



Introduction
Communication between the heart and kidney plays an important role in regulating fluid balance, metabolite excretion, and neuroendocrine function to maintain homeostasis [1]. The heart pumps fresh blood to maintain organ perfusion while the kidney filters the blood and concentrates metabolic waste, which is important for regulating blood volume and maintaining cardiac output. Cardiorenal syndrome (CRS) is defined as simultaneous bidirectional dysfunction of both the heart and kidney. The term ‘cardiorenal’ was first used by Thomas Lewis in a 1913 lecture entitled “Paroxysmal Dyspnoea in Cardiorenal Patients” in which he described his observation that patients with advanced kidney disease frequently developed dyspnea [1]. More recent studies have provided additional insight into the relationship between cardiac dysfunction and renal failure [2–4]. However, the etiology of CRS has not been fully elucidated [5]. Bongartz el al. expanded the definition of CRS to account for the role of renal dysfunction in heart failure in addition to the causal role of cardiac dysfunction in kidney disease [6]. The Acute Dialysis Quality Initiative (ADQI) currently defines CRS as a pathophysiological disorder in which acute or chronic dysfunction of one organ may induce acute or chronic dysfunction in the other organ [7].
Ronco et al. proposed a classification system that divides CRS into five subtypes based on acuity of onset and the primary organ involved (Table 1) [8]. CRS types 1 and 2 (CRS-1 and CRS-2) correspond to acute and chronic cardiorenal syndrome, respectively, whereas CRS types 3 and 4 (CRS-3 and CRS-4) correspond to acute and chronic renocardiac syndrome, respectively. CRS type 5 (CRS-5) is a secondary disease process that occurs in the context of other conditions such as diabetes, sepsis, and drug toxicity. In contrast, Hatamizadeh et al. proposed classifying CRS into seven distinct categories based on pathophysiologic mechanisms and the response to treatment strategies: 1) haemodynamic, 2) uraemic, 3) vascular, 4) neurohumoral, 5) anaemia and/or iron metabolism, 6) mineral metabolism, and 7) malnutrition-inflammation-cachexia (Table 2) [9].
Table 1. Cardiorenal syndrome classification system by Ronco et al.
| CRS General Definition: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A complex pathophysiologic disorder of the heart and kidneys whereby acute or chronic dysfunction in 1 organ may induce acute or chronic dysfunction in the other organ. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Types of CRS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CRS Type 1 (acute CRS) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description: Abrupt worsening of cardiac function leading to AKI | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Examples: Acute coronary syndrome, acute decompensated heart failure | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CRS Type 2 (chronic CRS) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description: Chronic abnormalities in cardiac function causing progressive and permanent CKD | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Examples: Chronic heart failure, ischemic heart disease, hypertension | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CRS Type 3 (acute renocardiac syndrome) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description: Abrupt worsening of kidney function causing acute cardiac disorder | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Examples: Postsurgery AKI, acute glomerulonephritis, rhabdomyolysis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CRS Type 4 (chronic renocardiac syndrome) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description: CKD contributing to decreased cardiac function, cardiac hypertrophy, fibrosis, and/or increased risk for adverse cardiovascular events | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Examples: Cardiac hypertrophy/fibrosis in CKD | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CRS Type 5 (secondary CRS) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description: Systemic condition causing both acute cardiac and kidney injury and dysfunction | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Examples: sepsis, diabetes mellitus | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: AKI: acute kidney injury; CKD: chronic kidney disease; CRS: cardiorenal syndrome. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 2. Cardiorenal syndrome classification system by Hatamizadeh et al.
| CRS category (subclassified) | Manifestation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Haemodynamic (acute/chronic) | Renal dysfunction due to cardiac output | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Uremic (acute/chronic) | Uremic cardiomyopathy, Uremic pleuritis, Uremic pericarditis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vascular (acute/chronic) | Coronary artery disease, Renal artery thrombosis, Renal artery stenosis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Neurohumoral (acute/chronic) | Abnormal serum calcium, potassium, magnesium and activated RAAS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Anemia and/or iron metabolism (acute/chronic) | Iron deficiency, Renal tubularinjury, Infection, Folate deficiency | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mineral metabolism (mostly chronic) | Vitamin D, Elevated FGF 23, Hypercalcemia, Hyperphosphatemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Malnutrition/inflammation-cachexia (mostly chronic) | Cachexia, malnutrition and inflammation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: RAAS: Renin-Angiotensin system, FGF-23: Fibroblast Growth Factor 23. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In this review, we describe recent insights into the pathophysiological mechanisms underlying acute kidney injury (AKI)-induced cardiac dysfunction (CRS-3). Additionally, we discuss current approaches in the management of patients with CRS-3 including the development of targeted therapeutics.
Epidemiology of AKI and CRS-3
AKI refers to acute kidney damage or failure. Although there is a consensus on the definition and diagnostic criteria for AKI, different terms are sometimes used to describe the pathology of the kidney injury. The primary criteria used to evaluate AKI stage are an increase in serum creatinine and a decrease in urine output (Table 3) [10]. Male sex, age, diabetes, blood pressure, a history of surgery, and atrial fibrillation are independent risk factors for AKI [11, 12].
Table 3. KDIGO classification criteria for acute kidney injury.
| Stage | Serum creatinine (Scr) | Urine output (UO) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Baseline increase of 1.5 to 2 times in 7 days | <0.5 mL/kg/hour for 6–12 hours | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Baseline increase of 2 to 3 times | <0.5 mL/kg/hour for ≥12 hours | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | ≥4 mg/dL or a baseline increase >3 times or initiation of renal replacement therapy | <0.3 mL/kg/hour for ≥24 hours or anuria for ≥12 hours | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviation: KDIGO: Kidney Disease Improving Global Outcomes. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The prevalence of AKI is increasing among hospitalized patients (4.9–7.2%) [13]. Severe AKI has been observed in over 40% of patients in the intensive care unit (ICU) [13]. The overall mortality for AKI patients is approximately 50%, but can be as high as 80% among ICU patients [14, 15]. AKI primarily results in acute tubular necrosis, a reduction in the glomerular filtration rate (GFR), and a decrease in renal perfusion. A previous multi-center study of AKI patients demonstrated that more than 60% of ICU patients also developed acute cardiovascular failure [16]. More recently, a multi-national, multi-center study of ICU patients with AKI indicated that the overall hospital mortality rate was 60.3% (95% confidence interval [CI]: 58.0–62.6%) and that cardiovascular-related death was the second leading cause of death [17]. Cardiogenic shock was shown to be an independent risk factor for hospital mortality (odds ratio [OR]: 1.41; 95% confidence interval [95% CI]: 1.05–1.90) [17].
The long-term effects of AKI on the cardiovascular system have also been investigated. A 5-year population-based study demonstrated that early- and late-onset post-operative AKI were independent risk factors for a composite cardiovascular endpoint that included myocardial infarction, heart failure, and other cardiovascular causes of mortality (hazard ratio [HR]: 1.41; 95% CI: 1.11–1.80) [18]. This finding was supported by a more recent study that explored the long-term risk of coronary events following AKI among 9,738 hospitalized patients who recovered from de novo dialysis-requiring AKI between 1999–2008 [19]. The incidence of coronary events was 19.8 per 1,000 person-years among AKI patients. They also found that AKI patients had an increased risk of coronary events (HR: 1.67; 95% CI: 1.36–2.04) and all-cause mortality (HR: 1.67; 95% CI: 1.57–1.79) after adjusting for chronic kidney disease and end-stage renal disease. Another study demonstrated that a single AKI event was associated with a two-fold and eight-fold increase in hospital mortality risk among patients with left or right ventricular dysfunction, respectively [20]. Several studies have indicated that AKI is an independent risk factor for 30-day readmission among heart failure patients (OR: 1.81; 95% CI: 1.35–2.39) [21]. ‘Frequent admitters’ were found to have longer lengths of stay (4.3 days vs. 4.0 days) and higher associated costs ($7,015 vs. $2,967) compared to non-frequent admitters among patients with repeat heart failure admissions [22].
The epidemiology of CRS-3 is not well understood. The incidence is likely underestimated as a consequence of a lack of early diagnostic criteria. Cardiac failure is typically diagnosed by echocardiography, but impaired cardiac function is frequently not observed until an advanced stage. However, early recognition of cardiac dysfunction is important given that AKI-mediated cardiac damage is frequently characterized by reduced diastolic function. The temporal relationship between AKI and cardiac damage has also not been fully elucidated. Patients with compensatory heart failure are likely to develop prerenal (functional) AKI as a consequence of reduced renal blood perfusion. If prerenal AKI persists for > 2 days, renal tubular cells will undergo cell death due to reduced perfusion. This can induce renal (structural) AKI, which can lead to decompensated heart failure. Thus, it can be difficult to determine the primary cause of CRS-3, particularly among elderly patients.
Pathophysiology of CRS-3
Hemodynamics
AKI can cause cardiovascular damage through direct and1/or indirect mechanisms. Both hemodynamic and non-hemodynamic mechanisms have been proposed to explain crosstalk between the heart and kidneys (Figure 1). Hemodynamic disorders are characterized by activation of the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS) [23, 24]. SNS activation has been observed in AKI patients [25]. One study found that renal sympathetic nerve activity was elevated during renal ischemia and further increased during reperfusion [26]. Renal venous plasma norepinephrine concentrations were primarily elevated following reperfusion [26]. These findings may be explained in that the renal ischemia time was relatively short (28–30 minutes), and renal damage predominantly occurs during reperfusion. Elevated norepinephrine concentrations may be a delayed response to increased SNS activity. Although activation of the SNS enhances cardiac output by increasing myocardial contractility, higher levels of norepinephrine can cause increased cardiomyocyte oxygen consumption, cardiomyocyte death, and dysregulation of intracellular calcium. Additionally, activation of the SNS is followed by vasoconstriction and a reduction in the blood supply to the renal tubules, which exacerbates necrosis. Feedback mechanisms involving SNS activation and AKI can promote cardiac failure. Renal denervation can reverse AKI-induced histological alterations in the kidney and is therefore a potential therapeutic approach for preserving cardiac function following AKI [26].
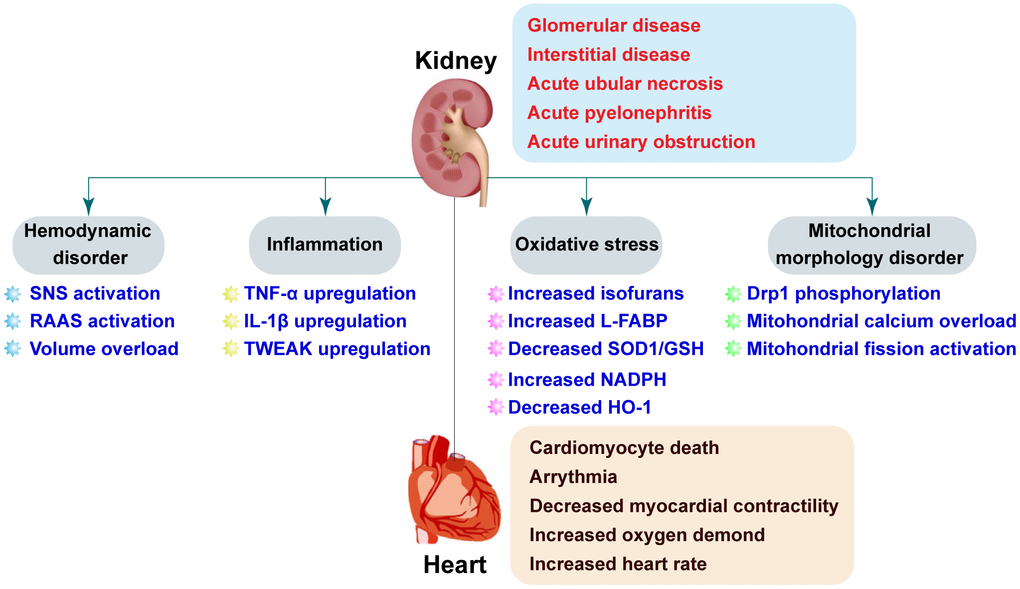
Figure 1. Pathophysiologic mechanisms underlying CRS-3. AKI is the initial insult in CRS-3 and has multiple potential etiologies. AKI may cause acute cardiac injury including heart failure, ischemia, and arrhythmia through both direct (e.g. SNS activation and RAAS) and indirect (e.g. volume overload, inflammation, oxidative stress, and mitochondrial dysfunction) effects.
RAAS activity is also elevated following AKI. Activation of the SNS stimulates β1-adrenergic receptors in the juxtaglomerular apparatus of the kidney resulting in reduced renal blood flow and activation of RAAS [27]. Juxtaglomerular cells in the juxtaglomerular apparatus secrete renin into the circulation in response to the reduction in renal perfusion. Renin cleaves angiotensinogen to yield angiotensin I (Ang I), which is then converted into angiotensin II (Ang II) by angiotensin converting enzyme. Increased Ang II concentrations are associated with systemic vascular resistance and water-sodium retention. These alterations can significantly augment cardiac preload and afterload. Ang II also exerts direct effects on cardiomyocytes by inducing hypertrophy and apoptosis [28, 29]. Interestingly, treatment with a RAAS inhibitor has been shown to protect against AKI and cardiac failure [30, 31].
AKI is characterized by a reduction in urine output and water-sodium retention. Increased blood volume, particularly venous congestion, results in an increase in cardiac preload. Volume overload causes myocardial edema, resulting in decreased myocardial contractility and left ventricular compliance [23, 24]. Right ventricular collagenase activity also increases in response to chronic interstitial edema, which contributes to myocardial remodeling [24]. Volume overload has also been shown to induce ventricular arrhythmias [32]. Although activation of the SNS and RAAS may be an adaptive response to AKI, dysregulated neurohumoral mechanisms also play an important role in promoting myocardial depression following AKI. Inhibition of SNS/RAAS activation is a standard treatment for patients with AKI.
Inflammation
Inflammation is thought to be the primary non-hemodynamic mechanism that contributes to CRS-3 (Figure 1). A previous study demonstrated that two cytokines (TNF-α and IL-1) were released from ischemic kidney tissue into the blood, which resulted in an increase in TNF-α levels in myocardial tissue in a bilateral renal ischemia-induced mouse model of AKI [33]. Excessive TNF-α accumulation contributed to cardiac damage, as evidenced by an increase in the left ventricular end-diastolic and end-systolic diameters, and fractional shortening measured by echocardiography. Interestingly, TNF-α inhibition attenuated AKI-induced cardiac damage. These results are supported by a recent study by Alarcon et al., who explored the molecular basis of AKI-induced cardiac arrhythmias [34]. They found that Nlrp3-/- and Casp1-/- mice had normal QJ intervals and fewer ventricular arrhythmias compared to wide-type mice following renal ischemia/reperfusion (I/R) injury [34]. AKI was shown to cause IL-1β overproduction. Treatment with an IL-1β antagonist rescued the duration and amplitude of the calcium transient thereby protecting against ventricular arrhythmias. However, Toldo et al. demonstrated that exogenously administered IL-1β was associated with depressed myocardial contractility [35]. AKI-mediated inflammation can induce apoptosis in cardiomyocytes and promote cardiac fibrosis [36]. Sanz et al. demonstrated that tumor necrosis factor-like weak inducer of apoptosis (TWEAK) levels were elevated during AKI and that pharmacologic inhibition of TWEAK inhibited cardiac remodeling [36]. Collectively, these data suggest that inflammation-associated cytokines may be involved in transmitting signals from damaged kidney tissue to the heart resulting in myocardial depression.
Given the critical role of TNF-α in triggering AKI-related cardiovascular events and regulating myocardial contractility, several large multi-center trials have been performed to evaluate whether the TNF-α inhibitor etanercept could prevent heart failure. However, the results have suggested that etanercept has limited clinical efficacy and potentially results in worse patient outcomes [37].
Oxidative stress
Oxidative stress results from an imbalance between the production and removal of reactive oxygen species (ROS). Four types of ROS have been defined: superoxide anion (O2·-), hydroxyl radical (·OH), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl). ROS are involved in the regulation of cardiomyocyte viability and function. At physiological concentrations, ROS act as second messengers that are required for intracellular signal transduction. However, high levels of ROS are toxic to cells because they can induce protein and lipid oxidation. ROS can directly damage DNA and cause cell death through oxidative stress-mediated apoptosis. Overproduction of ROS is caused by increased oxidative metabolism and decreased antioxidative capacity. ROS are derived from both endogenous and exogenous sources. Mitochondrial electron transport, xanthine oxidase (XO), and nicotinamide adenine dinucleotide phosphate (NADPH) are the primary generators of endogenous ROS [38, 39]. Radiation, xenobiotics, the inflammatory response, cigarettes, and alcohol are the primary exogenous agents that generate ROS.
Antioxidative capacity is regulated by a series of antioxidants including superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione (GSH) and oxidized glutathione (GSSG) [40]. Several studies have revealed that antioxidants and ROS-induced lipid peroxidation products can function as biomarkers of AKI. Ware et al. evaluated the levels of circulating lipid peroxidation products in ICU patients by mass spectroscopy [41]. They found that plasma levels of F2-isoprostanes and isofurans were highly correlated with renal damage, suggesting that lipid oxidation is predictive of acute kidney injury. Interestingly, both of F2-isoprostanes [42] and isofurans [43] have been monitored as markers of chronic and acute cardiac damage, respectively. Liver-type fatty acid-binding protein (L-FABP), a cytotoxic oxidation product secreted by proximal tubular epithelial cells, was identified as a predictive biomarker of AKI [44, 45]. Other biomarkers of kidney damage have also been shown to be predictive of acute and chronic cardiac damage (e.g. heart failure) [46, 47]. Cardiorenal “connectors” such as erythrocyte superoxide dismutase (SOD1), GSH, and NADPH have also been used to measure kidney and heart injury [48–53].
Parenica et al. performed a retrospective controlled study to investigate the role of AKI in ST-elevation myocardial infarction. They found that circulating biomarkers of the nitric oxide (NO) pathway were associated with AKI [54]. Additionally, they showed that increased NOx was correlated with 3-month cardiovascular mortality. Heme oxygenase 1 (HO-1) is the rate-limiting enzyme that catalyzes the oxidative degradation of cellular heme to liberate free iron, carbon monoxide (CO), and biliverdin in mammalian cells. HO-1 regulates oxidative stress, activates autophagy, suppresses inflammation, and promotes cell cycle progression [55]. HO-1 deficiency sensitizes kidneys to I/R injury whereas upregulation attenuates AKI. Loss of HO-1 results in an increase in IL-6, which induces post-transcriptional phosphorylation of STAT3 in the heart and kidney following injury [56]. Two other studies have demonstrated that HO-1 is overexpressed in the heart [57, 58]. Because oxidative stress-related molecules can induce damage in the heart and kidney, therapies that reduce oxidative stress could be effective in CRS-3 (Figure 1). The endothelial nitric oxide synthase (eNOS) system primarily regulates vascular tone, which is important for cardiac function. Interestingly, eNOS levels are downregulated in the kidney after AKI [59]. It is possible that signals transmitted from renal endothelial cells to cardiac endothelia cells may explain the connection between AKI and cardiac dysfunction.
Mitochondrial dysfunction
Mitochondria regulate cellular energy production through the tricarboxylic acid (TCA) cycle in which oxygen and glucose and broken down to generate ATP and H2O. Byproducts of the TCA cycle including ROS and lactic acid are released from the mitochondria into the cytoplasm where they regulate cell proliferation, pH, metabolism, and cell death. Mitochondria also play important roles in activating apoptosis. Mitochondrial dysfunction results in reduced ATP production and a reduction in cellular energy metabolism [60]. Damaged mitochondria can release pro-apoptotic factors into the cytoplasm and trigger apoptosis [61]. Additionally, interactions between mitochondria and other organelles such as the endoplasmic reticulum can activate apoptosis [62, 63].
The heart and kidney have high mitochondrial content compared to other organs, which makes them highly sensitive to the effects of mitochondrial dysfunction. Mitochondria-related oxidative stress can contribute to cardiorenal damage. Interestingly, alterations in mitochondrial morphology may play an important role in CRS-3. I/R injury can promote mitochondrial fission, which results in the division of a single mitochondria into two smaller units. Under normal physiological conditions, approximately 5% of renal tubules contain mitochondrial debris. However, approximately 50% or more of the tubules can be filled with fragmented mitochondria after AKI [64–66]. Mitochondrial fission is considered an early event in acute cardiac I/R injury that can induce cardiomyocyte apoptosis. Sumida et al. found that AKI induced mitochondrial fragmentation in heart tissue by promoting phosphorylation of dynamin-related protein 1 (Drp1) [67]. Inhibition of mitochondrial fission through administration of the Drp1 inhibitor Mdivi-1 attenuated I/R-mediated kidney damage and improved cardiac performance following AKI [67] (Figure 1).
Both oxidative stress and inflammation in the myocardium can induce mitochondrial fission [67]. Excessive mitochondrial fission results in a reduction in the mitochondrial membrane potential and the release of pro-apoptotic factors into the cytoplasm [68]. Mitochondrial fission can therefore function similarly to intracellular second messengers that sense extracellular signals (e.g. oxidative stress, inflammation, and hemodynamic changes) and then transmit these signals by undergoing changes in morphology. Alterations in mitochondrial shape suppress oxidative phosphorylation and activate mitochondria-dependent apoptosis, resulting in myocardial damage. Wang et al. demonstrated that mitochondrial calcium overload is also required for AKI-mediated mitochondrial fission in cardiomyocytes [68]. Additionally, Wang et al. reported that AKI induces phosphorylation of 1,4,5-trisphosphate receptor (IP3R) and upregulation of mitochondrial calcium uniporter (MCU) in cardiomyocytes [66]. Phosphorylation of IP3R and upregulation of MCU contributed to mitochondrial overload in cardiomyocytes, resulting in phosphorylation of Drp1 and mitochondrial fission [66]. These results indicate AKI activates Drp1-related mitochondrial fission in cardiomyocytes.
Conclusions
In this review, we have summarized recent insights into the pathophysiological mechanisms underlying CRS-3. We propose a three-step mechanism that could explain the pathophysiology of CRS-3. Following AKI, the damaged kidney tissue first releases pro-inflammatory factors and oxidative metabolites into the circulation. Alterations in the neuroendocrine system also result in the secretion of several hormones into the blood. Next, kidney-derived biomolecules directly interact with receptors or adaptors on the surfaces of cardiomyocytes. It is also possible that they exert indirect effects on cardiomyocytes through other mechanisms. Finally, mitochondria respond to the kidney-derived biomolecules by changing morphology, which results in a reduction in ATP production and activation of apoptosis in cardiomyocytes. Our hypothesis has several limitations. First, the receptors or adaptors expressed on the surfaces of cardiomyocytes have not been verified. Second, mitochondria are not the only determinants of pathological alterations in cells. Intracellular acidosis, disorders of calcium metabolism, and mechanical pressure resulting from fluid overload may also trigger cardiomyocyte damage. Therefore, further studies of the relationships between mitochondria and other intracellular stress responses are required in order to understand the sequence of events that lead to cardiac damage following AKI.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by supported by Military Medical Key projects (BLB19J009) and the National Key R&D Program of China (2017YFA0103200, 2018YFA0108803).
References
- 1. Lewis T. A clinical lecture on paroxysmal dyspnoea in cardiorenal patients: with special reference to “Cardiac” and “Uraemic” asthma: delivered at university college hospital, London, November 12th, 1913. Br Med J. 1913; 2:1417–20. https://doi.org/10.1136/bmj.2.2761.1417 [PubMed]
- 2. Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med. 1999; 341:577–85. https://doi.org/10.1056/NEJM199908193410806 [PubMed]
- 3. Merrill AJ. Edema and decreased renal blood flow in patients with chronic congestive heart failure; evidence of forward failure as the primary cause of edema. J Clin Invest. 1946; 25:389–400. [PubMed]
- 4. McCullough PA, Kellum JA, Haase M, Müller C, Damman K, Murray PT, Cruz D, House AA, Schmidt-Ott KM, Vescovo G, Bagshaw SM, Hoste EA, Briguori C, et al. Pathophysiology of the cardiorenal syndromes: executive summary from the eleventh consensus conference of the acute dialysis quality initiative (ADQI). Contrib Nephrol. 2013; 182:82–98. https://doi.org/10.1159/000349966 [PubMed]
- 5. Shlipak MG, Massie BM. The clinical challenge of cardiorenal syndrome. Circulation. 2004; 110:1514–17. https://doi.org/10.1161/01.CIR.0000143547.55093.17 [PubMed]
- 6. Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, Braam B. The severe cardiorenal syndrome: ‘Guyton revisited'. Eur Heart J. 2005; 26:11–17. https://doi.org/10.1093/eurheartj/ehi020 [PubMed]
- 7. Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, Bellomo R, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, et al, and Acute Dialysis Quality Initiative (ADQI) consensus group. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010; 31:703–11. https://doi.org/10.1093/eurheartj/ehp507 [PubMed]
- 8. Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008; 52:1527–39. https://doi.org/10.1016/j.jacc.2008.07.051 [PubMed]
- 9. Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, Kalantar-Zadeh K. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol. 2013; 9:99–111. https://doi.org/10.1038/nrneph.2012.279 [PubMed]
- 10. Kellum JA, Lameire N, and KDIGO AKI Guideline Work Group. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (part 1). Crit Care. 2013; 17:204. https://doi.org/10.1186/cc11454 [PubMed]
- 11. Dos Santos RP, Carvalho AR, Peres LA. Incidence and risk factors of acute kidney injury in critically ill patients from a single centre in Brazil: a retrospective cohort analysis. Sci Rep. 2019; 9:18141. https://doi.org/10.1038/s41598-019-54674-1 [PubMed]
- 12. Wang J, Yu W, Zhou Y, Yang Y, Li C, Liu N, Hou X, Wang L. Independent risk factors contributing to acute kidney injury according to updated valve academic research consortium-2 criteria after transcatheter aortic valve implantation: a meta-analysis and meta-regression of 13 studies. J Cardiothorac Vasc Anesth. 2017; 31:816–26. https://doi.org/10.1053/j.jvca.2016.12.021 [PubMed]
- 13. Bucuvic EM, Ponce D, Balbi AL. Risk factors for mortality in acute kidney injury. Rev Assoc Med Bras (1992). 2011; 57:158–63. https://doi.org/10.1590/s0104-42302011000200012 [PubMed]
- 14. Medeiros P, Nga HS, Menezes P, Bridi R, Balbi A, Ponce D. Acute kidney injury in septic patients admitted to emergency clinical room: risk factors and outcome. Clin Exp Nephrol. 2015; 19:859–66. https://doi.org/10.1007/s10157-014-1076-9 [PubMed]
- 15. Oweis AO, Alshelleh SA. Incidence and outcomes of acute kidney injury in octogenarians in Jordan. BMC Res Notes. 2018; 11:279. https://doi.org/10.1186/s13104-018-3397-3 [PubMed]
- 16. Liaño F, Junco E, Pascual J, Madero R, Verde E. The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The madrid acute renal failure study group. Kidney Int Suppl. 1998; 66:S16–24. [PubMed]
- 17. Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C, and Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005; 294:813–18. https://doi.org/10.1001/jama.294.7.813 [PubMed]
- 18. Hansen MK, Gammelager H, Jacobsen CJ, Hjortdal VE, Layton JB, Rasmussen BS, Andreasen JJ, Johnsen SP, Christiansen CF. Acute kidney injury and long-term risk of cardiovascular events after cardiac surgery: a population-based cohort study. J Cardiothorac Vasc Anesth. 2015; 29:617–25. https://doi.org/10.1053/j.jvca.2014.08.020 [PubMed]
- 19. Wu VC, Wu CH, Huang TM, Wang CY, Lai CF, Shiao CC, Chang CH, Lin SL, Chen YY, Chen YM, Chu TS, Chiang WC, Wu KD, et al, and NSARF Group. Long-term risk of coronary events after AKI. J Am Soc Nephrol. 2014; 25:595–605. https://doi.org/10.1681/ASN.2013060610 [PubMed]
- 20. Chen C, Lee J, Johnson AE, Mark RG, Celi LA, Danziger J. Right ventricular function, peripheral edema, and acute kidney injury in critical illness. Kidney Int Rep. 2017; 2:1059–65. https://doi.org/10.1016/j.ekir.2017.05.017 [PubMed]
- 21. Thakar CV, Parikh PJ, Liu Y. Acute kidney injury (AKI) and risk of readmissions in patients with heart failure. Am J Cardiol. 2012; 109:1482–86. https://doi.org/10.1016/j.amjcard.2012.01.362 [PubMed]
- 22. Go YY, Sellmair R, Allen JC
Jr , Sahlén A, Bulluck H, Sim D, Jaufeerally FR, MacDonald MR, Lim ZY, Chai P, Loh SY, Yap J, Lam CS. Defining a ‘Frequent admitter’ phenotype among patients with repeat heart failure admissions. Eur J Heart Fail. 2019; 21:311–18. https://doi.org/10.1002/ejhf.1348 [PubMed] - 23. Desai KV, Laine GA, Stewart RH, Cox CS
Jr , Quick CM, Allen SJ, Fischer UM. Mechanics of the left ventricular myocardial interstitium: effects of acute and chronic myocardial edema. Am J Physiol Heart Circ Physiol. 2008; 294:H2428–34. https://doi.org/10.1152/ajpheart.00860.2007 [PubMed] - 24. Davis KL, Laine GA, Geissler HJ, Mehlhorn U, Brennan M, Allen SJ. Effects of myocardial edema on the development of myocardial interstitial fibrosis. Microcirculation. 2000; 7:269–80. [PubMed]
- 25. Hering D, Winklewski PJ. Autonomic nervous system in acute kidney injury. Clin Exp Pharmacol Physiol. 2017; 44:162–71. https://doi.org/10.1111/1440-1681.12694 [PubMed]
- 26. Fujii T, Kurata H, Takaoka M, Muraoka T, Fujisawa Y, Shokoji T, Nishiyama A, Abe Y, Matsumura Y. The role of renal sympathetic nervous system in the pathogenesis of ischemic acute renal failure. Eur J Pharmacol. 2003; 481:241–48. https://doi.org/10.1016/j.ejphar.2003.09.036 [PubMed]
- 27. Saxena PR. Interaction between the renin-angiotensin-aldosterone and sympathetic nervous systems. J Cardiovasc Pharmacol. 1992 (Suppl 6); 19:S80–88. https://doi.org/10.1097/00005344-199219006-00013 [PubMed]
- 28. Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000; 52:11–34. [PubMed]
- 29. Kajstura J, Cigola E, Malhotra A, Li P, Cheng W, Meggs LG, Anversa P. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J Mol Cell Cardiol. 1997; 29:859–70. https://doi.org/10.1006/jmcc.1996.0333 [PubMed]
- 30. Tomson C, Tomlinson LA. Stopping RAS inhibitors to minimize AKI: more harm than good? Clin J Am Soc Nephrol. 2019; 14:617–19. https://doi.org/10.2215/CJN.14021118 [PubMed]
- 31. Chou YH, Chu TS, Lin SL. Role of renin-angiotensin system in acute kidney injury-chronic kidney disease transition. Nephrology (Carlton). 2018 (Suppl 4); 23:121–25. https://doi.org/10.1111/nep.13467 [PubMed]
- 32. Ip JE, Cheung JW, Park D, Hellawell JL, Stein KM, Iwai S, Liu CF, Lerman BB, Markowitz SM. Temporal associations between thoracic volume overload and Malignant ventricular arrhythmias: a study of intrathoracic impedance. J Cardiovasc Electrophysiol. 2011; 22:293–99. https://doi.org/10.1111/j.1540-8167.2010.01924.x [PubMed]
- 33. Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003; 14:1549–58. https://doi.org/10.1097/01.asn.0000064946.94590.46 [PubMed]
- 34. Alarcon MM, Trentin-Sonoda M, Panico K, Schleier Y, Duque T, Moreno-Loaiza O, de Yurre AR, Ferreira F, Caio-Silva W, Coury PR, Paiva CN, Medei E, Carneiro-Ramos MS. Cardiac arrhythmias after renal I/R depend on IL-1β. J Mol Cell Cardiol. 2019; 131:101–11. https://doi.org/10.1016/j.yjmcc.2019.04.025 [PubMed]
- 35. Toldo S, Mezzaroma E, O'Brien L, Marchetti C, Seropian IM, Voelkel NF, Van Tassell BW, Dinarello CA, Abbate A. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014; 306:H1025–31. https://doi.org/10.1152/ajpheart.00795.2013 [PubMed]
- 36. Sanz AB, Ruiz-Andres O, Sanchez-Niño MD, Ruiz-Ortega M, Ramos AM, Ortiz A. Out of the TWEAKlight: elucidating the role of Fn14 and TWEAK in acute kidney injury. Semin Nephrol. 2016; 36:189–98. https://doi.org/10.1016/j.semnephrol.2016.03.006 [PubMed]
- 37. Anker SD, Coats AJ. How to recover from renaissance? the significance of the results of recover, renaissance, renewel and attach. Int J Cardiol. 2002; 86:123–30. https://doi.org/10.1016/s0167-5273(02)00470-9 [PubMed]
- 38. Zhou H, Toan S. Pathological roles of mitochondrial oxidative stress and mitochondrial dynamics in cardiac microvascular ischemia/reperfusion injury. Biomolecules. 2020; 10:85. https://doi.org/10.3390/biom10010085 [PubMed]
- 39. Biernacki M, Ambrożewicz E, Gęgotek A, Toczek M, Bielawska K, Skrzydlewska E. Redox system and phospholipid metabolism in the kidney of hypertensive rats after FAAH inhibitor URB597 administration. Redox Biol. 2018; 15:41–50. https://doi.org/10.1016/j.redox.2017.11.022 [PubMed]
- 40. Lee BW, Ghode P, Ong DS. Redox regulation of cell state and fate. Redox Biol. 2019; 25:101056. https://doi.org/10.1016/j.redox.2018.11.014 [PubMed]
- 41. Ware LB, Fessel JP, May AK, Roberts LJ
2nd . Plasma biomarkers of oxidant stress and development of organ failure in severe sepsis. Shock. 2011; 36:12–17. https://doi.org/10.1097/SHK.0b013e318217025a [PubMed] - 42. Azizi-Namini P, Ahmed M, Yan AT, Desjardins S, Al-Hesayen A, Mangat I, Keith M. Prevalence of thiamin deficiency in ambulatory patients with heart failure. J Acad Nutr Diet. 2019; 119:1160–67. https://doi.org/10.1016/j.jand.2019.01.015 [PubMed]
- 43. Lopez MG, Pandharipande P, Morse J, Shotwell MS, Milne GL, Pretorius M, Shaw AD, Roberts LJ
2nd , Billings FT4th . Intraoperative cerebral oxygenation, oxidative injury, and delirium following cardiac surgery. Free Radic Biol Med. 2017; 103:192–98. https://doi.org/10.1016/j.freeradbiomed.2016.12.039 [PubMed] - 44. Katayama M, Miyazaki T, Ohata K, Oikawa T, Kamiie J, Sugaya T, Miyazaki M. Temporal changes in urinary excretion of liver-type fatty acid binding protein (L-FABP) in acute kidney injury model of domestic cats: a preliminary study. J Vet Med Sci. 2019; 81:1868–72. https://doi.org/10.1292/jvms.19-0325 [PubMed]
- 45. Yoneyama F, Okamura T, Takigiku K, Yasukouchi S. Novel Urinary Biomarkers for Acute Kidney Injury and Prediction of Clinical Outcomes After Pediatric Cardiac Surgery. Pediatr Cardiol. 2019. [Epub ahead of print]. https://doi.org/10.1007/s00246-019-02280-3 [PubMed]
- 46. Okubo Y, Sairaku A, Morishima N, Ogi H, Matsumoto T, Kinoshita H, Kihara Y. Increased urinary liver-type fatty acid-binding protein level predicts worsening renal function in patients with acute heart failure. J Card Fail. 2018; 24:520–24. https://doi.org/10.1016/j.cardfail.2018.07.003 [PubMed]
- 47. Nishida M, Kubo S, Morishita Y, Nishikawa K, Ikeda K, Itoi T, Hosoi H. Kidney injury biomarkers after cardiac angiography in children with congenital heart disease. Congenit Heart Dis. 2019; 14:1087–93. https://doi.org/10.1111/chd.12853 [PubMed]
- 48. Xing JJ, Hou JG, Liu Y, Zhang RB, Jiang S, Ren S, Wang YP, Shen Q, Li W, Li XD, Wang Z. Supplementation of saponins from leaves of Panax quinquefolius mitigates cisplatin-evoked cardiotoxicity via inhibiting oxidative stress-associated inflammation and apoptosis in mice. Antioxidants (Basel). 2019; 8:347. https://doi.org/10.3390/antiox8090347 [PubMed]
- 49. Islam MS, Miao L, Yu H, Han Z, Sun H. Ethanol extract of Illicium henryi attenuates LPS-induced acute kidney injury in mice via regulating inflammation and oxidative stress. Nutrients. 2019; 11:1412. https://doi.org/10.3390/nu11061412 [PubMed]
- 50. Wei H, Zhang R, Su Y, Bi Y, Li X, Zhang X, Li J, Bao J. Effects of acute cold stress after long-term cold stimulation on antioxidant status, heat shock proteins, inflammation and immune cytokines in broiler heart. Front Physiol. 2018; 9:1589. https://doi.org/10.3389/fphys.2018.01589 [PubMed]
- 51. Li J, Xu S, Zhu JB, Song J, Luo B, Song YP, Zhang ZH, Chen YH, Zhang ZQ, Xie DD, Yu DX, Xu DX. Pretreatment with cholecalciferol alleviates renal cellular stress response during ischemia/reperfusion-induced acute kidney injury. Oxid Med Cell Longev. 2019; 2019:1897316. https://doi.org/10.1155/2019/1897316 [PubMed]
- 52. Sampaio TL, Menezes RR, Lima DB, Costa Silva RA, de Azevedo IE, Magalhães EP, Marinho MM, Dos Santos RP, Martins AM. Involvement of NADPH-oxidase enzyme in the nephroprotective effect of (-)-α-bisabolol on HK2 cells exposed to ischemia - reoxygenation. Eur J Pharmacol. 2019; 855:1–9. https://doi.org/10.1016/j.ejphar.2019.04.044 [PubMed]
- 53. Wen SY, Tamilselvi S, Shen CY, Day CH, Chun LC, Cheng LY, Ou HC, Chen RJ, Viswanadha VP, Kuo WW, Huang CY. Protective effect of HDL on NADPH oxidase-derived super oxide anion mediates hypoxia-induced cardiomyocyte apoptosis. PLoS One. 2017; 12:e0179492. https://doi.org/10.1371/journal.pone.0179492 [PubMed]
- 54. Parenica J, Kala P, Mebazaa A, Littnerova S, Benesova K, Tomandl J, Goldbergová Pavkova M, Jarkovský J, Spinar J, Tomandlova M, Dastych M, Ince C, Helanova K, et al, and GREAT Network. Activation of the nitric oxide pathway and acute myocardial infarction complicated by acute kidney injury. Cardiorenal Med. 2020; 10:85–96. https://doi.org/10.1159/000503718 [PubMed]
- 55. Bolisetty S, Zarjou A, Agarwal A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am J Kidney Dis. 2017; 69:531–45. https://doi.org/10.1053/j.ajkd.2016.10.037 [PubMed]
- 56. Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, Kane GC, Terzic A, Griffin MD, Nath KA. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007; 72:1073–80. https://doi.org/10.1038/sj.ki.5002471 [PubMed]
- 57. Jain K, Suryakumar G, Ganju L, Singh SB. Amelioration of ER stress by 4-phenylbutyric acid reduces chronic hypoxia induced cardiac damage and improves hypoxic tolerance through upregulation of HIF-1α. Vascul Pharmacol. 2016; 83:36–46. https://doi.org/10.1016/j.vph.2016.03.004 [PubMed]
- 58. Chen G, Liu G, Cao D, Jin M, Guo D, Yuan X. Polydatin protects against acute myocardial infarction-induced cardiac damage by activation of Nrf2/HO-1 signaling. J Nat Med. 2019; 73:85–92. https://doi.org/10.1007/s11418-018-1241-7 [PubMed]
- 59. Ohkita M, Hayashi H, Ito K, Shigematsu N, Tanaka R, Tsutsui H, Matsumura Y. Preventive effects of grape extract on ischemia/reperfusion-induced acute kidney injury in mice. Biol Pharm Bull. 2019; 42:1883–90. https://doi.org/10.1248/bpb.b19-00462 [PubMed]
- 60. Bartnik E, Tońska K, Rusecka J. [Mitochondrial diseases 2018]. Postepy Biochem. 2018; 64:300–03. https://doi.org/10.18388/pb.2018_143 [PubMed]
- 61. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018; 25:1080–93. https://doi.org/10.1038/s41418-018-0086-7 [PubMed]
- 62. Zhou H, Wang S, Hu S, Chen Y, Ren J. ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front Physiol. 2018; 9:755. https://doi.org/10.3389/fphys.2018.00755 [PubMed]
- 63. Li J, Zhang D, Brundel BJ, Wiersma M. Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells. 2019; 8:1617. https://doi.org/10.3390/cells8121617 [PubMed]
- 64. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017; 13:629–46. https://doi.org/10.1038/nrneph.2017.107 [PubMed]
- 65. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020; 30:101415. https://doi.org/10.1016/j.redox.2019.101415 [PubMed]
- 66. Wang J, Zhu P, Toan S, Li R, Ren J, Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol Toxicol. 2020. [Epub ahead of print]. https://doi.org/10.1007/s10565-020-09513-9 [PubMed]
- 67. Sumida M, Doi K, Ogasawara E, Yamashita T, Hamasaki Y, Kariya T, Takimoto E, Yahagi N, Nangaku M, Noiri E. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol. 2015; 26:2378–87. https://doi.org/10.1681/ASN.2014080750 [PubMed]
- 68. Wang J, Zhu P, Li R, Ren J, Zhang Y, Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020; 10:384–97. https://doi.org/10.7150/thno.40098 [PubMed]