



Introduction
The blood is a circulating representation of all body tissues and hence carries a plethora of information about the overall pathophysiology of an individual. Age-related changes in the blood have been investigated by metabolomics or proteomics, with the goal of understanding the mechanisms of aging and age-related pathogenesis, and also to identify candidate diagnostic and prognostic biomarkers for diseases [1–4]. Longitudinal metabolomics data from human plasma have revealed that plasma metabolite levels are influenced by aging, and that numerous metabolites are correlated with age [1, 5]. Using the SomaScan aptamer technology, two studies identified a conserved aging signature in human plasma proteomic data [2, 3]. These observations offer a deeper understanding of the aging process and provide new insight into the molecular mechanisms underlying human health and aging.
Changes in plasma proteins contribute to the aging process, as well as age-related diseases. Parabiosis models, aimed at elucidating the effect of young or old blood on aging, revealed that young blood rejuvenates aged tissues in multiple organs, including brain, heart, pancreas, bone, skeletal muscle [6–10]. These studies strongly support the notion that both pro-aging and rejuvenating factors are present in the circulation, and a wide range of efforts are underway to identify these factors. Initial studies in parabiosis models identified GDF11 as a molecule capable of rejuvenating cerebral, cardiac, and skeletal muscle functions [7, 11, 12]. Similarly, the C-C motif chemokine 11 (CCL11) and β2 microglobulin (B2M) negatively regulate neurogenesis and cognitive function in the hippocampus. In addition, several plasma proteins have been identified as rejuvenating factors that provide beneficial effects in diverse tissues. Based on the identification of these circulating proteins, anti-aging startups have started to develop new therapies targeting age-related diseases [13].
The discovery and validation of rejuvenating factors have been investigated so far with diverse approaches. In this review, we highlight the potential roles of selected plasma proteins that are present at different levels between young and old blood, and describe the results of many fascinating studies reporting pro-aging or rejuvenating factors in the blood (Table 1 and Figure 1).
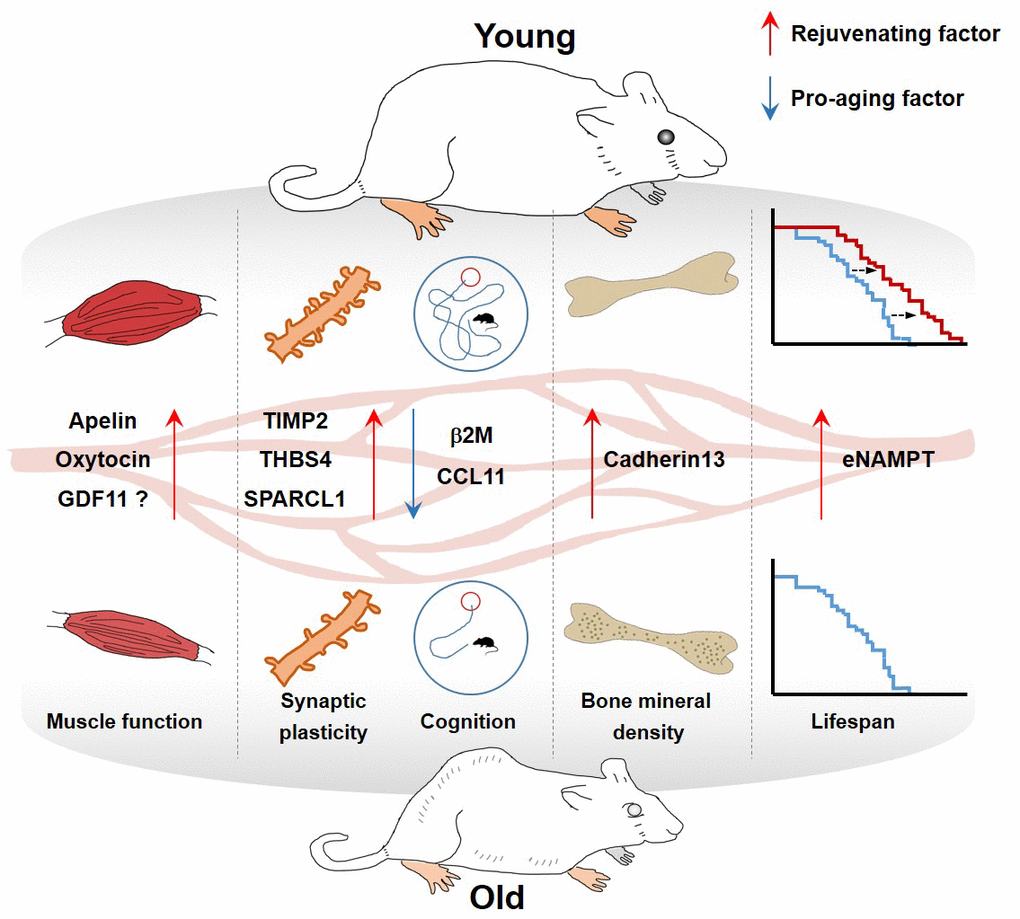
Figure 1. Effect of blood factors on organs.
Table 1. Summary of circulating plasma factors involved in rejuvenation.
| Circulating factor | Plasma levels in aged mice | Class | Functions | References |
| Apelin | Decline | Hormone (Rejuvenating factor) | • Extends the murine healthspan and promotes mitochondriogenesis and protein synthesis | Vinel et al., Nature Medicine (2018) |
| • Reverses age-associated muscle wasting and delays stress-induced cellular senescence | Rai et al., Cell Reports (2017) | |||
| β2-microglobulin | Increase | component of MHC class I molecules (Pro-aging factor) | • Impairs hippocampus-dependent cognitive function and neurogenesis | Smith et al., Nature Medicine (2016) |
| Cadherin13 | Decline | cell adhesion protein (Rejuvenating factor) | • Inhibits osteoclast differentiation and ameliorates age-associated bone loss | Yang et al. Aging (2020) |
| CCL11 | Increase | Cytokine (Pro-aging factor) | • Reduced synaptic plasticity, and impairment of contextual fear conditioning and spatial learning and memory. | Villeda et al., Nature (2011) |
| GDF11 | Decline | Cytokine (Rejuvenating factor) | • Reverses age-related cardiac hypertrophy by suppressing phosphorylation Forkhead transcription factor | Loffredo et al., Cell (2013) |
| Not verified | • Restores the functionality of the vasculature of the neurogenic niches | Katsimpardi et. al., Science (2014) | ||
| • Improves muscle physiology and physical function by improving satellite cell competency | Sinha et al., Science (2014) | |||
| • Shows calorie restriction–like phenotype | Katsimpardi et al., Aging Cell (2020) | |||
| • Increases bone mass by stimulating the BMP signaling pathway, thereby promoting osteogenic differentiation | Suh et al., PNAS (2020) | |||
| Increase | (Pro-aging factor) | • Impairs muscle regeneration | Egerman et al. Cell Metab (2015) | |
| • Impairs liver regeneration | Liu et al. Faseb J (2018) | |||
| Not verified | • Induces skeletal muscle atrophy | Hammers et al. EMBO Mol Med (2017) | ||
| • Decrease bone mass | Liu et al. Nat Commun (2016) | |||
| eNAMPT | Decline | enzyme | • Improves physical activity and extends mouse lifespan by promoting NAD+ biosynthesis | Mitsukuni et al. Cell Metab. (2019) |
| Oxytocin | Decline | hormone | • Prevent skeletal muscle aging by promoting proliferation of myogenic progenitor cells | Elabd et al. Nat. Comm. (2014) |
| SPARCL1 | Decline | matricellular extracellular matrix protein | • Elevated spontaneous synaptic responses, synapse density, and dendritic branching; enhancement of evoked neurotransmission | Kathlyn et al. PNAS (2019) |
| TIMP2 | Decline | Protease inhibitor | • Increases synaptic plasticity and hippocampus-dependent cognition | Castellano et al. Nature (2017) |
| THBS4 | Decline | matricellular extracellular matrix protein | • Increased spontaneous synaptic responses, synapse density, and dendritic branching; enhancement of evoked neurotransmission | Kathlyn et al. PNAS (2019) |
Apelin
Apelin, a 13-amino acid peptide, was initially identified as an endogenous ligand that regulates gastric acid production through a G protein–coupled receptor, APJ [14, 15]. The human APLN gene encodes a proprotein consisting of 77 amino acid residues, which can be cleaved into a 55–amino acid fragment and then into several active fragments. Apelin-13, which is highly bioactive, is responsible for APJ binding and biological activity [16, 17]. Apelin and APJ are widely expressed in the body and play important roles in many organs. In addition, apelin ameliorates the symptoms of many diseases, including, neurological disorders [18], hypertension [19], metabolic disorders [16], gastrointestinal diseases [20], and hepatic diseases [21]. Interestingly, expression of apelin and APJ in tissues decline with age, and stress-induced senescence downregulates endogenous apelin and APJ [22]. Consistent with this, the circulating levels of apelin decrease with age in mice [23, 24]. Although Apln−/− and Aplnr−/− mice appear healthy, they undergo accelerated multi-organ aging. Genetic or pharmacological inhibition of apelin-mediated signaling leads to cellular senescence, whereas systemic restoration of apelin attenuates age-related pathologies and extends healthspan in mice [22].
Physical exercise ameliorates age-related muscle wasting [25], and restoration of myokines is the main mechanism by which physical activity prevents muscle wasting. Besse-Patin et al. identified apelin as an exercise-regulated myokine in humans [26]. In particular, they found that 8 weeks of endurance exercise training in obese male subjects upregulated APLN expression in skeletal muscle. Consistent with this, exercise stimulates secretion of apelin into the bloodstream in both mice and humans. Plasma apelin levels are associated with sarcopenia, defined as a gradual loss of function and muscle with age [23]. Accordingly, chronic apelin administration in old mice increases muscle mass and function by promoting mitochondriogenesis and protein synthesis. In addition, apelin targets muscle stem cells and increases their regenerative capacity. The phenotypes of Apln−/− and Aplnr−/− mice support the idea that these factors play critical roles muscle physiology during aging. Collectively, these findings suggest that apelin and its receptor are therapeutic targets for the rejuvenation of aged skeletal muscle [23].
β2-microglobulin
Beta 2–microglobulin (β2M) is a secretory protein that serves as coreceptor for antigen presentation by major histocompatibility complex (MHC) class I to the immune system [27]. β2M is produced by all nucleated cells and is present in the circulation under normal physiological conditions. β2M has no transmembrane region and contains a distinctive molecular structure called a constant-1 Ig superfamily domain. β2M-deficient mice exhibit a wide variety of immunological aberrations, such as antigen-specific IgG production [28], the catabolism of IgG [29], and greater susceptibility to pathogen infections [30–33].
Serum and plasma β2M levels are elevated in many pathological conditions, including renal disease, immunodeficiency, autoimmune diseases, and tumor burden [34–39]. Accelerated aging also affects the concentration of circulating β2M. In the general population of older adults, serum β2M concentration is a predictor of total mortality [40]. Furthermore, among geriatric inpatients, higher levels of β2M are associated with frailty [41]. High levels of plasma β2M are observed in elderly humans and aged mice [42], and circulating β2M levels are elevated in the younger partners in heterochronic parabiosis experiments [8]. Villeda et al. found that β2M acts as pro-aging factor by contributing to age-related decline in adult neurogenesis and impairments in hippocampal-dependent cognitive functions. Systemic administration of β2M or local injection of the protein into the brain impairs hippocampal-dependent neurogenesis and cognitive functions. Aged B2m-/- mice exhibit superior cognitive function and neurogenesis in comparison aged WT mice [42]. These observations raise the possibility that targeting β2M could attenuate age-related decline in cognition and regenerative functions in old age.
Cadherin-13
Cadherin-13 (also known as T-cadherin or H-cadherin) is an atypical member of the cadherin superfamily, whose members are membrane proteins that mediate calcium-dependent cellular adhesion. Cadherin-13 lacks transmembrane and cytoplasmic domains, but rather contains a glycosylphosphatidylinositol moiety that anchors it to the plasma membrane [43, 44]. Thus, cadherin-13 acts by interacting with other membrane-bound molecules to regulate cellular functions. It is highly expressed in developing and adult brain [45], and is known as an ADHD-risk gene [46]. Studies have reported that the cadherin-13 protein inhibits neuronal outgrowth, cell migration, and axon guidance [44, 47–49]. Cadherin-13 is also found in the heart, pancreatic β-cells, liver, and skeletal muscle, and studies have shown that it contributes to regulating stress-induced pathological cardiac remodeling and metabolism through binding to adiponectin and insulin granules [50–52]. Although cadherin-13 is attached to the cell membrane, it is detected in the blood. Reduced levels of circulating cadherin-13 have been associated with coronary artery disease [53]. Plasma proteomic profiling revealed that cadherin-13 is expressed at lower levels in old mice compared to young mice [54]. Cadherin-13 inhibits osteoclast differentiation by blocking the activation of RANKL-associated signaling cascades, and cadherin-13 treatment of aged mice was found to delay age-associated bone loss [54]. These results suggest that cadherin-13 is an age-related bone factor that contributes to osteoporosis or osteopenia during aging, and that it could potentially be used as a novel therapeutic molecule for the treatment of bone loss. However, the underlying molecular mechanisms remain to be elucidated.
CCL11/Eotaxin-1
CCL11, a chemokine that can bind the chemokine receptors CCR2, CCR3, and CCR5 [55, 56], was first identified as a potent eosinophil chemoattractant. Thus, it participates in multiple inflammatory diseases including allergic reactions, allergic reactions, atopic dermatitis, and inflammatory bowel diseases; accordingly, its serum levels are elevated in each of these diseases [57–59]. In addition to its role in the immune response, CCL11 has multiple functions in diverse tissues and organs. Moreover, CCL11 plays roles in diseases, and has been implicated in cancer, atherosclerosis, neurogenesis, and neurodegeneration, and myocardial diseases [8, 60–63]. Notably, in this regard Villeda et al. showed that the plasma levels of CCL11 increase with aging and contribute to impaired neurogenesis and cognitive function in mice; the reduction in neurogenesis could be prevented by CCL11 neutralizing antibodies [8]. Injection of CCL11 into young mice mimicked hippocampal aging, with a reduction in synapses and an increase in microglial activation [64]. Interestingly, plasma levels of CCL11 are negatively associated with memory function in patients with Alzheimer’s dementia and older adults dwelling in rural communities [65, 66]. Furthermore, plasma CCL11 levels are elevated in patients with psychiatric conditions such as schizophrenia, bipolar disorder, dysthymia, and autism spectrum disorder [67–70]. However, the mechanism by which circulating CCL11 affects neuronal dysfunction is currently not well understood. Several studies have suggested that CCL11 may play a neurotoxic role in the pathophysiology of AD and PD [71, 72]. CCL11 is now considered to be a pro-aging factor that promotes aging-related neuronal dysfunction independent of eosinophil recruitment. Inspired by such findings, a startup called Alkahest has initiated a Phase II clinical trial investigating ALK4290, the first non–plasma-derived product targeting CCL11, for treatment of wet age-related macular degeneration (AMD) and Parkinson’s disease.
eNAMPT
Nicotinamide adenine dinucleotide (NAD+) an essential pyridine nucleotide that is present in all living cells. NAD+ is an abundant cofactor that participates in multiple aspects of biological processes, including energy metabolism, DNA repair, gene expression, cellular signaling, and stress response [73, 74]. NAD+ levels decrease with age, and that elevated intracellular NAD+ levels extend lifespan in model organisms [75–81]. Several studies have confirmed that administration of NMN, an intermediate in NAD+ biosynthesis, ameliorates age-related dysfunction [76, 81–83]. Nicotinamide phosphoribosyltransferase (NAMPT), a key enzyme in the biosynthesis of nicotinamide adenine dinucleotide (NAD+), converts nicotinamide and 5′-phosphoribosyl-pyrophosphate (PRPP) to nicotinamide mononucleotide (NMN) [84]. Stimulation of NAMPT activity has been proposed as a strategy for preventing and treating age-related diseases [85, 86]. In mammals, NAMPT exists in intracellular (iNAMPT) and extracellular (eNAMPT) forms [87]. In addition to its enzymatic function, eNAMPT (also known as pre–B-cell enhancing factor (PBEF)/visfatin) acts as a cytokine in circulation. Remarkably, Yoshida et al. found that circulating eNAMPT is carried in extracellular vesicles and correlates significantly with age in both mice and humans. In mice, increasing eNAMPT levels via adipose-specific overexpression maintains NAD+ levels during aging, prevents age-associated physiological decline, and extends lifespan. eNAMPT carried by EVs is internalized into target cells, resulting in upregulation of NMN/NAD+ biosynthesis [88]. Based on these findings, NAD+ metabolism has emerged as a potential target for treatment of age-related disorders. Indeed, several clinical studies have been planned or are already ongoing in the US and Europe.
GDF11
Growth differentiation factor 11 (GDF11), a secreted protein, is also known as bone morphogenetic protein 11 (BMP-11). GDF11 is a member of TGF-β/BMP superfamily that mainly activates Smad and non-Smad signaling pathways via binding to Activin receptor I and II, thereby regulating expression of its target genes [89]. GDF11 was initially synthesized as a precursor protein, and then cleaved by pro-protein convertase subtilisin/kexin type 5 (PCSK5), forming a non-covalent latent complex consisting of an N-terminal inhibitory pro-domain and two disulfide-linked carboxyl-terminal active domains [90, 91]. BMP1/Tolloid matrix metalloproteinase further cleaves the latent complex to generate active GDF11 [92].
Over the past few decades, a series of studies revealed that GDF11 participates in embryonic development by regulating anterior/posterior patterning of the axial skeleton, kidney organogenesis, spinal cord neurogenesis, pancreatic beta-cell differentiation, and retinal formation [93–97]. In addition, it plays a critical role in multiple fundamental homeostatic processes including osteogenesis, myogenesis, neurogenesis, and erythropoiesis [7, 12, 98–101]. Therefore, many studies have demonstrated that GDF11-mediated physiological processes have implications in human development and diseases.
Loffredo et al. hypothesized that anti-aging factors might reverse age-related cardiac hypertrophy, and identified GDF11 as anti-aging factor with antihypertrophic properties [11]. Aptamer-based proteomic analysis revealed several circulating proteins present at different levels between young and old mice. Among these candidates, GDF11 level declined with age and administration of GDF11 reversed age-related cardiac hypertrophy in old mice [11]. In addition to cardiac aging, GDF11 reverses skeletal muscle aging: systemic delivery of GDF11 protein restores genomic integrity in aged muscle stem cells and improves muscle physiology and physical functions [12]. GDF11 also exerts positive effects in the aged brain by promoting vascular remodeling and neurogenesis [7]. In contrast to these observations, however, a series of follow-up studies reported that GDF11 negatively affects aged tissues. In particular, GDF11 inhibits myoblast differentiation in mice, thereby impairing muscle regeneration [100]. Moreover, Hinken et al. reported that GDF11 does not affect outgrowth of muscle stem cells [102]; indeed, systemic overexpression of GDF11 induces skeletal and cardiac muscle atrophy in mice [103]. GDF11 inhibits bone formation, and inhibition of GDF11 functions prevents age-related osteoporosis [99]. Furthermore, it remains unclear whether the levels of circulating GDF11 are related to age. Multiple studies using different detection methods have reported conflicting age-related changes in GDF11 levels. The mature active region of GDF11 is highly similar to that of myostatin (GDF8). Consequently, commercially available ELISAs are available using antibodies with cross-reactivity with myostatin. Egerman et al. found that a previously used GDF11 SOMAmer and GDF11 antibodies bind to both GDF11 and myostatin [11, 12, 100], and GDF11-specific immunoassays revealed that serum GDF11 levels are elevated with age in human and rats [100]. Importantly, using a highly specific LC-MS/MS assay, Schafer et al. found that the levels of GDF11 do not decline in men or women throughout the lifespan and are instead positively associated with frailty and other morbidities [104]. The controversy about the relationship between GDF11 and aging may be caused by differences in GDF11 detection methods, the source of recombinant GDF11 protein, experimental design, or other factors. Although the biology of GDF11 remains controversial, the protein has important implications for physiological and pathological processes related to diagnosis and therapy of human diseases.
Oxytocin
Oxytocin is a hormone produced mainly in the hypothalamus and secreted by the pituitary gland. Oxytocin plays well-known roles in female reproduction, including uterine contraction and milk ejection. The oxytocin receptor, a typical member of the class I G protein–coupled receptor superfamily, is expressed in a variety of tissues, such as the ovary, testis, adrenals, uterus, mammary glands, bone, brain, liver, and adipose [105]. Because the oxytocin receptor is present on diverse cell types, oxytocin has multiple positive physiological and psychological effects [106]. In particular, it influences a wide array of social behaviors through direct projections to other brain regions such as the nucleus accumbens, olfactory bulb, amygdala, and brain stem [107–110]. Although several studies of postmortem neural tissues have investigated whether aging affects the oxytocin system, some controversy persists regarding the number of oxytonergic cells in the brain of elderly subjects [111, 112]. In addition, reduced levels of oxytocin have been detected in postmenopausal women with osteoporosis [113]. Consistent with these observations, ovariectomized mice and rats have significantly lower plasma oxytocin levels than sham-operated mice. Oxytocin enhances osteoblast differentiation, and supplementation of oxytocin reverses bone loss induced by ovariectomy in rodents [114]. Interestingly, circulating oxytocin levels decline with age in rhesus macaques and mice [115, 116]. Elabd et al. [116] suggested that this age-related decline in oxytocin contributes to defects in muscle regeneration. They found that oxytocin rejuvenates muscle stem cells by promoting their proliferation after muscle injury. Furthermore, aged Oxt-/- mice exhibit premature sarcopenia. Because several lines of evidence have revealed that oxytocin improves social deficits associated with various psychiatric disorders, numerous clinical trials have investigated the effect of this protein on social dysfunction [117]. In addition to improvement of social behavioral dysfunction, oxytocin and the oxytocin-mediated signaling pathway represent new clinical targets for rejuvenation of aged skeletal muscle.
TIMP2
Tissue inhibitor of metalloproteinases (TIMPs) are a family of secretory proteins consisting of four members: TIMP-1, -2, -3, and -4. TIMPs are endogenous inhibitors of metalloproteinases, including the matrix metalloproteinases (MMPs), a disintegrin and metalloproteinases (ADAMs), and ADAMs with thrombospondin motifs (ADAMTSs) [118]. Generally, TIMPs participate in extracellular matrix (ECM) catabolism, which is essential for many biological processes such as embryonic development, morphogenesis, tissue repair, and regeneration. In addition to ability to inhibit metalloproteinases, TIMPs are involved in multiple biological functions such as cell proliferation, apoptosis, and synaptic plasticity, many of which are independent of MMP inhibitory activity [119]. Notably in this regard, TIMP2 expression in plasma and hippocampus decreases with age in mice [120]. Interestingly, Castellano et al. reported that human umbilical cord plasma enriched with TIMP2 improved hippocampal function, and systemic supplementation with TIMP2 increase synaptic plasticity and cognition in aged mice. On the basis of these findings, they suggested that plasma TIMP2 reverses age-related neuronal dysfunction. However, little is known regarding the molecular mechanism underlying this effect.
SPARCL1 and THBS4
Ever since parabiosis experiments revealed that blood from young animals could rejuvenate neural function in old mice, many scientists used a wide range of approaches to identify rejuvenating factors in the blood. In a mass spectrometry study of plasma from young and old mice, Gan et al. found a series of proteins enriched in young or old blood [121]. Among the proteins they identified, SPARC-like protein 1 (SPARCL1) and thrombospondin-4 (THBS4) are abundant in young serum. Both proteins boost synaptic responses, synapse density, and dendritic branching in neurons transdifferentiated from human embryonic stem cells. Remarkably, these proteins are extracellular matrix-associated proteins and synaptogenic factors secreted from astrocytes. Astrocytes are essential for the function of the nervous system, as they regulate neurons by providing metabolic and trophic support [122]. THBS1 and 2 are astrocyte-secreted proteins that promotes synaptogenesis, whereas THBS4 enhance neurite adhesion and outgrowth [123]. SPARCL1 regulates CNS synaptogenesis [124]. Although THBS4 and SPARCL1, which are enriched in young mouse blood, increase synapse formation in vitro, their effects on brain rejuvenation in animal models remain unclear.
Conclusions
Multiple studies have reported that young blood can reverse aspects of aging in various organs. Although young blood transfusion is effective for rejuvenation in aged mice, it remains unclear whether young blood transfusion into older people has clinical benefits. Young blood may contain multiple factors that contribute to the aging process, and studies using diverse approaches have identified several candidate anti-aging and pro-aging factors. The use of recombinant proteins as rejuvenating factors may enable the revitalization of aged organs in clinical application. Conboy’s team demonstrated that young blood by itself does not have positive effects on rejuvenating old tissues. [125]. Furthermore, the team found that replacing half of the blood plasma of old mice with a mixture of saline and albumin. is sufficient to rejuvenate the brain, liver, and muscle [126]. The authors suggested that young blood or rejuvenating factors are not required for rejuvenating effect, and removing pro-aging factors in old blood is effective way to rejuvenate old tissues. Thus, blockade of specific pro-aging factors or their receptors could contribute to treating age-related diseases. Although the mechanism by which these proteins act is far from being fully understood, multiple studies have demonstrated that these factors play important roles in age-related diseases, and may therefore have clinical applications in the future.
Conflicts of Interest
The authors declare that there is no conflicts of interest.
Funding
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean Government (2017M3A9D8048709 and 2019R1C1C1006801) and by the KRIBB initiative program.
References
- 1. Darst BF, Koscik RL, Hogan KJ, Johnson SC, Engelman CD. Longitudinal plasma metabolomics of aging and sex. Aging (Albany NY). 2019; 11:1262–82. https://doi.org/10.18632/aging.101837 [PubMed]
- 2. Lehallier B, Gate D, Schaum N, Nanasi T, Lee SE, Yousef H, Moran Losada P, Berdnik D, Keller A, Verghese J, Sathyan S, Franceschi C, Milman S, et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat Med. 2019; 25:1843–50. https://doi.org/10.1038/s41591-019-0673-2 [PubMed]
- 3. Tanaka T, Biancotto A, Moaddel R, Moore AZ, Gonzalez-Freire M, Aon MA, Candia J, Zhang P, Cheung F, Fantoni G, Semba RD, Ferrucci L, and CHI consortium. Plasma proteomic signature of age in healthy humans. Aging Cell. 2018; 17:e12799. https://doi.org/10.1111/acel.12799 [PubMed]
- 4. Chaleckis R, Murakami I, Takada J, Kondoh H, Yanagida M. Individual variability in human blood metabolites identifies age-related differences. Proc Natl Acad Sci USA. 2016; 113:4252–59. https://doi.org/10.1073/pnas.1603023113 [PubMed]
- 5. Menni C, Kastenmüller G, Petersen AK, Bell JT, Psatha M, Tsai PC, Gieger C, Schulz H, Erte I, John S, Brosnan MJ, Wilson SG, Tsaprouni L, et al. Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int J Epidemiol. 2013; 42:1111–19. https://doi.org/10.1093/ije/dyt094 [PubMed]
- 6. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005; 433:760–64. https://doi.org/10.1038/nature03260 [PubMed]
- 7. Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, Rubin LL. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014; 344:630–34. https://doi.org/10.1126/science.1251141 [PubMed]
- 8. Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011; 477:90–94. https://doi.org/10.1038/nature10357 [PubMed]
- 9. Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014; 20:659–63. https://doi.org/10.1038/nm.3569 [PubMed]
- 10. Baht GS, Silkstone D, Vi L, Nadesan P, Amani Y, Whetstone H, Wei Q, Alman BA. Exposure to a youthful circulaton rejuvenates bone repair through modulation of β-catenin. Nat Commun. 2015; 6:7131. https://doi.org/10.1038/ncomms8131 [PubMed]
- 11. Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall’Osso C, Khong D, Shadrach JL, Miller CM, Singer BS, Stewart A, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013; 153:828–39. https://doi.org/10.1016/j.cell.2013.04.015 [PubMed]
- 12. Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, Hirshman MF, Lebowitz J, Shadrach JL, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014; 344:649–52. https://doi.org/10.1126/science.1251152 [PubMed]
- 13. de Magalhães JP, Stevens M, Thornton D. The business of anti-aging science. Trends Biotechnol. 2017; 35:1062–73. https://doi.org/10.1016/j.tibtech.2017.07.004 [PubMed]
- 14. O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993; 136:355–60. https://doi.org/10.1016/0378-1119(93)90495-o [PubMed]
- 15. Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998; 251:471–76. https://doi.org/10.1006/bbrc.1998.9489 [PubMed]
- 16. Dray C, Knauf C, Daviaud D, Waget A, Boucher J, Buléon M, Cani PD, Attané C, Guigné C, Carpéné C, Burcelin R, Castan-Laurell I, Valet P. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metab. 2008; 8:437–45. https://doi.org/10.1016/j.cmet.2008.10.003 [PubMed]
- 17. Hwangbo C, Wu J, Papangeli I, Adachi T, Sharma B, Park S, Zhao L, Ju H, Go GW, Cui G, Inayathullah M, Job JK, Rajadas J, et al. Endothelial APLNR regulates tissue fatty acid uptake and is essential for apelin’s glucose-lowering effects. Sci Transl Med. 2017; 9:eaad4000. https://doi.org/10.1126/scitranslmed.aad4000 [PubMed]
- 18. Duan J, Cui J, Yang Z, Guo C, Cao J, Xi M, Weng Y, Yin Y, Wang Y, Wei G, Qiao B, Wen A. Neuroprotective effect of apelin 13 on ischemic stroke by activating AMPK/GSK-3β/Nrf2 signaling. J Neuroinflammation. 2019; 16:24. https://doi.org/10.1186/s12974-019-1406-7 [PubMed]
- 19. Zhang Q, Yao F, Raizada MK, O’Rourke ST, Sun C. Apelin gene transfer into the rostral ventrolateral medulla induces chronic blood pressure elevation in normotensive rats. Circ Res. 2009; 104:1421–28. https://doi.org/10.1161/CIRCRESAHA.108.192302 [PubMed]
- 20. Ge Y, Li Y, Chen Q, Zhu W, Zuo L, Guo Z, Gong J, Cao L, Gu L, Li J. Adipokine apelin ameliorates chronic colitis in Il-10-/- mice by promoting intestinal lymphatic functions. Biochem Pharmacol. 2018; 148:202–12. https://doi.org/10.1016/j.bcp.2018.01.011 [PubMed]
- 21. Melgar-Lesmes P, Pauta M, Reichenbach V, Casals G, Ros J, Bataller R, Morales-Ruiz M, Jiménez W. Hypoxia and proinflammatory factors upregulate apelin receptor expression in human stellate cells and hepatocytes. Gut. 2011; 60:1404–11. https://doi.org/10.1136/gut.2010.234690 [PubMed]
- 22. Rai R, Ghosh AK, Eren M, Mackie AR, Levine DC, Kim SY, Cedernaes J, Ramirez V, Procissi D, Smith LH, Woodruff TK, Bass J, Vaughan DE. Downregulation of the apelinergic axis accelerates aging, whereas its systemic restoration improves the mammalian healthspan. Cell Rep. 2017; 21:1471–80. https://doi.org/10.1016/j.celrep.2017.10.057 [PubMed]
- 23. Vinel C, Lukjanenko L, Batut A, Deleruyelle S, Pradère JP, Le Gonidec S, Dortignac A, Geoffre N, Pereira O, Karaz S, Lee U, Camus M, Chaoui K, et al. The exerkine apelin reverses age-associated sarcopenia. Nat Med. 2018; 24:1360–71. https://doi.org/10.1038/s41591-018-0131-6 [PubMed]
- 24. Sauvant J, Delpech JC, Palin K, De Mota N, Dudit J, Aubert A, Orcel H, Roux P, Layé S, Moos F, Llorens-Cortes C, Nadjar A. Mechanisms involved in dual vasopressin/apelin neuron dysfunction during aging. PLoS One. 2014; 9:e87421. https://doi.org/10.1371/journal.pone.0087421 [PubMed]
- 25. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013; 17:162–84. https://doi.org/10.1016/j.cmet.2012.12.012 [PubMed]
- 26. Besse-Patin A, Montastier E, Vinel C, Castan-Laurell I, Louche K, Dray C, Daviaud D, Mir L, Marques MA, Thalamas C, Valet P, Langin D, Moro C, Viguerie N. Effect of endurance training on skeletal muscle myokine expression in obese men: identification of apelin as a novel myokine. Int J Obes (Lond). 2014; 38:707–13. https://doi.org/10.1038/ijo.2013.158 [PubMed]
- 27. Pérarnau B, Siegrist CA, Gillet A, Vincent C, Kimura S, Lemonnier FA. Beta 2-microglobulin restriction of antigen presentation. Nature. 1990; 346:751–54. https://doi.org/10.1038/346751a0 [PubMed]
- 28. Spriggs MK, Koller BH, Sato T, Morrissey PJ, Fanslow WC, Smithies O, Voice RF, Widmer MB, Maliszewski CR. Beta 2-microglobulin-, CD8+ t-cell-deficient mice survive inoculation with high doses of vaccinia virus and exhibit altered IgG responses. Proc Natl Acad Sci USA. 1992; 89:6070–74. https://doi.org/10.1073/pnas.89.13.6070 [PubMed]
- 29. Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol. 1996; 26:690–96. https://doi.org/10.1002/eji.1830260327 [PubMed]
- 30. Tarleton RL, Koller BH, Latour A, Postan M. Susceptibility of beta 2-microglobulin-deficient mice to trypanosoma cruzi infection. Nature. 1992; 356:338–40. https://doi.org/10.1038/356338a0 [PubMed]
- 31. Hildeman D, Salvato M, Whitton JL, Muller D. Vaccination protects beta 2 microglobulin deficient mice from immune mediated mortality but not from persisting viral infection. Vaccine. 1996; 14:1223–29. https://doi.org/10.1016/s0264-410x(96)00028-x [PubMed]
- 32. Klingel K, Schnorr JJ, Sauter M, Szalay G, Kandolf R. Beta2-microglobulin-associated regulation of interferon-gamma and virus-specific immunoglobulin G confer resistance against the development of chronic coxsackievirus myocarditis. Am J Pathol. 2003; 162:1709–20. https://doi.org/10.1016/s0002-9440(10)64305-2 [PubMed]
- 33. Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. Major histocompatibility complex class i-restricted T cells are required for resistance to mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 1992; 89:12013–17. https://doi.org/10.1073/pnas.89.24.12013 [PubMed]
- 34. Astor BC, Shafi T, Hoogeveen RC, Matsushita K, Ballantyne CM, Inker LA, Coresh J. Novel markers of kidney function as predictors of ESRD, cardiovascular disease, and mortality in the general population. Am J Kidney Dis. 2012; 59:653–62. https://doi.org/10.1053/j.ajkd.2011.11.042 [PubMed]
- 35. Zissis M, Afroudakis A, Galanopoulos G, Palermos L, Boura X, Michopoulos S, Archimandritis A. B2 microglobulin: is it a reliable marker of activity in inflammatory bowel disease? Am J Gastroenterol. 2001; 96:2177–83. https://doi.org/10.1111/j.1572-0241.2001.03881.x [PubMed]
- 36. Cassuto JP, Krebs BP, Viot G, Dujardin P, Masseyeff R. Beta 2 microglobulin, a tumour marker of lymphoproliferative disorder. Lancet. 1978; 2:950. https://doi.org/10.1016/s0140-6736(78)91677-x [PubMed]
- 37. Prizment AE, Linabery AM, Lutsey PL, Selvin E, Nelson HH, Folsom AR, Church TR, Drake CG, Platz EA, Joshu C. Circulating beta-2 microglobulin and risk of cancer: the atherosclerosis risk in communities study (ARIC). Cancer Epidemiol Biomarkers Prev. 2016; 25:657–64. https://doi.org/10.1158/1055-9965.EPI-15-0849 [PubMed]
- 38. Cheung CL, Lam KS, Cheung BM. Serum β-2 microglobulin predicts mortality in people with diabetes. Eur J Endocrinol. 2013; 169:1–7. https://doi.org/10.1530/EJE-13-0003 [PubMed]
- 39. Amighi J, Hoke M, Mlekusch W, Schlager O, Exner M, Haumer M, Pernicka E, Koppensteiner R, Minar E, Rumpold H, Schillinger M, Wagner O. Beta 2 microglobulin and the risk for cardiovascular events in patients with asymptomatic carotid atherosclerosis. Stroke. 2011; 42:1826–33. https://doi.org/10.1161/STROKEAHA.110.600312 [PubMed]
- 40. Shinkai S, Chaves PH, Fujiwara Y, Watanabe S, Shibata H, Yoshida H, Suzuki T. Beta2-microglobulin for risk stratification of total mortality in the elderly population: comparison with cystatin C and c-reactive protein. Arch Intern Med. 2008; 168:200–06. https://doi.org/10.1001/archinternmed.2007.64 [PubMed]
- 41. Annweiler C, Bataille R, Ferrière N, Douillet D, Fantino B, Beauchet O. Plasma beta-2 microglobulin as a marker of frailty in older adults: a pilot study. J Gerontol A Biol Sci Med Sci. 2011; 66:1077–79. https://doi.org/10.1093/gerona/glr104 [PubMed]
- 42. Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, Gontier G, Wabl R, Plambeck KE, Udeochu J, Wheatley EG, Bouchard J, Eggel A, et al. Β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat Med. 2015; 21:932–37. https://doi.org/10.1038/nm.3898 [PubMed]
- 43. Philippova M, Joshi MB, Kyriakakis E, Pfaff D, Erne P, Resink TJ. A guide and guard: the many faces of t-cadherin. Cell Signal. 2009; 21:1035–44. https://doi.org/10.1016/j.cellsig.2009.01.035 [PubMed]
- 44. Ranscht B, Dours-Zimmermann MT. T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. Neuron. 1991; 7:391–402. https://doi.org/10.1016/0896-6273(91)90291-7 [PubMed]
- 45. Takeuchi T, Misaki A, Liang SB, Tachibana A, Hayashi N, Sonobe H, Ohtsuki Y. Expression of t-cadherin (CDH13, h-cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells. J Neurochem. 2000; 74:1489–97. https://doi.org/10.1046/j.1471-4159.2000.0741489.x [PubMed]
- 46. Rivero O, Selten MM, Sich S, Popp S, Bacmeister L, Amendola E, Negwer M, Schubert D, Proft F, Kiser D, Schmitt AG, Gross C, Kolk SM, et al. Cadherin-13, a risk gene for ADHD and comorbid disorders, impacts GABAergic function in hippocampus and cognition. Transl Psychiatry. 2015; 5:e655. https://doi.org/10.1038/tp.2015.152 [PubMed]
- 47. Fredette BJ, Ranscht B. T-cadherin expression delineates specific regions of the developing motor axon-hindlimb projection pathway. J Neurosci. 1994; 14:7331–46. https://doi.org/10.1523/JNEUROSCI.14-12-07331.1994 [PubMed]
- 48. Fredette BJ, Miller J, Ranscht B. Inhibition of motor axon growth by t-cadherin substrata. Development. 1996; 122:3163–71. [PubMed]
- 49. Hayano Y, Zhao H, Kobayashi H, Takeuchi K, Norioka S, Yamamoto N. The role of t-cadherin in axonal pathway formation in neocortical circuits. Development. 2014; 141:4784–93. https://doi.org/10.1242/dev.108290 [PubMed]
- 50. Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci USA. 2004; 101:10308–13. https://doi.org/10.1073/pnas.0403382101 [PubMed]
- 51. Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest. 2010; 120:4342–52. https://doi.org/10.1172/JCI43464 [PubMed]
- 52. Tyrberg B, Miles P, Azizian KT, Denzel MS, Nieves ML, Monosov EZ, Levine F, Ranscht B. T-cadherin (Cdh13) in association with pancreatic β-cell granules contributes to second phase insulin secretion. Islets. 2011; 3:327–37. https://doi.org/10.4161/isl.3.6.17705 [PubMed]
- 53. Pfaff D, Schoenenberger AW, Dasen B, Erne P, Resink TJ, Philippova M. Plasma t-cadherin negatively associates with coronary lesion severity and acute coronary syndrome. Eur Heart J Acute Cardiovasc Care. 2015; 4:410–18. https://doi.org/10.1177/2048872614557229 [PubMed]
- 54. Yang YR, Kabir MH, Park JH, Park JI, Kang JS, Ju S, Shin YJ, Lee SM, Lee J, Kim S, Lee KP, Lee SY, Lee C, Kwon KS. Plasma proteomic profiling of young and old mice reveals cadherin-13 prevents age-related bone loss. Aging (Albany NY). 2020; 12:8652–68. https://doi.org/10.18632/aging.103184 [PubMed]
- 55. Kitaura M, Nakajima T, Imai T, Harada S, Combadiere C, Tiffany HL, Murphy PM, Yoshie O. Molecular cloning of human eotaxin, an eosinophil-selective CC chemokine, and identification of a specific eosinophil eotaxin receptor, CC chemokine receptor 3. J Biol Chem. 1996; 271:7725–30. https://doi.org/10.1074/jbc.271.13.7725 [PubMed]
- 56. Ye J, Kohli LL, Stone MJ. Characterization of binding between the chemokine eotaxin and peptides derived from the chemokine receptor CCR3. J Biol Chem. 2000; 275:27250–57. https://doi.org/10.1074/jbc.M003925200 [PubMed]
- 57. Mishra A, Hogan SP, Lee JJ, Foster PS, Rothenberg ME. Fundamental signals that regulate eosinophil homing to the gastrointestinal tract. J Clin Invest. 1999; 103:1719–27. https://doi.org/10.1172/JCI6560 [PubMed]
- 58. Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996; 2:449–56. https://doi.org/10.1038/nm0496-449 [PubMed]
- 59. Spergel JM, Mizoguchi E, Oettgen H, Bhan AK, Geha RS. Roles of TH1 and TH2 cytokines in a murine model of allergic dermatitis. J Clin Invest. 1999; 103:1103–11. https://doi.org/10.1172/JCI5669 [PubMed]
- 60. Miyagaki T, Sugaya M, Murakami T, Asano Y, Tada Y, Kadono T, Okochi H, Tamaki K, Sato S. CCL11-CCR3 interactions promote survival of anaplastic large cell lymphoma cells via ERK1/2 activation. Cancer Res. 2011; 71:2056–65. https://doi.org/10.1158/0008-5472.CAN-10-3764 [PubMed]
- 61. Zhu F, Liu P, Li J, Zhang Y. Eotaxin-1 promotes prostate cancer cell invasion via activation of the CCR3-ERK pathway and upregulation of MMP-3 expression. Oncol Rep. 2014; 31:2049–54. https://doi.org/10.3892/or.2014.3060 [PubMed]
- 62. Haley KJ, Lilly CM, Yang JH, Feng Y, Kennedy SP, Turi TG, Thompson JF, Sukhova GH, Libby P, Lee RT. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation. 2000; 102:2185–89. https://doi.org/10.1161/01.cir.102.18.2185 [PubMed]
- 63. Li Y, Zhao Y, Qiu C, Yang Y, Liao G, Wu X, Zhang X, Zhang Q, Zhang R, Wang Z. Role of eotaxin-1/CCL11 in sepsis-induced myocardial injury in elderly patients. Aging (Albany NY). 2020; 12:4463–73. https://doi.org/10.18632/aging.102896 [PubMed]
- 64. Das MM, Godoy M, Chen S, Moser VA, Avalos P, Roxas KM, Dang I, Yáñez A, Zhang W, Bresee C, Arditi M, Liu GY, Svendsen CN, Goodridge HS. Young bone marrow transplantation preserves learning and memory in old mice. Commun Biol. 2019; 2:73. https://doi.org/10.1038/s42003-019-0298-5 [PubMed]
- 65. Bettcher BM, Fitch R, Wynn MJ, Lalli MA, Elofson J, Jastrzab L, Mitic L, Miller ZA, Rabinovici GD, Miller BL, Kao AW, Kosik KS, Kramer JH. MCP-1 and eotaxin-1 selectively and negatively associate with memory in MCI and Alzheimer’s disease dementia phenotypes. Alzheimers Dement (Amst). 2016; 3:91–97. https://doi.org/10.1016/j.dadm.2016.05.004 [PubMed]
- 66. Butcher L, Pérès K, André P, Morris RH, Walter S, Dartigues JF, Rodriguez-Mañas L, Feart C, Erusalimsky JD, and FRAILOMIC consortium. Association between plasma CCL11 (eotaxin-1) and cognitive status in older adults: differences between rural and urban dwellers. Exp Gerontol. 2018; 113:173–79. https://doi.org/10.1016/j.exger.2018.10.004 [PubMed]
- 67. Panizzutti B, Gubert C, Schuh AL, Ferrari P, Bristot G, Fries GR, Massuda R, Walz J, Rocha NP, Berk M, Teixeira AL, Gama CS. Increased serum levels of eotaxin/CCL11 in late-stage patients with bipolar disorder: an accelerated aging biomarker? J Affect Disord. 2015; 182:64–69. https://doi.org/10.1016/j.jad.2014.12.010 [PubMed]
- 68. Teixeira AL, Reis HJ, Nicolato R, Brito-Melo G, Correa H, Teixeira MM, Romano-Silva MA. Increased serum levels of CCL11/eotaxin in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2008; 32:710–14. https://doi.org/10.1016/j.pnpbp.2007.11.019 [PubMed]
- 69. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol Psychiatry. 2015; 20:440–46. https://doi.org/10.1038/mp.2014.59 [PubMed]
- 70. Ho PS, Yen CH, Chen CY, Huang SY, Liang CS. Changes in cytokine and chemokine expression distinguish dysthymic disorder from major depression and healthy controls. Psychiatry Res. 2017; 248:20–27. https://doi.org/10.1016/j.psychres.2016.12.014 [PubMed]
- 71. Chandra G, Rangasamy SB, Roy A, Kordower JH, Pahan K. Neutralization of RANTES and eotaxin prevents the loss of dopaminergic neurons in a mouse model of parkinson disease. J Biol Chem. 2016; 291:15267–81. https://doi.org/10.1074/jbc.M116.714824 [PubMed]
- 72. Parajuli B, Horiuchi H, Mizuno T, Takeuchi H, Suzumura A. CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia. 2015; 63:2274–84. https://doi.org/10.1002/glia.22892 [PubMed]
- 73. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014; 24:464–71. https://doi.org/10.1016/j.tcb.2014.04.002 [PubMed]
- 74. Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, Bohr VA. NAD+ in aging: molecular mechanisms and translational implications. Trends Mol Med. 2017; 23:899–916. https://doi.org/10.1016/j.molmed.2017.08.001 [PubMed]
- 75. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011; 6:e19194. https://doi.org/10.1371/journal.pone.0019194 [PubMed]
- 76. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011; 14:528–36. https://doi.org/10.1016/j.cmet.2011.08.014 [PubMed]
- 77. Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA. 2015; 112:2876–81. https://doi.org/10.1073/pnas.1417921112 [PubMed]
- 78. Camacho-Pereira J, Tarragó MG, Chini CC, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016; 23:1127–39. https://doi.org/10.1016/j.cmet.2016.05.006 [PubMed]
- 79. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013; 154:430–41. https://doi.org/10.1016/j.cell.2013.06.016 [PubMed]
- 80. Frederick DW, Loro E, Liu L, Davila A
Jr , Chellappa K, Silverman IM, Quinn WJ3rd , Gosai SJ, Tichy ED, Davis JG, Mourkioti F, Gregory BD, Dellinger RW, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016; 24:269–82. https://doi.org/10.1016/j.cmet.2016.07.005 [PubMed] - 81. Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013; 155:1624–38. https://doi.org/10.1016/j.cell.2013.11.037 [PubMed]
- 82. Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, Qi N, Imai S, Yoshino J. NAMPT-mediated NAD+ biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 2016; 16:1851–60. https://doi.org/10.1016/j.celrep.2016.07.027 [PubMed]
- 83. de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S, Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016; 15:522–30. https://doi.org/10.1111/acel.12461 [PubMed]
- 84. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004; 279:50754–63. https://doi.org/10.1074/jbc.M408388200 [PubMed]
- 85. Yoshino J, Baur JA, Imai SI. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018; 27:513–28. https://doi.org/10.1016/j.cmet.2017.11.002 [PubMed]
- 86. Gardell SJ, Hopf M, Khan A, Dispagna M, Hampton Sessions E, Falter R, Kapoor N, Brooks J, Culver J, Petucci C, Ma CT, Cohen SE, Tanaka J, et al. Boosting NAD+ with a small molecule that activates NAMPT. Nat Commun. 2019; 10:3241. https://doi.org/10.1038/s41467-019-11078-z [PubMed]
- 87. Kitani T, Okuno S, Fujisawa H. Growth phase-dependent changes in the subcellular localization of pre-B-cell colony-enhancing factor. FEBS Lett. 2003; 544:74–78. https://doi.org/10.1016/s0014-5793(03)00476-9 [PubMed]
- 88. Yoshida M, Satoh A, Lin JB, Mills KF, Sasaki Y, Rensing N, Wong M, Apte RS, Imai SI. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 2019; 30:329–42.e5. https://doi.org/10.1016/j.cmet.2019.05.015 [PubMed]
- 89. Nakashima M, Toyono T, Akamine A, Joyner A. Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech Dev. 1999; 80:185–89. https://doi.org/10.1016/s0925-4773(98)00205-6 [PubMed]
- 90. Essalmani R, Zaid A, Marcinkiewicz J, Chamberland A, Pasquato A, Seidah NG, Prat A. In vivo functions of the proprotein convertase PC5/6 during mouse development: Gdf11 is a likely substrate. Proc Natl Acad Sci USA. 2008; 105:5750–55. https://doi.org/10.1073/pnas.0709428105 [PubMed]
- 91. Tsuda T, Iwai N, Deguchi E, Kimura O, Ono S, Furukawa T, Sasaki Y, Fumino S, Kubota Y. PCSK5 and GDF11 expression in the hindgut region of mouse embryos with anorectal malformations. Eur J Pediatr Surg. 2011; 21:238–41. https://doi.org/10.1055/s-0031-1273691 [PubMed]
- 92. Ge G, Hopkins DR, Ho WB, Greenspan DS. GDF11 forms a bone morphogenetic protein 1-activated latent complex that can modulate nerve growth factor-induced differentiation of PC12 cells. Mol Cell Biol. 2005; 25:5846–58. https://doi.org/10.1128/MCB.25.14.5846-5858.2005 [PubMed]
- 93. McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999; 22:260–64. https://doi.org/10.1038/10320 [PubMed]
- 94. Oh SP, Yeo CY, Lee Y, Schrewe H, Whitman M, Li E. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes Dev. 2002; 16:2749–54. https://doi.org/10.1101/gad.1021802 [PubMed]
- 95. Esquela AF, Lee SJ. Regulation of metanephric kidney development by growth/differentiation factor 11. Dev Biol. 2003; 257:356–70. https://doi.org/10.1016/s0012-1606(03)00100-3 [PubMed]
- 96. Shi Y, Liu JP. Gdf11 facilitates temporal progression of neurogenesis in the developing spinal cord. J Neurosci. 2011; 31:883–93. https://doi.org/10.1523/JNEUROSCI.2394-10.2011 [PubMed]
- 97. Harmon EB, Apelqvist AA, Smart NG, Gu X, Osborne DH, Kim SK. GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development. 2004; 131:6163–74. https://doi.org/10.1242/dev.01535 [PubMed]
- 98. Dussiot M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, Grapton D, Paubelle E, Payen E, Beuzard Y, Leboulch P, Ribeil JA, Arlet JB, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat Med. 2014; 20:398–407. https://doi.org/10.1038/nm.3468 [PubMed]
- 99. Liu W, Zhou L, Zhou C, Zhang S, Jing J, Xie L, Sun N, Duan X, Jing W, Liang X, Zhao H, Ye L, Chen Q, Yuan Q. GDF11 decreases bone mass by stimulating osteoclastogenesis and inhibiting osteoblast differentiation. Nat Commun. 2016; 7:12794. https://doi.org/10.1038/ncomms12794 [PubMed]
- 100. Egerman MA, Cadena SM, Gilbert JA, Meyer A, Nelson HN, Swalley SE, Mallozzi C, Jacobi C, Jennings LL, Clay I, Laurent G, Ma S, Brachat S, et al. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab. 2015; 22:164–74. https://doi.org/10.1016/j.cmet.2015.05.010 [PubMed]
- 101. Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL. Autoregulation of neurogenesis by GDF11. Neuron. 2003; 37:197–207. https://doi.org/10.1016/s0896-6273(02)01172-8 [PubMed]
- 102. Hinken AC, Powers JM, Luo G, Holt JA, Billin AN, Russell AJ. Lack of evidence for GDF11 as a rejuvenator of aged skeletal muscle satellite cells. Aging Cell. 2016; 15:582–84. https://doi.org/10.1111/acel.12475 [PubMed]
- 103. Hammers DW, Merscham-Banda M, Hsiao JY, Engst S, Hartman JJ, Sweeney HL. Supraphysiological levels of GDF11 induce striated muscle atrophy. EMBO Mol Med. 2017; 9:531–44. https://doi.org/10.15252/emmm.201607231 [PubMed]
- 104. Schafer MJ, Atkinson EJ, Vanderboom PM, Kotajarvi B, White TA, Moore MM, Bruce CJ, Greason KL, Suri RM, Khosla S, Miller JD, Bergen HR
3rd , LeBrasseur NK. Quantification of GDF11 and myostatin in human aging and cardiovascular disease. Cell Metab. 2016; 23:1207–15. https://doi.org/10.1016/j.cmet.2016.05.023 [PubMed] - 105. Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001; 81:629–83. https://doi.org/10.1152/physrev.2001.81.2.629 [PubMed]
- 106. Zingg HH, Laporte SA. The oxytocin receptor. Trends Endocrinol Metab. 2003; 14:222–27. https://doi.org/10.1016/s1043-2760(03)00080-8 [PubMed]
- 107. Oettl LL, Ravi N, Schneider M, Scheller MF, Schneider P, Mitre M, da Silva Gouveia M, Froemke RC, Chao MV, Young WS, Meyer-Lindenberg A, Grinevich V, Shusterman R, Kelsch W. Oxytocin enhances social recognition by modulating cortical control of early olfactory processing. Neuron. 2016; 90:609–21. https://doi.org/10.1016/j.neuron.2016.03.033 [PubMed]
- 108. Knobloch HS, Charlet A, Hoffmann LC, Eliava M, Khrulev S, Cetin AH, Osten P, Schwarz MK, Seeburg PH, Stoop R, Grinevich V. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron. 2012; 73:553–66. https://doi.org/10.1016/j.neuron.2011.11.030 [PubMed]
- 109. Takayanagi Y, Yoshida M, Takashima A, Takanami K, Yoshida S, Nishimori K, Nishijima I, Sakamoto H, Yamagata T, Onaka T. Activation of supraoptic oxytocin neurons by secretin facilitates social recognition. Biol Psychiatry. 2017; 81:243–51. https://doi.org/10.1016/j.biopsych.2015.11.021 [PubMed]
- 110. Herpertz SC, Bertsch K. A new perspective on the pathophysiology of borderline personality disorder: a model of the role of oxytocin. Am J Psychiatry. 2015; 172:840–51. https://doi.org/10.1176/appi.ajp.2015.15020216 [PubMed]
- 111. Wierda M, Goudsmit E, Van der Woude PF, Purba JS, Hofman MA, Bogte H, Swaab DF. Oxytocin cell number in the human paraventricular nucleus remains constant with aging and in Alzheimer’s disease. Neurobiol Aging. 1991; 12:511–16. https://doi.org/10.1016/0197-4580(91)90081-t [PubMed]
- 112. Calzà L, Pozza M, Coraddu F, Farci G, Giardino L. Hormonal influences on brain ageing quality: focus on corticotropin releasing hormone-, vasopressin- and oxytocin-immunoreactive neurones in the human brain. J Neural Transm (Vienna). 1997; 104:1095–100. https://doi.org/10.1007/BF01273321 [PubMed]
- 113. Breuil V, Amri EZ, Panaia-Ferrari P, Testa J, Elabd C, Albert-Sabonnadière C, Roux CH, Ailhaud G, Dani C, Carle GF, Euller-Ziegler L. Oxytocin and bone remodelling: relationships with neuropituitary hormones, bone status and body composition. Joint Bone Spine. 2011; 78:611–15. https://doi.org/10.1016/j.jbspin.2011.02.002 [PubMed]
- 114. Elabd C, Basillais A, Beaupied H, Breuil V, Wagner N, Scheideler M, Zaragosi LE, Massiéra F, Lemichez E, Trajanoski Z, Carle G, Euller-Ziegler L, Ailhaud G, et al. Oxytocin controls differentiation of human mesenchymal stem cells and reverses osteoporosis. Stem Cells. 2008; 26:2399–407. https://doi.org/10.1634/stemcells.2008-0127 [PubMed]
- 115. Parker KJ, Hoffman CL, Hyde SA, Cummings CS, Maestripieri D. Effects of age on cerebrospinal fluid oxytocin levels in free-ranging adult female and infant rhesus macaques. Behav Neurosci. 2010; 124:428–33. https://doi.org/10.1037/a0019576 [PubMed]
- 116. Elabd C, Cousin W, Upadhyayula P, Chen RY, Chooljian MS, Li J, Kung S, Jiang KP, Conboy IM. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat Commun. 2014; 5:4082. https://doi.org/10.1038/ncomms5082 [PubMed]
- 117. Erdozain AM, Peñagarikano O. Oxytocin as treatment for social cognition, not there yet. Front Psychiatry. 2020; 10:930. https://doi.org/10.3389/fpsyt.2019.00930 [PubMed]
- 118. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010; 1803:55–71. https://doi.org/10.1016/j.bbamcr.2010.01.003 [PubMed]
- 119. Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008; 1:re6. https://doi.org/10.1126/scisignal.127re6 [PubMed]
- 120. Castellano JM, Mosher KI, Abbey RJ, McBride AA, James ML, Berdnik D, Shen JC, Zou B, Xie XS, Tingle M, Hinkson IV, Angst MS, Wyss-Coray T. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature. 2017; 544:488–92. https://doi.org/10.1038/nature22067 [PubMed]
- 121. Gan KJ, Südhof TC. Specific factors in blood from young but not old mice directly promote synapse formation and NMDA-receptor recruitment. Proc Natl Acad Sci USA. 2019; 116:12524–33. https://doi.org/10.1073/pnas.1902672116 [PubMed]
- 122. Banker GA. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science. 1980; 209:809–10. https://doi.org/10.1126/science.7403847 [PubMed]
- 123. Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J Cell Biol. 1995; 131:1083–94. https://doi.org/10.1083/jcb.131.4.1083 [PubMed]
- 124. Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc Natl Acad Sci USA. 2011; 108:E440–49. https://doi.org/10.1073/pnas.1104977108 [PubMed]
- 125. Rebo J, Mehdipour M, Gathwala R, Causey K, Liu Y, Conboy MJ, Conboy IM. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat Commun. 2016; 7:13363. https://doi.org/10.1038/ncomms13363 [PubMed]
- 126. Mehdipour M, Skinner C, Wong N, Lieb M, Liu C, Etienne J, Kato C, Kiprov D, Conboy MJ, Conboy IM. Rejuvenation of three germ layers tissues by exchanging old blood plasma with saline-albumin. Aging (Albany NY). 2020; 12:8790–819. https://doi.org/10.18632/aging.103418 [PubMed]