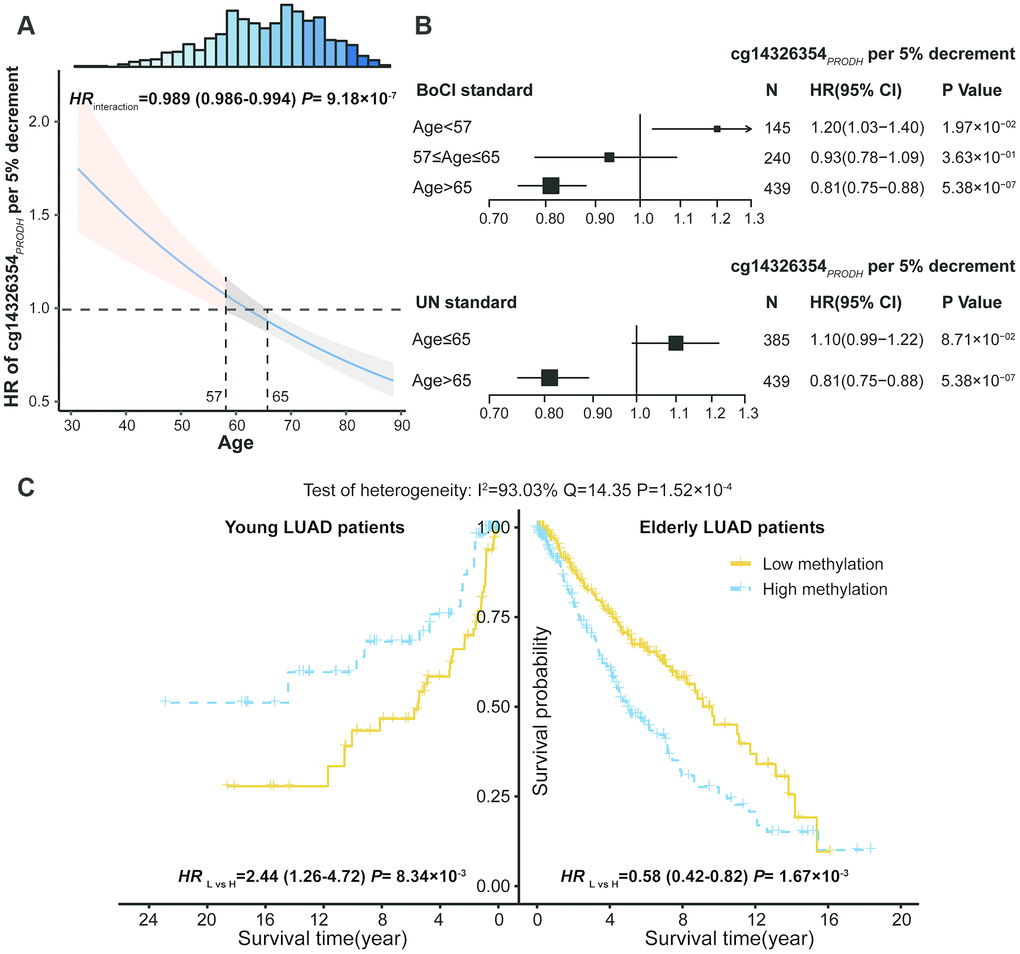
Figure 2. DNA methylation and age interaction on survival of lung adenocarcinoma (LUAD) patients. (A) Hazard ratio (HR) of cg14326354PRODH 5% per decrement of methylation level among different aged patients. The 95% confidence interval (95% CI) band of HR for patients aged <57 or >65 years is statistically significant. Top histogram shows distribution of age. (B) Forest plots of HR of cg14326354PRODH 5% per decrement of methylation level in young and elderly LUAD patients, categorized based on boundary of 95% CI (BoCI) and 1956 United Nations standard. (C) Kaplan-Meier survival curves of low and high methylation groups (categorized by median value) among young and elderly LUAD patients defined using BoCI standard. Pheterogeneity was used to evaluate heterogeneity of HRs across age groups.
In addition, we evaluated the joint effect of cg14326354PRODH methylation level (low vs high) and age (elderly vs young) on LUAD survival (Table 1). The group with the best survival (young patients with high methylation) was used as the reference to evaluate effects of low methylation, elderly age, and their interaction. The main effect of low cg14326354PRODH methylation was HR = 2.84 (95% CI: 1.59–5.08, P = 4.35×10−4), and the main effect of elderly age was HR = 3.18 (95% CI: 1.85–5.46, P = 2.64×10−5). However, the joint effect was HR = 1.86 (95% CI: 1.08–3.19, P = 2.42×10−2), which was less than the product of the two main effects (2.84×3.18 = 9.03). This result indicates an antagonistic interaction between low cg14326354PRODH methylation and elderly age (HRinteraction = 0.21; 95% CI: 0.11–0.40; P = 2.20×10−6).
Table 1. Joint effect and interaction of low methylation and elderly age on the prognosis of early-stage lung adenocarcinoma (LUAD).
| Effect type a | Elderly b | Low methylation | Number | Death | Crude mortality | HR (95% CI) a | Pa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No | No | 75 | 17 | 22.67% | Ref. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Main effect 1 | No | Yes | 70 | 33 | 47.14% | 2.8398 (1.5876,5.0798) | 4.35×10-4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Main effect 2 | Yes | No | 217 | 98 | 45.16% | 3.1804 (1.8542,5.4553) | 2.64×10-5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Joint effect | Yes | Yes | 222 | 70 | 31.53% | 1.8590 (1.0840,3.1890) | 0.0242 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Interaction c | 0.2058 (0.1070,0.3961) | 2.20×10-6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Patients categorized into two groups (low vs high) by medium of cg14326354PRODH methylation level. Classification criteria of age were based on boundary of 95% confidence interval (CI) standard (young: <57 years; elderly >65 years). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| b Main effects of low methylation and elderly age and their joint effect and interaction were derived from Cox proportional hazards model adjusted for covariates. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| c Interaction = Joint effect ÷ (Main effect 1 × Main effect 2). 0.2058 ≈ 1.8590 ÷ (3.1804 × 2.8398). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Further, cis-regulation and genome-wide trans-regulation analyses were conducted in the TCGA cohort. We observed significant correlation between cg14326354PRODH and PRODH expression (r = –0.23; P = 3.38 × 10-5) in LUAD patients (Figure 3A), indicating that cg14326354PRODHcis-regulated gene expression. Moreover, genome-wide trans-regulation analysis revealed that expression of 821 genes was significantly correlated with methylation level of cg14326354PRODH (Supplementary Material 3, Figure 3B). KEGG enrichment analysis based on 821 trans-regulated genes showed several significant immune- or inflammation-related pathways, such as chemokine signaling, T cell receptor and B cell receptor signaling, and cellular pathways such as cell differentiation and cell cycle (Figure 3C). Notably, these trans-regulated genes were also enriched in senescence-related pathways (e.g., cellular senescence) and cancer-related pathways (e.g., NF-κB signaling).
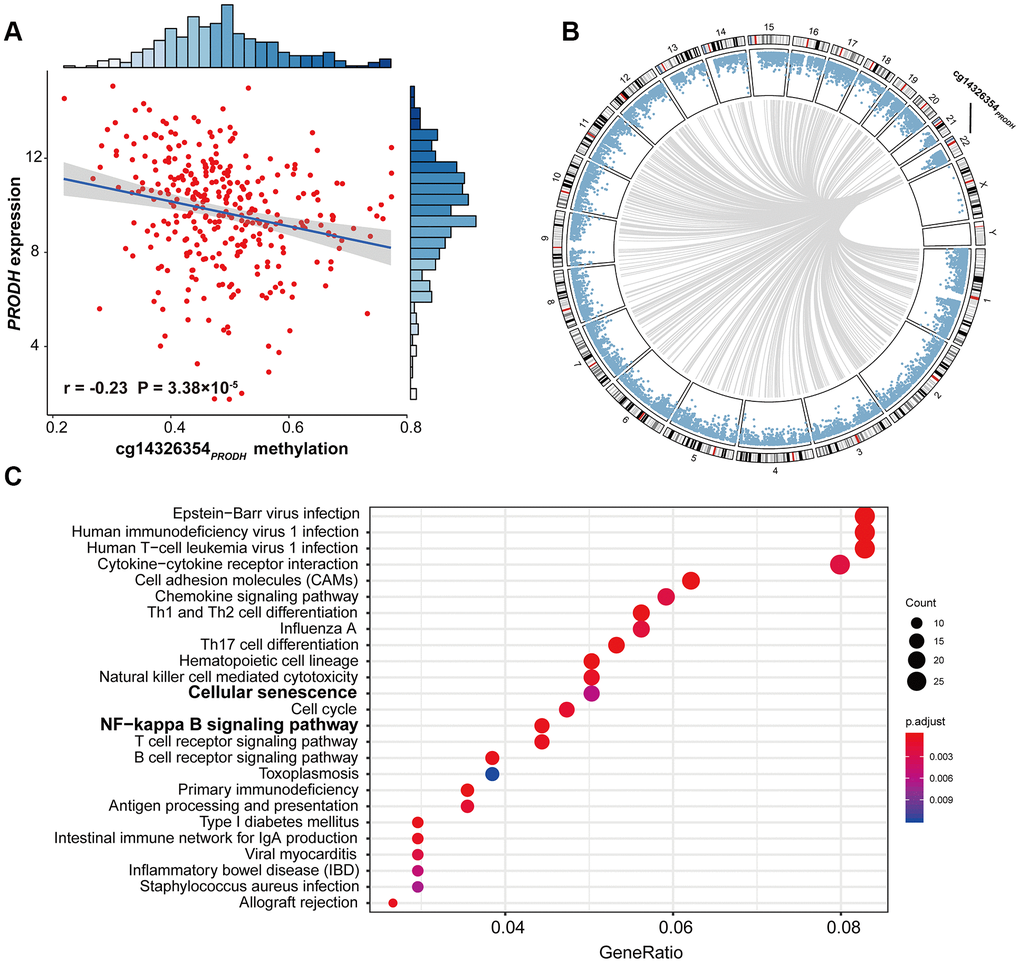
Figure 3. Scatter plot of cis-regulation, circos plot of genome-wide trans-regulation analysis, and significant pathways of gene enrichment pathway analysis. (A) Correlation between DNA methylation of cg14326354PRODH and expression of PRODH. The r coefficient and P-value were derived from Pearson correlation analysis. Gene expression was log2-transformed before correlation analysis. (B) Circos plot of genome-wide trans-regulation analysis in the TCGA cohort. Blue points ordered by genomic position represent P-values of correlation between gene expression and methylation at cg14326354PRODH. Grey lines represent significant correlations with Bonferroni-adjusted P ≤ 0.05. (C) KEGG gene enrichment analysis of 821 trans-regulated genes correlated with cg14326354PRODH methylation.
Because tumor mutational burden (TMB) serves as a biomarker to select patients who might benefit from immune checkpoint inhibitors [15], we also evaluated association between TMB and cg14326354PRODH as well as PRODH expression. TMB of each sample is shown in Supplementary Material 4. cg14326354PRODH was positively correlated with TMB (r = 0.23; P = 4.04×10-5), while PRODH expression was negatively correlated with TMB (r = –0.22; P = 6.62×10-5) (Supplementary Figure 5).
Discussion
In this two-stage study using five independent cohorts, we systematically investigated methylation–age interactions on an epigenome-wide scale. Our results show an antagonistic interaction between elderly age and low methylation of cg14326354PRODH, indicating opportunities for epi-drug intervention due to the inherent reversibility of epigenetic events [16] and increasing treatment efficiency based on age-specific drug targeting.
PRODH is located in chromosome 22q11.2, a region often deleted in various human tumors. This gene encodes a mitochondrial inner membrane-associated enzyme that acts as a tumor suppressor in vitro and in vivo [17]. However, PRODH plays a paradoxical role in tumors. Hypoxia and nutrient depletion are important characteristics of the tumor microenvironment, where PRODH may serve as a tumor survival factor [18]. Indeed, PRODH supports tumor metastasis formation, and inhibiting its activity impairs cancer cell growth, indicating PRODH is a potential drug target [19]. A metabolic enzyme, PRODH can catabolize proline to pyrroline-5-carboxylate (P5C). The process can donate electrons that enter the electron transport chain to produce reactive oxygen species (ROS), which then participate in protective autophagy rather than apoptotic cell death [18].
Autophagy, a self-digestion process, plays an important role in maintaining intracellular homeostasis. Autophagy can clear intracellular abnormally folded protein and dysfunctional organelles, inhibit cell stress response, and prevent genetic damage in early phases of tumorigenesis. However, autophagy helps tumor cells survive nutritional deficiencies and hypoxic conditions when tumors develop and accumulate more mutations to promote malignant progression [20]. Further, tumor-surrounding normal cells, which are active and essential parts of the microenvironment, support tumor proliferation by autophagy. Besides, autophagy in distant organs may also support growth of tumor tissue [21]. Additionally, autophagy can act as a mechanism of tumor resistance to chemotherapy agents and lead to antagonistic effects of gefitinib combined with cisplatin in NSCLC treatment, which may contribute to poor therapeutic effectiveness and patient prognosis [22, 23]. Further, our results suggest that low methylation of cg14326354PRODH may potentially promote PRODH expression, further heighten autophagy to some extent [24], and then result in poor prognosis (Figure 4).
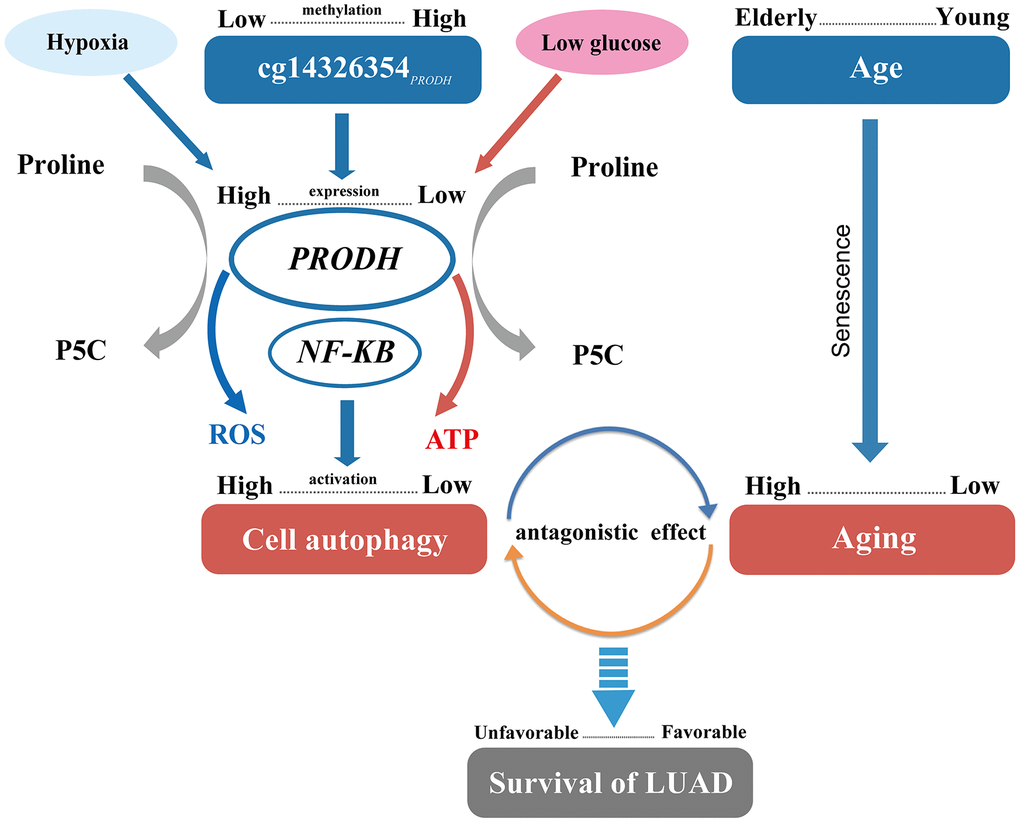
Figure 4. Pathway of DNA methylation–age interaction effect on survival of lung adenocarcinomas (LUAD) patients.
Age is an independent risk factor for lung cancer survival [25]. Individual aging implies a higher abundance of senescent cells in aged tissues and reflects an increase in the generation of senescent cells [26]. At old age, senescent cells generate a pro-tumorigenic microenvironment, though at young age these cells may protect against transformation into primary tumors [27]. A previous study also shows that p53 function declines during aging [28] and might promote tumor growth and decrease cancer survival [29]. Moreover, senescent cells can promote reprogramming of tumor stem cells, increase cancer stemness, and accelerate tumor growth [30]. Thus, combined with our results, increased generation of senescent may be relevant to poor NSCLC prognosis for elderly patients (Figure 4).
Autophagy is reduced in aging, likely through several mechanisms [31]. Lipofuscin produced during aging can destroy the function of lysosomes, restricting binding between autophages and lysosomes [32]. In addition, expression of lysosome-associated membrane glycoprotein (LAMP2a), which assists autophagy, decreases during aging and thus can inhibit autophagy [33]. Further, by guaranteeing stability of the cellular proteome and proper organelle turnover, autophagy can prevent or slow down aging and extend lifespan [34]. The antagonistic effect exists between aging and the autophagy level resulting from low methylation of cg14326354PRODH, in spite of the harmful effect of both, which could provide a possible mechanism of the cg14326354PRODH–age interaction (Figure 4).
The 821 significant trans-regulated genes we identified were enriched in KEGG pathways including inflammation and immune-related pathways. Notably, cellular senescence was involved in these pathways, again indicting potential indirect induction of cg14326354PRODH on senescence. Meanwhile, the NF-κB pathway, with the ability to upregulate genes responsible for inflammation, cell survival, proliferation, invasion, angiogenesis, and metastasis, often plays a critical role in initiation, promotion, progression, and therapy resistance of cancers [35, 36]. Further, NF-κB family members can activate or inhibit signaling pathways, leading to induction of autophagy or transcription of certain pro-autophagic-regulating genes [35], and can induce senescence [37]. Because cell proliferation can be associated with both senescence and survival [38, 39], we also analyzed several proliferation-associated genes retrieved from the KEGG database. Expression of these genes were significantly correlated with cg14326354PRODH methylation and affected LUAD survival, including MKI67, BTG2, KIAA1524, and CDC123 (Supplementary Table 7). Our previous study of BTG2 expression and methylation already indicated it is a prognostic biomarker of NSCLC [7]. These results also indicate the potential role of cg14326354PRODH in indirect induction of autophagy, senescence, and survival. Further functional studies are warranted to elucidate the mechanism of cg14326354PRODH and age interaction on LUAD survival.
Age represents a complex surrogate for a host of underlying phenomena, although its measurement is simple and accurate [40]. A previous study suggested that gene–age interactions may partially be surrogates for gene–gene and gene–environment interactions [41]. In a study investigating the efficacy of metronomic vinorelbine to treat patients with advanced unresectable NSCLC, age was an important factor that decreased treatment efficacy [42]. Our study might provide a novel explanation of age effects on treatment efficacy from the cg14326354PRODH–age interaction perspective. Further clinical studies will provide additional insight into cg14326354PRODH and its age-specific effects in tumors, which may lead to new age-specific biomarkers and therapeutic strategies that improve prediction accuracy and treatment efficacy.
Our study has several strengths. First, this is the first epigenome-wide study to investigate the interaction between DNA methylation and age on NSCLC survival, which provides new evidence to account for the missing heritability of complex diseases [43] and may further reveal the role of age in heterogeneity of NSCLC prognosis and treatment efficacy. Second, to identify stable and reliable biomarkers, a two-stage study design along with Bonferroni correction and sensitivity analysis was used to exhaustively search for interactions, which is quite conservative in controlling for false positives. Finally, with a large sample size to analyze DNA methylation–age interactions, our study has improved statistical power to identify complex associations with small–medium effect size.
Nonetheless, several limitations also need to be acknowledged. First, we did not observe robustly significant methylation–age interactions on survival for LUSC, which may be due to limited sample size and thus insufficient power. However, there was no significant heterogeneous effect between LUAD and LUSC groups (Supplementary Table 8). Second, the association was no longer significant in young LUAD patients when the analysis used UN standards to define age groups. However, we still observed a significant association in patients <57 years old. This effect might be because >62% (240/385) of young patients defined using the UN standard (57–65 years) attenuated the effect of cg14326354PRODH methylation. Therefore, high methylation of cg14326354PRODH might benefit survival of young LUAD patients. Third, although widespread methylation–age interactions may exist, we only identified one interaction, which may be due to the most conservative correction method used in the discovery phase and limited statistical power in the validation phase due to low event rate of survival time in the TCGA population. We may need longer time to follow-up early-stage patients in TCGA for their events to occur. Nevertheless, the interaction between cg14326354PRODH and age was successfully confirmed, indicating it was a conservative and robust association. Fourth, our analysis was based on the assumption of linear additive interaction, and new statistical models can be used to properly capture non-linear methylation–age interactions. Last, the cis-regulation pattern of cg14326354PRODH requires more biological evidence, although methylation is believed to play a crucial role in regulating gene expression [44] and further influence disease gene function [45]. Therefore, our findings should be interpreted with caution, and functional experiments are warranted to confirm these associations.
In conclusion, low methylation of cg14326354PRODH benefited survival of elderly LUAD patients. Our results have implications for not only age-specific prediction of cancer survival, but also possible methylation-specific drug targeting.
Materials and Methods
Lung cancer study populations
Only early-stage (stage I–II) LUAD and LUSC patients were included in our study. DNA methylation data was harmonized from five cohorts: Harvard, Spain, Norway, Sweden, and TCGA.
Harvard
Since 1992, patients have been recruited at Massachusetts General Hospital (MGH) and histologically confirmed as primary NSCLC [46]. We profiled 151 early-stage patients from this cohort. During curative surgery, tumor specimens were collected with complete resection and snap-frozen. A MGH pathologist evaluated each specimen for tumor cell amount (tumor cellularity > 70%) and quality. Specimens were classified histologically according to World Health Organization (WHO) criteria. The Institutional Review Boards at Harvard T.H. Chan School of Public Health and MGH approved the study. All patients provided written informed consent.
Spain
The Spain cohort included 226 early-stage NSCLC patients recruited from eight sub-centers in 1991–2009 [47]. Tumor DNA was extracted from fresh-frozen tumor specimens and further checked for integrity and quantity. Patients provided written consent, and tumors were surgically collected. The study was approved by the Bellvitge Biomedical Research Institute Institutional Review Boards.
Norway
The Norway study population consisted of 133 early-stage NSCLC patients from Oslo University Hospital-Riks Hospitalet, Norway, in 2006–2011 [48]. Tumor tissues were snap-frozen in liquid nitrogen and stored at –80°C until DNA isolation. The project was developed with approval of the Oslo University Institutional Review Board and Regional Ethics Committee (S-05307). All patients provided informed consent.
Sweden
Tumor DNA was collected from 103 early-stage NSCLC patients, including 80 LUAD and 23 LUSC patients, at the Skane University Hospital in Lund, Sweden [49]. The study was developed under the approval of the Regional Ethical Review Board in Lund, Sweden (registration no. 2004/762 and 2008/702). All patients provided written informed consent.
TCGA
A total of 332 LUAD and 285 LUSC cases with full DNA methylation, survival time, and covariate data were included. Level-1 HumanMethylation450 DNA methylation data of early-stage NSCLC patient were downloaded on October 01, 2015.
Quality control for DNA methylation data
DNA methylation was assessed using Illumina Infinium HumanMethylation450 BeadChips (Illumina Inc.). Raw image data were imported into Genome Studio Methylation ModuleV1.8 (Illumina Inc.) to calculate methylation signals and to perform normalization, background subtraction, and QC. Unqualified probes were excluded if they fit any of the following criteria: (i) failed detection (P > 0.05) in ≥5% samples, (ii) coefficient of variance (CV) <5%, (iii) all samples methylated or all unmethylated, (iv) common single nucleotide polymorphisms located in probe sequence or in 10-bp flanking regions, (v) cross-reactive probes [50], or (vi) data did not pass QC in all cohorts. Samples with >5% undetectable probes were excluded. Methylation signals were further processed for quantile normalization (betaqn function in R package minfi [51]), type I and II probe correction (BMIQ function in R package lumi [52]), and adjusted for batch effects (ComBat function in R package sva) [53]. Details of QC process are described Supplementary Figure 6.
Quality control for gene expression data
The TCGA workgroup completed mRNA sequencing data processing and QC. Raw counts were normalized using RNA-sequencing by expectation maximization (RSEM). Level-3 gene quantification data were downloaded from the TCGA data portal and were further checked for quality. Expression of genes was extracted and log2-transformed before analysis. Normalization results were then evaluated through boxplots of the distribution of gene expression across all samples (Supplementary Figure 7).
Statistical analysis
Statistical analysis flow is presented in Figure 1. Patients from Harvard, Spain, Norway, and Sweden study cohorts were assigned to the discovery phase, while patients in TCGA were assigned to the validation phase.
In the discovery phase, histology-stratified analysis and Cox proportional hazards model adjusted for age, smoking status, sex, clinical stage, and study center were applied to test the methylation–age interaction effect on overall survival in LUAD and LUSC patients using the R package survival [54]. Hazard ratio (HR) and 95% confidence interval (CI) were described per 5% level of methylation decrement. The P-value threshold for multiple testing was established using the Bonferroni method, which set the significance level to 0.05 divided by number of tests. This way, overall type I error was controlled at the 0.05 level. In our study, significance level of interaction analysis was defined as 1.60×10-07 = 0.05/311,891. Interactions with P ≤ 1.60×10-07 were screened out and then further confirmed in the validation phase. Robustly significant probes were retained if they fit both of the following criteria: (i) interaction P ≤ 0.05, and (ii) direction of interaction effects was consistent across both phases. In sensitivity analysis, patients were excluded if their methylation values were out of range mean ± 3×standard deviation (SD) on logit2-transformed scale. Genome-wide expression correlation analysis was performed to identify potential trans-regulation genes in TCGA. KEGG enrichment analysis of potential trans-regulation genes (Bonferroni adjusted P < 0.05) was conducted using R package clusterProfiler [55].
Continuous variables were summarized as mean ± SD; categorical variables were described as n (%). Kaplan-Meier survival curves were used to illustrate survival difference between patients in low and high methylation groups (categorized by median value). We used two classification criteria to define young and elderly patients: (i) the UN standard (1956) of 65 years old as the cut-off value to distinguish elderly and young people [56], (ii) and a cut-off value calculated based on BoCI of HR of the CpG probe. All statistical analyses were performed in R version 3.5.1 (The R Foundation).
Supplementary Materials
Abbreviations
NSCLC: non-small cell lung cancer; LUSC: lung squamous cell carcinoma; LUAD: lung adenocarcinoma; TCGA: The Cancer Genome Atlas; BoCI: boundary of 95% CI; UN: United Nations; QC: quality control; HR: hazard ratio; CI: confidence interval; SD: standard deviation; PRODH: proline dehydrogenase.
Author Contributions
C.C., Y.W., R.Z., F.C., and D.C.C. contributed to the study design. R.Z., L.S., A.S., T.F., M.M.B., A.K., M.P., J.S., Å.H., M.E., and D.C.C. contributed to data collection. C.C., Y.W., L.Lai, R.Z., and D.C.C. performed statistical analysis and interpretation and drafted the manuscript. L.W., J.C., X.C., X.D., J.H., L.Lin, Y.Z., H.H., D.Y., S.S., W.D., L.S., and R.W. revised the manuscript. All authors contributed to critical revision of the manuscript and approved its final version. Financial support and study supervision were provided by R.Z., Y.W., F.C., and D.C.C.
Acknowledgments
The authors thank all participants enrolled in this study.
Conflicts of Interest
The authors declare no potential conflicts of interest.
Funding
This study was funded by National Key Research and Development Program of China (2016YFE0204900 to F.C.), US National Institutes of Health (CA209414, CA092824, and ES000002 to D.C.C.), National Natural Science Foundation of China (81530088 to F.C. and 81973142 to Y.W.), Natural Science Foundation of Jiangsu Province (BK20191354 to R.Z.), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJB310011 to R.Z.), China Postdoctoral Science Foundation (2018M633767 to R.Z.), and Priority Academic Program Development of Jiangsu Higher Education Institutions. R.Z. was partially supported by the Outstanding Young Teachers Training Program of Nanjing Medical University.
References
- 1. Miao R, Ge C, Zhang X, He Y, Ma X, Xiang X, Gu J, Fu Y, Qu K, Liu C, Wu Q, Lin T. Combined eight-long noncoding RNA signature: a new risk score predicting prognosis in elderly non-small cell lung cancer patients. Aging (Albany NY). 2019; 11:467–79. https://doi.org/10.18632/aging.101752 [PubMed]
- 2. Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016; 66:271–89. https://doi.org/10.3322/caac.21349 [PubMed]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019; 69:7–34. https://doi.org/10.3322/caac.21551 [PubMed]
- 4. Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY). 2015; 7:690–700. https://doi.org/10.18632/aging.100809 [PubMed]
- 5. Liu L, van Groen T, Kadish I, Li Y, Wang D, James SR, Karpf AR, Tollefsbol TO. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin Epigenetics. 2011; 2:349–60. https://doi.org/10.1007/s13148-011-0042-6 [PubMed]
- 6. Wei Y, Liang J, Zhang R, Guo Y, Shen S, Su L, Lin X, Moran S, Helland Å, Bjaanæs MM, Karlsson A, Planck M, Esteller M, et al. Epigenetic modifications in KDM lysine demethylases associate with survival of early-stage NSCLC. Clin Epigenetics. 2018; 10:41. https://doi.org/10.1186/s13148-018-0474-3 [PubMed]
- 7. Shen S, Zhang R, Guo Y, Loehrer E, Wei Y, Zhu Y, Yuan Q, Moran S, Fleischer T, Bjaanaes MM, Karlsson A, Planck M, Staaf J, et al. A multi-omic study reveals BTG2 as a reliable prognostic marker for early-stage non-small cell lung cancer. Mol Oncol. 2018; 12:913–24. https://doi.org/10.1002/1878-0261.12204 [PubMed]
- 8. Zhang R, Lai L, He J, Chen C, You D, Duan W, Dong X, Zhu Y, Lin L, Shen S, Guo Y, Su L, Shafer A, et al. EGLN2 DNA methylation and expression interact with HIF1A to affect survival of early-stage NSCLC. Epigenetics. 2019; 14:118–29. https://doi.org/10.1080/15592294.2019.1573066 [PubMed]
- 9. Zhang R, Lai L, Dong X, He J, You D, Chen C, Lin L, Zhu Y, Huang H, Shen S, Wei L, Chen X, Guo Y, et al. SIPA1L3 methylation modifies the benefit of smoking cessation on lung adenocarcinoma survival: an epigenomic-smoking interaction analysis. Mol Oncol. 2019; 13:1235–48. https://doi.org/10.1002/1878-0261.12482 [PubMed]
- 10. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016; 17:630–41. https://doi.org/10.1038/nrg.2016.93 [PubMed]
- 11. Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013; 14:R102. https://doi.org/10.1186/gb-2013-14-9-r102 [PubMed]
- 12. Zheng SC, Widschwendter M, Teschendorff AE. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics. 2016; 8:705–19. https://doi.org/10.2217/epi-2015-0017 [PubMed]
- 13. Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007; 23:413–18. https://doi.org/10.1016/j.tig.2007.05.008 [PubMed]
- 14. Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, Sugarbaker DJ, Yeh RF, Wiencke JK, Kelsey KT. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009; 5:e1000602. https://doi.org/10.1371/journal.pgen.1000602 [PubMed]
- 15. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N, Hollmann T, Schalper KA, Gainor JF, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018; 36:633–41. https://doi.org/10.1200/JCO.2017.75.3384 [PubMed]
- 16. Wright J. Epigenetics: reversible tags. Nature. 2013; 498:S10–11. https://doi.org/10.1038/498S10a [PubMed]
- 17. Liu W, Phang JM. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy. 2012; 8:1407–09. https://doi.org/10.4161/auto.21152 [PubMed]
- 18. Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012; 72:3677–86. https://doi.org/10.1158/0008-5472.CAN-12-0080 [PubMed]
- 19. Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, Verfaillie C, Grünewald TG, Fendt SM. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun. 2017; 8:15267. https://doi.org/10.1038/ncomms15267 [PubMed]
- 20. Sun K, Deng W, Zhang S, Cai N, Jiao S, Song J, Wei L. Paradoxical roles of autophagy in different stages of tumorigenesis: protector for normal or cancer cells. Cell Biosci. 2013; 3:35. https://doi.org/10.1186/2045-3701-3-35 [PubMed]
- 21. Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhász G, Bilder D, Brech A, Stenmark H, Rusten TE. Microenvironmental autophagy promotes tumour growth. Nature. 2017; 541:417–20. https://doi.org/10.1038/nature20815 [PubMed]
- 22. Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011; 17:654–66. https://doi.org/10.1158/1078-0432.CCR-10-2634 [PubMed]
- 23. Liu JT, Li WC, Gao S, Wang F, Li XQ, Yu HQ, Fan LL, Wei W, Wang H, Sun GP. Autophagy inhibition overcomes the antagonistic effect between gefitinib and cisplatin in epidermal growth factor receptor mutant non–small-cell lung cancer cells. Clin Lung Cancer. 2015; 16:e55–66. https://doi.org/10.1016/j.cllc.2015.03.006 [PubMed]
- 24. Liu W, Phang JM. Proline dehydrogenase (oxidase) in cancer. Biofactors. 2012; 38:398–406. https://doi.org/10.1002/biof.1036 [PubMed]
- 25. Torre LA, Siegel RL, Jemal A. Lung Cancer Statistics. In: Ahmad A, Gadgeel S, eds. Lung Cancer and Personalized Medicine: Current Knowledge and Therapies. Cham: Springer International Publishing, 2016. pp. 1–19.
- 26. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007; 130:223–33. https://doi.org/10.1016/j.cell.2007.07.003 [PubMed]
- 27. Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. 2017; 7:278. https://doi.org/10.3389/fonc.2017.00278 [PubMed]
- 28. Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA. 2007; 104:16633–38. https://doi.org/10.1073/pnas.0708043104 [PubMed]
- 29. Wörmann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Görgülü K, Ruess D, Campbell A, Doglioni C, Jodrell D, Neesse A, Demir IE, Karpathaki AP, et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016; 151:180–193.e12. https://doi.org/10.1053/j.gastro.2016.03.010 [PubMed]
- 30. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, Mendoza-Parra MA, Kanashova T, Metzner M, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018; 553:96–100. https://doi.org/10.1038/nature25167 [PubMed]
- 31. Zhang Y, Wang C, Zhou J, Sun A, Hueckstaedt LK, Ge J, Ren J. Complex inhibition of autophagy by mitochondrial aldehyde dehydrogenase shortens lifespan and exacerbates cardiac aging. Biochim Biophys Acta Mol Basis Dis. 2017; 1863:1919–32. https://doi.org/10.1016/j.bbadis.2017.03.016 [PubMed]
- 32. Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010; 12:503–35. https://doi.org/10.1089/ars.2009.2598 [PubMed]
- 33. Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000; 275:31505–13. https://doi.org/10.1074/jbc.M002102200 [PubMed]
- 34. Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013; 4:2300. https://doi.org/10.1038/ncomms3300 [PubMed]
- 35. Trocoli A, Djavaheri-Mergny M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am J Cancer Res. 2011; 1:629–49. [PubMed]
- 36. Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-κB addiction and its role in cancer: ‘One size does not fit all’. Oncogene. 2011; 30:1615–30. https://doi.org/10.1038/onc.2010.566 [PubMed]
- 37. Salminen A, Kaarniranta K. NF-kappaB signaling in the aging process. J Clin Immunol. 2009; 29:397–405. https://doi.org/10.1007/s10875-009-9296-6 [PubMed]
- 38. Mondal AM, Horikawa I, Pine SR, Fujita K, Morgan KM, Vera E, Mazur SJ, Appella E, Vojtesek B, Blasco MA, Lane DP, Harris CC. P53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J Clin Invest. 2013; 123:5247–57. https://doi.org/10.1172/JCI70355 [PubMed]
- 39. Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, Gascoyne RD, Grogan TM, Muller-Hermelink HK, Smeland EB, Chiorazzi M, Giltnane JM, Hurt EM, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003; 3:185–97. https://doi.org/10.1016/s1535-6108(03)00028-x [PubMed]
- 40. Shi G, Gu CC, Kraja AT, Arnett DK, Myers RH, Pankow JS, Hunt SC, Rao DC. Genetic effect on blood pressure is modulated by age: the hypertension genetic epidemiology network study. Hypertension. 2009; 53:35–41. https://doi.org/10.1161/HYPERTENSIONAHA.108.120071 [PubMed]
- 41. Bae HT, Sebastiani P, Sun JX, Andersen SL, Daw EW, Terracciano A, Ferrucci L, Perls TT. Genome-wide association study of personality traits in the long life family study. Front Genet. 2013; 4:65. https://doi.org/10.3389/fgene.2013.00065 [PubMed]
- 42. D’Ascanio M, Pezzuto A, Fiorentino C, Sposato B, Bruno P, Grieco A, Mancini R, Ricci A. Metronomic chemotherapy with vinorelbine produces clinical benefit and low toxicity in frail elderly patients affected by advanced non-small cell lung cancer. Biomed Res Int. 2018; 2018:6278403. https://doi.org/10.1155/2018/6278403 [PubMed]
- 43. Trerotola M, Relli V, Simeone P, Alberti S. Epigenetic inheritance and the missing heritability. Hum Genomics. 2015; 9:17. https://doi.org/10.1186/s40246-015-0041-3 [PubMed]
- 44. Bird A. Perceptions of epigenetics. Nature. 2007; 447:396–98. https://doi.org/10.1038/nature05913 [PubMed]
- 45. Schübeler D. Function and information content of DNA methylation. Nature. 2015; 517:321–26. https://doi.org/10.1038/nature14192 [PubMed]
- 46. Heist RS, Zhou W, Chirieac LR, Cogan-Drew T, Liu G, Su L, Neuberg D, Lynch TJ, Wain JC, Christiani DC. MDM2 polymorphism, survival, and histology in early-stage non-small-cell lung cancer. J Clin Oncol. 2007; 25:2243–47. https://doi.org/10.1200/JCO.2006.08.8914 [PubMed]
- 47. Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A, Sanchez-Cespedes M, Assenov Y, Müller F, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013; 31:4140–47. https://doi.org/10.1200/JCO.2012.48.5516 [PubMed]
- 48. Bjaanæs MM, Fleischer T, Halvorsen AR, Daunay A, Busato F, Solberg S, Jørgensen L, Kure E, Edvardsen H, Børresen-Dale AL, Brustugun OT, Tost J, Kristensen V, Helland Å. Genome-wide DNA methylation analyses in lung adenocarcinomas: association with EGFR, KRAS and TP53 mutation status, gene expression and prognosis. Mol Oncol. 2016; 10:330–43. https://doi.org/10.1016/j.molonc.2015.10.021 [PubMed]
- 49. Karlsson A, Jönsson M, Lauss M, Brunnström H, Jönsson P, Borg Å, Jönsson G, Ringnér M, Planck M, Staaf J. Genome-wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin Cancer Res. 2014; 20:6127–40. https://doi.org/10.1158/1078-0432.CCR-14-1087 [PubMed]
- 50. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the illumina infinium HumanMethylation450 microarray. Epigenetics. 2013; 8:203–09. https://doi.org/10.4161/epi.23470 [PubMed]
- 51. Fortin JP, Triche TJ
Jr , Hansen KD. Preprocessing, normalization and integration of the illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017; 33:558–60. https://doi.org/10.1093/bioinformatics/btw691 [PubMed] - 52. Du P, Kibbe WA, Lin SM. Lumi: a pipeline for processing illumina microarray. Bioinformatics. 2008; 24:1547–48. https://doi.org/10.1093/bioinformatics/btn224 [PubMed]
- 53. Marabita F, Almgren M, Lindholm ME, Ruhrmann S, Fagerström-Billai F, Jagodic M, Sundberg CJ, Ekström TJ, Teschendorff AE, Tegnér J, Gomez-Cabrero D. An evaluation of analysis pipelines for DNA methylation profiling using the illumina HumanMethylation450 BeadChip platform. Epigenetics. 2013; 8:333–46. https://doi.org/10.4161/epi.24008 [PubMed]
- 54. Therneau T. (2015). A Package for Survival Analysis in S. version 2.38.
- 55. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–87. https://doi.org/10.1089/omi.2011.0118 [PubMed]
- 56. United Nations Department of Economic and Social Affairs (1956). The aging of populations and its economic and social implications. New York: United Nations.