



Introduction
Lung cancer is the most common malignant tumor and the main cause of cancer-related mortality worldwide. Non-small cell lung cancer (NSCLC) accounts for about 80% of all lung cancers, including adenocarcinoma, squamous cell carcinoma and large cell carcinoma. The incidence of lung adenocarcinoma (LUAD), the most common subtype of NSCLC, is increasing year by year [1]. Different from lung squamous cell carcinoma, LUAD is more likely to occur in women and non-smokers and is a heterogeneous disease characterized by a high rate of gene mutations [2]. Although, targeted therapy and immunotherapy have greatly improved the outcomes and prognosis of patients with LUAD in recent years, the emergence of drug resistance is still inevitable, and the long-term survival rate is still low [3, 4]. Especially for a large proportion of female patients with lung adenocarcinoma, there is still a lack of targeted risk prediction system. Therefore, it is still an arduous task to evaluate and improve the prognosis of female patients with LUAD. In recent years, with the deepening of tumor research, it is recognized that the expression of oncogene depends not only on the gene itself, but also on epigenetic modification without changing the gene sequence [5, 6].
With the extensive study of epigenetic changes in tumor progression, researchers finally focus on DNA, RNA and histone modifications. As a post-transcriptional regulation strategy, RNA modification occurs widely in all kinds of RNA [7], especially in mRNAs. Among the RNA modifications, 6-methyladenine (M6A) is the most common methyl modification in mRNAs that regulate RNA splicing, translocation, stability, and transformation. The level of M6A is dynamically regulated by a methyltransferase (encoder), a binding protein (reader), and a demethylase (decoder), so, it is a dynamic and reversible process. Related studies have shown that changes in M6A regulatory genes could promote the progression of breast cancer, liver cancer, and hematological malignant tumors by inducing the formation of cancer stem cells and their abnormal differentiation [8–10]. It has also been reported that miR-33a can inhibit the proliferation of non-small cell lung cancer (NSCLC) cells by targeting M6A regulatory factor METTL3 mRNA 3'UTR binding site [11]. Through the description of the genetic variation of M6A regulatory factor in lung adenocarcinoma, it was found that FTO and YTHDF3 mutations were related to the decrease of overall survival rate [12]. A series of studies have shown that once the components involved in the regulation of M6A modification have been lost or abnormal, it would lead to abnormal physiological functions such as cell differentiation, and gene expression would be abnormally activated or inhibited. Whether the level of M6A can be reversed or enhanced to improve the clinical efficacy of tumor patients is worthy of further exploration.
Various types of genomic changes, including CNV, played an important role in promoting the occurrence and development of cancer, and were also key factors leading to genetic and epigenetic abnormalities [13]. Through the study of 270individuals, Redon and colleagues constructed the first generation CNV map of the human genome, and identified 1447 copy number variable regions in these populations, accounting for 12% of the genome. Further exploration found that the nucleotide content of CNV region was more than single nucleotide polymorphism, so CNV played an more important role in genetic diversity [14]. CNV, the amplification or deletion of DNA caused by genomic instability, can be used as an important therapeutic target, as it is related to drug resistance and tumor biology in many cancers, such as breast cancer and non-small cell lung cancer (NSCLC) [15, 16]. Li et al found that two subgroups of NSCLC, LUAD and lung squamous cell carcinoma, can be distinguished by comparing their CNV patterns [17]. Therefore, the full analysis of the clinicopathological features and prognostic value of the key M6A regulatory factors and CNV through the sample data was very important for the further exploration of LUAD.
As a powerful resource of genomic data on various cancers, The Cancer Genome Atlas (TCGA) is very helpful in studying the molecular mechanisms of cancer and identifying prognostic markers. For example, Shukla et al. obtained a four-gene signature that significantly stratified the overall survival rate for LUAD through the analysis of EGFR wild type and EGFR mutation subgroups of LUAD [18]. In this study, we analyzed the clinical and sequencing data on the female LUAD cohort from TCGA, evaluated the expression profiles of 16 M6A regulatory genes in the cohort and the association between genetic changes and clinical characteristics, and developed an M6A-related risk scoring system to evaluate the prognosis of female patients with LUAD. The copy number variations (CNVs) of the related genes in the samples were analyzed and verified using the cBioPortal platform. Notably, compared with other clinical factors, this risk scoring system showed a higher predictive sensitivity and specificity.
Results
Extraction and analysis of M6A-related genes
M6A-related protein is an important regulator of tumorigenesis, and its expression level often directly determines the pathological process of tumor. In this study, we systematically analyzed the expression of 16 widely reported M6A RNA regulatory factors in female LUAD and normal tissues. These 16 widely reported M6A related genes were not only selected in lung cancer related articles, but also selected as key genes for RNA M6A methylation in other tumor types [19–22]. By analyzing the mRNA expression in samples from female LUAD patients in TCGA, 16 M6A-related genes were screened, and their differential expression in normal and female LUAD samples were analyzed using the edger package. With P<0.05 as the cutoff value, there were some differences in the expression of these 16 genes between the normal and female LUAD samples, especially for LRPPR, YTHDF1, HNRNPC, METTL3, METTL14, RBM15, HNRNPA2B1, FTO, KIAA1429, and YTHDF2 (Figure 1).
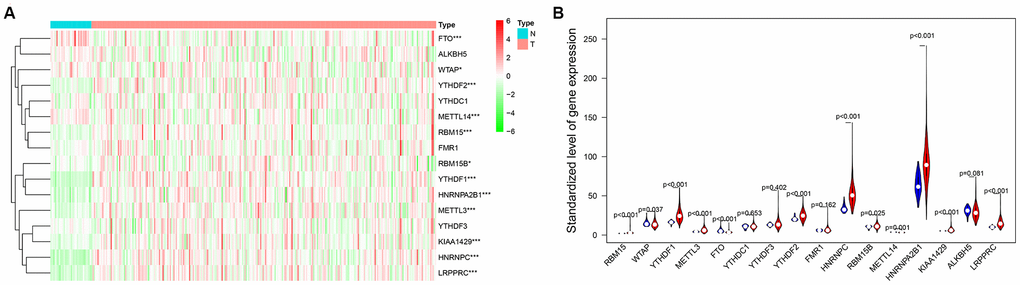
Figure 1. (A) The heat map of differential expression of M6A-related genes in normal samples and female lung adenocarcinoma samples. The color from green to red shows a trend from low expression to high expression. (B) The violin diagram of differential expression of M6A-related genes in normal samples and female lung adenocarcinoma samples. The Y axis is the standardized level of gene expression. Red represents high expression, blue represents low expression. P<0.05 as the statistical cutoff value.
Derivation of the risk scoring system for female LUAD
The risk scoring system for female LUAD was developed based on the analysis of the 16 M6A-related genes. Firstly, we incorporate 16 M6A-related genes (METTL3, METTL14, WTAP, KIAA1429, RBM15, RBM15B, YTHDC1, YTHDF1, YTHDF2, YTHDF3, HNRNPC, HNRNPA2B1, FMR1, LRPPRC, ALKBH5 and FTO.) into the multivariate model. Then, based on the code cox <-coxph (Surv (futime, fustat) ~., data = rt), multivariate Cox analysis was performed to output five genes (METTL3, YTHDF1, HNRNPC, HNRNPA2B1, FTO) and correlation coefficients that can be used to build a risk scoring system (Table 1). The risk score for each sample was determined according to the coefficient analysis of the five genes, as follows:
Table 1. The specific baseline clinicopathological characteristics of 266 female LUAD samples.
| Clinical character | 266 female LUAD samples with prognostic information |
| Age | |
| <= 60 years | 89 |
| > 60 years | 177 |
| Stage | |
| I | 156 |
| II | 52 |
| III | 43 |
| IV | 12 |
| Unknown | 3 |
| Pathologic T stage | |
| T1-2 | 236 |
| T3-4 | 28 |
| Unknown | 2 |
| Pathologic N stage | |
| N0-1 | 219 |
| N2-3 | 38 |
| Unknown | 9 |
| Pathologic M stage | |
| M0 | 167 |
| M1 | 11 |
| Unknown | 88 |
| Survival time | |
| <= 1 years | 57 |
| 1 years < | 160 |
| <= 3 years | |
| 3 years < | 32 |
| <= 5 years | |
| > 5 years | 17 |
Risk score = (- 0.4356 × METTL3 expression) + (- 0.5791 × YTHDF1 expression) + (1.1551 × HNRNPC expression) + (0.5820 × HNRNPA2B1 expression) + (0.6329 × FTO expression)
Using the median risk score as the cut-off value, 266 female LUAD samples with prognostic information were divided into high-risk and low-risk groups, and the overall survival (OS) of the low-risk group was significantly longer than that of the high-risk group (Figure 2). In addition, HNRNPC, HNRNPA2B1, and FTO showed positive coefficients, indicating that these genes were closely related to the high prognostic risk of female LUAD, that is, their higher expression levels corresponds to shorter OS. However, METTL3 and YTHDF1 showed negative coefficients, indicating that the lower their expression, the shorter the OS.
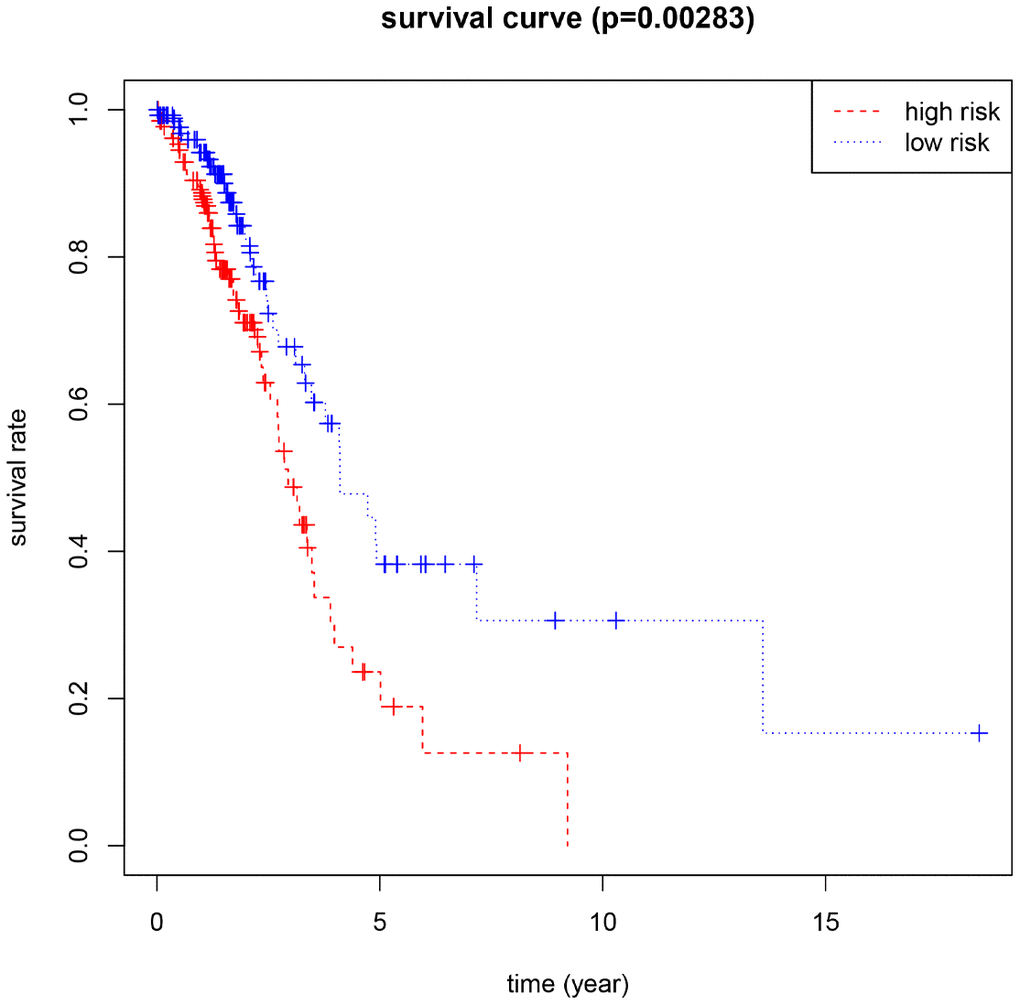
Figure 2. Kaplan-Meier for the risk scoring system based on 5 M6A-related genes (P<0.05 as the statistical cutoff value).
Correlation between risk score and clinical traits
To further evaluate the predictive performance of the risk scoring system that we developed, we included relevant clinical factors in this study (Table 2). Univariate Cox analysis showed that risk score, stage, and N stage were all closely related to OS in female patients with LUAD. Multivariate Cox analysis showed that with p<0.05 as the cutoff value, risk score could be used as an independent prognostic factor for evaluating patient survival time compared with clinical characteristics such as age and stage (Figure 3). Whether through univariate or multivariate Cox analysis, the risk scoring system could effectively evaluate the prognosis of female patients with LUAD, which further demonstrates the value of this model. In addition, we evaluated the relationship between the risk score and different clinical traits, and found that staging and N and M stages were all correlated with the risk score (P <0.05). And the higher the stage, N, and M stages, the higher the risk score, which is consistent with the current clinical prognosis, and further illustrates the accuracy of the risk scoring system we constructed. (Figure 4).
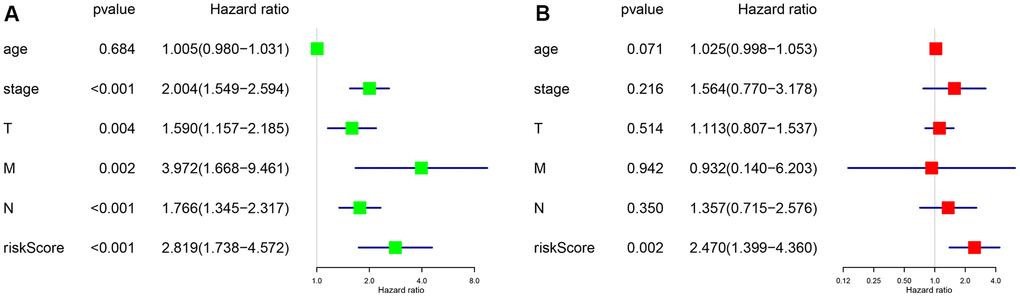
Figure 3. P <0.05 as the statistical cutoff value. (A) Univariate analysis of risk scores and clinical traits in female lung adenocarcinoma samples from TCGA database. (B) Multivariate analysis of risk scores and clinical traits in female lung adenocarcinoma samples from TCGA database.
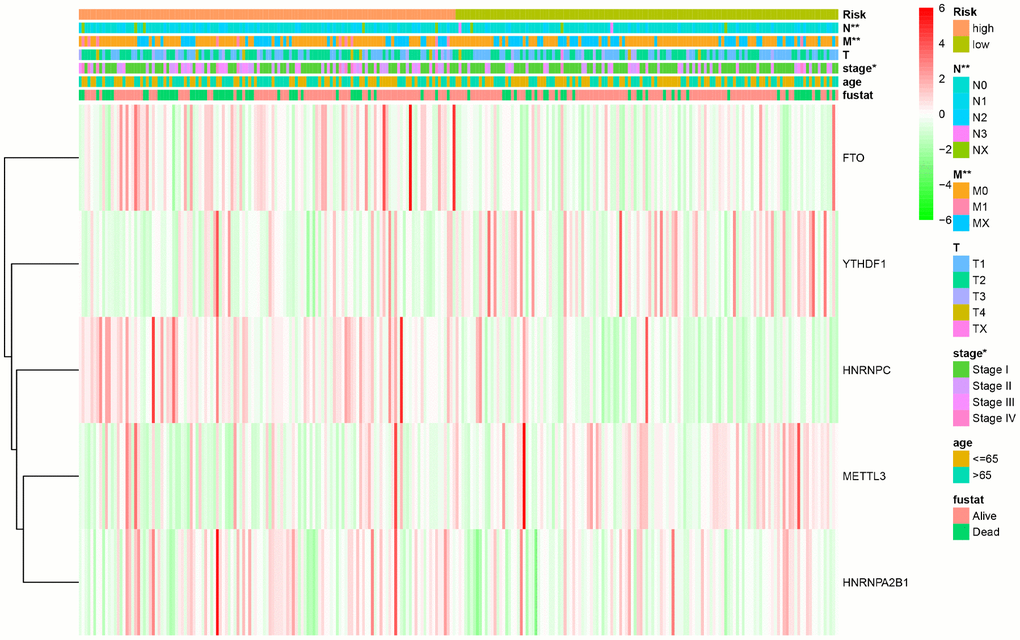
Figure 4. The relationship between the risk score and different clinical traits (* represents p < 0.05, ** represents p < 0.01, and *** represents p < 0.001).
Table 2. Multivariate Cox analysis of M6A related genes in female patients with lung adenocarcinoma.
| Gene | coef | exp(coef) | se(coef) | z | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| YTHDF1 | -0.5791 | 0.5604 | 0.3024 | -1.915 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| METTL3 | -0.4356 | 0.6469 | 0.2118 | -2.056 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FTO | 0.6329 | 1.8830 | 0.3016 | 2.098 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HNRNPC | 1.1551 | 3.1743 | 0.3335 | 3.463 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HNRNPA2B1 | 0.5820 | 1.7897 | 0.3126 | 1.862 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *Coef : the regression coefficient, exp (coef) : the risk ratio, se (coef) : the standard error, z : the Wald statistical value. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
CNVs of M6A-related genes in the risk scoring system
Combined with the CNV data analysis of female LUAD samples, it was found that compared with normal samples, the five M6A-related genes used to construct the risk scoring system in female LUAD samples were prone to CNV (Table 3), and there was a correlation between CNV and the expression of these genes, and they all showed a trend of increasing expression with increase in their copy numbers (Figure 5). Analysis of the cBioportal platform-related data also revealed the proportion of CNVs of these genes, which further verified the results obtained in the TCGA data analysis (Figure 6). Pathway enrichment analysis showed that METTL3, YTHDF1, HNRNPC, HNRNPA2B1, and FTO could participate in genetic information processing, which is related to the biological process of mRNAs.
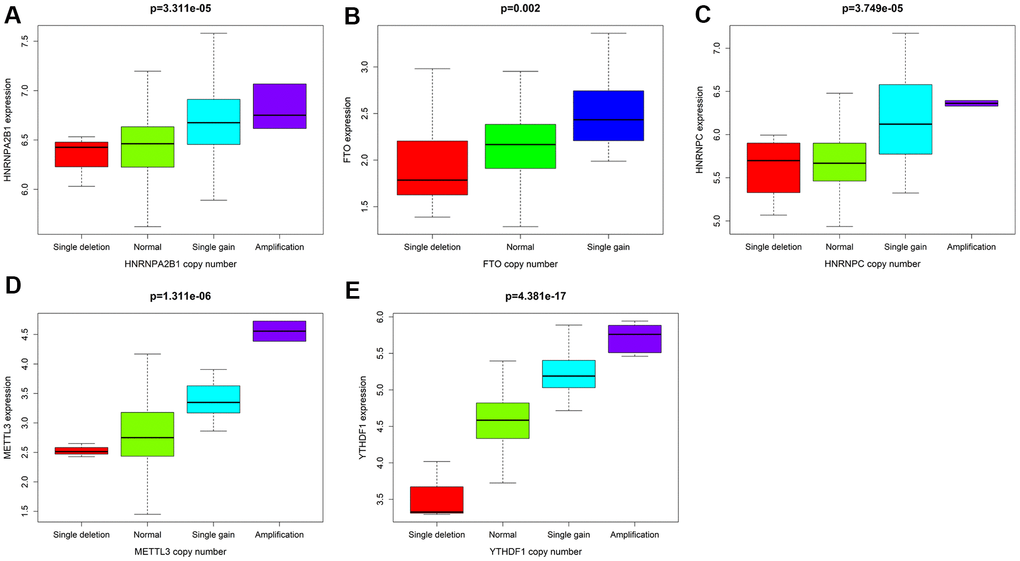
Figure 5. With P<0.05 as the cutoff value, the correlation analysis between CNV mutations and the expression of 5 M6A-related genes. (A) HNRNPA2B1 (B) FTO (C) HNRNPC (D) METTL3 (E) YTHDF1.

Figure 6. Analysis of CNV mutations in 5 M6A-related genes in lung adenocarcinoma data of cBioportal platform. Red represents an amplification in the number of copies, blue represents a deletion in the number of copies, green represents missense mutation, and gray represents truncating mutation.
Table 3. The main chromosomes and related sites where CNV occurs in the 5 M6A-related genes in the risk scoring system.
| Chromosome | chromStart | chromEnd | Gene |
| chr7 | 26189927 | 26201529 | HNRNPA2B1 |
| chr14 | 21209136 | 21269494 | HNRNPC |
| chr14 | 21498133 | 21511375 | METTL3 |
| chr16 | 53703963 | 54121941 | FTO |
| chr20 | 63195429 | 63216234 | YTHDF1 |
Discussion
Lung adenocarcinoma (LUAD) is the most common pathological subtype of non-small cell lung cancer (NSCLC), with an average 5-year survival rate of patients with LUAD is only 18%. Different from lung squamous cell carcinoma, LUAD is more likely to occur in women and non-smokers. Although great progress has been made in neoadjuvant radiotherapy, chemotherapy, molecular targeted therapy and immunotherapy methods, and greatly increase clinical expectations for the successful treatment of malignant diseases [23–26], local recurrence and distant metastasis caused by tumor heterogeneity and drug resistance are still threatening the lives of patients, resulting in a low long-term survival rate [27, 28]. There is an urgent need to identify effective biomarkers to improve treatment strategies and predict the prognosis of patients.
Cancer is a complex genetic and epigenetic disease, which involves mutations and dysregulation of oncogenes and tumor suppressor genes [29, 30]. Among them, epigenetic modifications can participate in the occurrence and progression of tumors by inhibiting the expression of tumor suppressor genes and enhancing that of oncogenes [31, 32]. The inherently reversible epigenetic changes make them a potential target for drug therapy [33]. They showed great potential in clinical personalized treatment and in prolonging the survival time of patients. M6A played a key role in different diseases including cancer and the expression was related to the activation or inhibition of many carcinogenic pathways [34]. For example, Xinyao Lin et al found that M6A could regulate the progression of epithelial-mesenchymal transformation in vitro and in vivo and the M6A modification of mRNAs increases in cancer cells were an important step in cancer cell metastasis. In addition, they also found that the up-regulated M6A regulatory factors METTL3 and YTHDF1 were important factors leading to the poor prognosis of hepatocellular carcinoma [35]. Qiang Wang et al found that high expression of M6A regulatory factors METTL3 in gastric cancer could play a carcinogenic role by regulating different targets or pathways and was closely related to the poor prognosis of patients with gastric cancer [36].
M6A regulatory factors are closely related to the activation and inhibition of cancer-related pathways and are potentially useful prognostic markers. In different types of cancer, M6A regulatory factors have been shown to undergo a wide range of genetic changes, including mutations and CNVs [37]. In this study, 16 widely reported M6A RNA regulatory factors were selected. Through multivariate COX analysis, five M6A-related gene markers, namely HNRNPC, HNRNPA2B1, FTO, METTL3, and YTHDF1, with high predictive ability were determined as a prognostic model for female LUAD. We divided 266 female LUAD samples into two high-risk and the low-risk groups according to the median risk scores of the five gene markers, and confirmed a significant difference in OS between the two groups. Combined with the CNV data analysis of female LUAD samples, it was found that compared with normal samples, the five M6A-related genes used to construct the risk scoring system in female LUAD samples were prone to CNV. Further, we observed a correlation between the CNVs and the corresponding gene expression. Our analyses emphasize the importance of M6A modulators in cancer development and lay the foundation for the development of novel therapeutic strategies based on RNA methylation.
HNRNPC, YTHDF1, and HNRNPA2B1 are all involved in the formation of M6A as readers. Among them, HNRNPC can regulate the stability and translation levels of bound mRNA molecules [38], while M6A can promote the binding of mRNAs and HNRNPC through the "m6A-switch" mechanism, thus regulating mRNA splicing and affecting the expression of the corresponding proteins [39]. Huang et al found that HNRNPC can promote cell proliferation, migration, and invasion [40], which was closely related to the occurrence and progression of cancer and was an important M6A regulatory factor that caused poor prognosis of patients. YTHDF1 can recognize and bind the M6A methylation of mRNA, thus inhibiting the translation of mRNA. It was found that the tumor size of YTHDF1 knockout mice is much smaller than that of wild type, and the immunoreactivity of M6A knockout mice was stronger than that of wild type [41], indicating that YTHDF1 can be used as an important target of immunotherapy against clinical tumors. In addition, Shi Y et al observed that high expression of YTHDF1 was associated with better clinical outcomes, and its depletion lead to cancer cells becoming resistant to cisplatin therapy [42]. In our researchers, we found that the increased expression of YTHDF1 was negatively correlated with risk score, and the down-regulated expression of YTHDF1 was associated with longer survival in female patients with LUAD, which was consistent with the results of previous studies.
HNRNPA2B1 plays a role in transcription, mRNA pretreatment, RNA nuclear output, mRNA translation, and mature mRNA stability [43]. Studies have shown that HNRNPA2B1 was up-regulated by CACNA1G-AS1, thus participating in the progression of NSCLC [44]. METTL3 participates in the formation of M6A as an encoder. The study found that the OS and progression-free survival of colorectal cancer patients with high expression of M6A regulatory factor METTL3 were shorter on average [45]. In addition, the expression of METTL3 is up-regulated in lung cancer, wherein simvastatin treatment can attenuate this increased expression, which can in turn inhibit the malignant progression of lung cancer [46]. Our results also found that the expression of METTL3 was up-regulated in female LUAD samples, which can also affect the prognosis of the patients. The demethylase FTO, as a decoder, can regulate the stability of cellular mRNA by removing M6A residues. Studies have shown that FTO can activate the migration of LUAD cells through M6A demethylation and promote the progression of LUAD [47]. The above analysis showed that the five M6A related genes in the risk scoring system were closely related to the occurrence and progression of tumor, which further proved the reliability of the results of this study.
In our study, the expression of HNRNPC, YTHDF1, HNRNPA2B1, and METTL3 in female LUAD was higher than that in normal tissues, but that of FTO was lower. Taking the median risk score as the cut-off value, the risk scoring system could divide female LUAD samples into high-risk group and low-risk group. The overall survival time (OS) of low-risk group was significantly longer than that of high-risk group. The M6A-related risk scoring system developed in this study help to improve the screening of high-risk female patients with LUAD and provides important theoretical bioinformatics support for evaluating the prognosis of such patients. Notably, compared with normal samples, the five M6A-related genes in female LUAD samples were prone to CNV, and have been verified using the sequencing and CNV data in cBioPortal. Further analysis showed a correlation between the copy numbers of HNRNPC, YTHDF1, HNRNPA2B1, METTL3, FTO and the corresponding gene expression, that is, with an increase in copy number, the expression of these genes increased. In addition, univariate and multivariate analyses combined with clinical factors (such as age and TNM) showed that the performance of the risk scoring system developed in this study in predicting specificity and sensitivity was effective. The M6A-related genes involved in the risk scoring system can be considered new targets in LUAD, and may form the basis for further exploring the pathogenesis and treatment strategies of female LUAD. Although this study may be of vital clinical importance, it undoubtedly has some limitations. More information needs to be collected to verify its accuracy and to conduct further studies in vivo and in vitro to explore the role and mechanism of action of several M6A-related biomarkers in female LUAD.
Conclusions
In this study, we developed a female LUAD risk scoring system based on five M6A-related genes to evaluate the prognosis of patients. The scoring system can classify female LUAD patients into different risk levels according to their expression of these M6A-related genes. In addition, we observed the correlation between the CNVs of five M6A-related genes in the risk scoring system and the corresponding gene expression. We expect that this study will be helpful for early diagnosis and personalized treatment of female LUAD in the future. Although the results of the study need to be further confirmed, the risk scoring system we established may be of great value for the timely prevention of female LUAD and provide important theoretical bioinformatics support for evaluating the prognosis of patients.
Materials and Methods
Data source
In this study, the mRNA expression, clinical and CNV data used to construct and analysis the female LUAD risk score system were obtained from the TCGA database, and verified by the CNV data of LUAD patients on the cBioPortal platform. We downloaded data related to mRNA expression in 320 female LUADs from the database, including those for 34 normal samples and 286 LUAD samples. Samples without clinical survival data were removed. Finally, data for 266 female LUAD samples were used to develop a risk scoring system. In addition, the CNV data on 623 samples of female LUAD were considered, including those for 323 normal cases and 299 cancer cases. Both TCGA database and the cBioportal platform are open to the public and thus, no further ethical approval was required for this study.
Data processing and analysis
The level of M6A is dynamically regulated by its encoder (methyltransferase), reader, and decoder. The main genes involved in encoding include METTL3, METTL14, WTAP, KIAA1429, RBM15, and RBM15B. For reader, namely binding protein, the main genes include YTHDC1, YTHDF1, YTHDF2, YTHDF3, HNRNPC, HNRNPA2B1, FMR1, and LRPPRC. Decoder finger demethylase, and the main genes for decoding include ALKBH5 and FTO. We screened the expression of 16 genes related to M6A from the mRNA expression data of female LUAD. Using the R package of edger software, the expression data of these genes were standardized to elucidate their differential expression in normal samples and female LUAD samples. Then the related genes were looped by FOR sentence, and the violin map was drawn to visualize the results of differential expression. Then, the downloaded CNV data were also analyzed for the normal samples and female LUAD samples.
Analysis of correlation between risk score and clinical traits
To further evaluate the predictive performance of the risk scoring system that we developed, we included relevant clinical factors in this study. The clinical data of the female patients with LUAD downloaded from TCGA database included age, staging, tumor-node-metastasis (TNM) staging, survival time, and survival status. In order to test the correlation between risk score and clinical characteristics and whether the prognosis of the risk scoring system is independent of other clinical parameters, we included risk score and clinical traits into the variable model, further analyzed the independent prognosis by univariate and multivariate Cox, and further obtained the heat map of the correlation between risk score and clinical traits for visual display. P < 0.05 was considered to indicate statistical significance.
Correlation and pathway analysis of CNVs in the scoring system
Combined with the CNV data of female LUAD patients in TCGA database, we analyzed the CNVs of M6A-related genes in the risk scoring system, and determined the relationship between CNVs and corresponding gene expression. The expression data and CNV data of five M6A-related genes in the risk scoring system were read in turn by R language, tested and analyzed by ksTest. Then the CNV of related genes was analyzed and verified by using the relevant LUAD data in cBioportal platform. In addition, the KEGG website (https://www.kegg.jp/) was used to analyze the pathway enrichment of M6A-related genes in the scoring system, in order to further understand the possible mechanism of abnormal expression of these genes in female LUADs.
Author Contributions
CG and CS conceived and designed the study; JZ, HL and FF performed data analysis; CL, CZ, LL contributed analysis tools; CG and JZ was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work is supported by the grants from National Natural Science Foundation of China (81673799) and National Natural Science Foundation of China (81703915).
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492 [PubMed]
- 2. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–18. https://doi.org/10.1038/nature12213 [PubMed]
- 3. Park CK, Cho HJ, Choi YD, Oh IJ, Kim YC. A phase II trial of osimertinib in the second-line treatment of non-small cell lung cancer with the EGFR T790M mutation, detected from circulating tumor DNA: LiquidLung-O-cohort 2. Cancer Res Treat. 2019; 51:777–87. https://doi.org/10.4143/crt.2018.387 [PubMed]
- 4. Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, Mazieres J, Zalcman G, Brosseau S, Le Moulec S, Leroy L, Duchemann B, Lefebvre C, et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol. 2018; 4:1543–52. https://doi.org/10.1001/jamaoncol.2018.3676 [PubMed]
- 5. Verma M. The role of epigenomics in the study of cancer biomarkers and in the development of diagnostic tools. Adv Exp Med Biol. 2015; 867:59–80. https://doi.org/10.1007/978-94-017-7215-0_5 [PubMed]
- 6. Wang KC, Chang HY. Epigenomics: technologies and applications. Circ Res. 2018; 122:1191–99. https://doi.org/10.1161/CIRCRESAHA.118.310998 [PubMed]
- 7. Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011; 39:D195–201. https://doi.org/10.1093/nar/gkq1028 [PubMed]
- 8. Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, Semenza GL. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016; 7:64527–42. https://doi.org/10.18632/oncotarget.11743 [PubMed]
- 9. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, Weitman S, Tomlinson GE, Rao MK, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014; 28:1171–74. https://doi.org/10.1038/leu.2014.16 [PubMed]
- 10. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, Wong CC, Ng IO, Wong CM. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018; 67:2254–70. https://doi.org/10.1002/hep.29683 [PubMed]
- 11. Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue Q, Wang D, Huang J, Gao S, Gao Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. 2017; 482:582–89. https://doi.org/10.1016/j.bbrc.2016.11.077 [PubMed]
- 12. Li Y, Gu J, Xu F, Zhu Q, Chen Y, Ge D, Lu C. Molecular characterization, biological function, tumor microenvironment association and clinical significance of m6A regulators in lung adenocarcinoma. Brief Bioinform. 2020. [Epub ahead of print]. https://doi.org/10.1093/bib/bbaa225 [PubMed]
- 13. Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013; 14:703–18. https://doi.org/10.1038/nrg3539 [PubMed]
- 14. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, et al. Global variation in copy number in the human genome. Nature. 2006; 444:444–54. https://doi.org/10.1038/nature05329 [PubMed]
- 15. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. https://doi.org/10.1038/nature17676 [PubMed]
- 16. Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, Nogami N, Ohe Y, Mann H, Rukazenkov Y, Ghiorghiu S, Stetson D, Markovets A, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018; 36:841–49. https://doi.org/10.1200/JCO.2017.74.7576 [PubMed]
- 17. Li BQ, You J, Huang T, Cai YD. Classification of non-small cell lung cancer based on copy number alterations. PLoS One. 2014; 9:e88300. https://doi.org/10.1371/journal.pone.0088300 [PubMed]
- 18. Shukla S, Evans JR, Malik R, Feng FY, Dhanasekaran SM, Cao X, Chen G, Beer DG, Jiang H, Chinnaiyan AM. Development of a RNA-seq based prognostic signature in lung adenocarcinoma. J Natl Cancer Inst. 2016; 109:djw200. https://doi.org/10.1093/jnci/djw200 [PubMed]
- 19. Zhuang Z, Chen L, Mao Y, Zheng Q, Li H, Huang Y, Hu Z, Jin Y. Diagnostic, progressive and prognostic performance of m6A methylation RNA regulators in lung adenocarcinoma. Int J Biol Sci. 2020; 16:1785–97. https://doi.org/10.7150/ijbs.39046 [PubMed]
- 20. Zhang Y, Liu X, Liu L, Li J, Hu Q, Sun R. Expression and prognostic significance of m6A-related genes in lung adenocarcinoma. Med Sci Monit. 2020; 26:e919644. https://doi.org/10.12659/MSM.919644 [PubMed]
- 21. Du J, Hou K, Mi S, Ji H, Ma S, Ba Y, Hu S, Xie R, Chen L. Malignant evaluation and clinical prognostic values of m6A RNA methylation regulators in glioblastoma. Front Oncol. 2020; 10:208. https://doi.org/10.3389/fonc.2020.00208 [PubMed]
- 22. Ji L, Chen S, Gu L, Zhang X. Exploration of potential roles of m6A regulators in colorectal cancer prognosis. Front Oncol. 2020; 10:768. https://doi.org/10.3389/fonc.2020.00768 [PubMed]
- 23. Christofi T, Baritaki S, Falzone L, Libra M, Zaravinos A. Current Perspectives in Cancer Immunotherapy. Cancers (Basel). 2019; 11:1472. https://doi.org/10.3390/cancers11101472 [PubMed]
- 24. Bao X, Shi R, Zhao T, Wang Y. Immune landscape and a novel immunotherapy-related gene signature associated with clinical outcome in early-stage lung adenocarcinoma. J Mol Med (Berl). 2020; 98:805–18. https://doi.org/10.1007/s00109-020-01908-9 [PubMed]
- 25. Leonardi GC, Candido S, Falzone L, Spandidos DA, Libra M. Cutaneous melanoma and the immunotherapy revolution (review). Int J Oncol. 2020; 57:609–18. https://doi.org/10.3892/ijo.2020.5088 [PubMed]
- 26. Falzone L, Salomone S, Libra M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front Pharmacol. 2018; 9:1300. https://doi.org/10.3389/fphar.2018.01300 [PubMed]
- 27. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150:1107–20. https://doi.org/10.1016/j.cell.2012.08.029 [PubMed]
- 28. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018; 68:7–30. https://doi.org/10.3322/caac.21442 [PubMed]
- 29. Mafficini A, Scarpa A. Genetics and epigenetics of gastroenteropancreatic neuroendocrine neoplasms. Endocr Rev. 2019; 40:506–36. https://doi.org/10.1210/er.2018-00160 [PubMed]
- 30. Zhou S, Treloar AE, Lupien M. Emergence of the noncoding cancer genome: a target of genetic and epigenetic alterations. Cancer Discov. 2016; 6:1215–29. https://doi.org/10.1158/2159-8290.CD-16-0745 [PubMed]
- 31. Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016; 76:3446–50. https://doi.org/10.1158/0008-5472.CAN-15-3278 [PubMed]
- 32. Vizoso M, Puig M, Carmona FJ, Maqueda M, Velásquez A, Gómez A, Labernadie A, Lugo R, Gabasa M, Rigat-Brugarolas LG, Trepat X, Ramírez J, Moran S, et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis. 2015; 36:1453–63. https://doi.org/10.1093/carcin/bgv146 [PubMed]
- 33. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011; 7:885–87. https://doi.org/10.1038/nchembio.687 [PubMed]
- 34. Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014; 15:313–26. https://doi.org/10.1038/nrm3785 [PubMed]
- 35. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, Luo G, Tauler J, Du J, Lin S, He C, Wang H. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of snail. Nat Commun. 2019; 10:2065. https://doi.org/10.1038/s41467-019-09865-9 [PubMed]
- 36. Wang Q, Geng W, Guo H, Wang Z, Xu K, Chen C, Wang S. Emerging role of RNA methyltransferase METTL3 in gastrointestinal cancer. J Hematol Oncol. 2020; 13:57. https://doi.org/10.1186/s13045-020-00895-1 [PubMed]
- 37. Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, Wang Q, Li X, Zhang Y, Xu J. Molecular characterization and clinical relevance of m6A regulators across 33 cancer types. Mol Cancer. 2019; 18:137. https://doi.org/10.1186/s12943-019-1066-3 [PubMed]
- 38. Huang M, Rech JE, Northington SJ, Flicker PF, Mayeda A, Krainer AR, LeStourgeon WM. The C-protein tetramer binds 230 to 240 nucleotides of pre-mRNA and nucleates the assembly of 40S heterogeneous nuclear ribonucleoprotein particles. Mol Cell Biol. 1994; 14:518–33. https://doi.org/10.1128/mcb.14.1.518 [PubMed]
- 39. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015; 518:560–64. https://doi.org/10.1038/nature14234 [PubMed]
- 40. Huang GZ, Wu QQ, Zheng ZN, Shao TR, Chen YC, Zeng WS, Lv XZ. M6A-related bioinformatics analysis reveals that HNRNPC facilitates progression of OSCC via EMT. Aging (Albany NY). 2020; 12:11667–84. https://doi.org/10.18632/aging.103333 [PubMed]
- 41. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, Huang X, Liu Y, Wang J, Dougherty U, Bissonnette MB, Shen B, Weichselbaum RR, et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. 2019; 566:270–74. https://doi.org/10.1038/s41586-019-0916-x [PubMed]
- 42. Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, Shen Q, Xu P, Zeng L, Zhou Y, Huang Y, Yang Z, Zhou J, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019; 10:4892. https://doi.org/10.1038/s41467-019-12801-6 [PubMed]
- 43. He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci. 2009; 66:1239–56. https://doi.org/10.1007/s00018-008-8532-1 [PubMed]
- 44. Yu PF, Kang AR, Jing LJ, Wang YM. Long non-coding RNA CACNA1G-AS1 promotes cell migration, invasion and epithelial-mesenchymal transition by HNRNPA2B1 in non-small cell lung cancer. Eur Rev Med Pharmacol Sci. 2018; 22:993–1002. https://doi.org/10.26355/eurrev_201802_14381 [PubMed]
- 45. Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, Chen D, Li B, Kang TB, et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019; 18:112. https://doi.org/10.1186/s12943-019-1038-7 [PubMed]
- 46. Chen WW, Qi JW, Hang Y, Wu JX, Zhou XX, Chen JZ, Wang J, Wang HH. Simvastatin is beneficial to lung cancer progression by inducing METTL3-induced m6A modification on EZH2 mRNA. Eur Rev Med Pharmacol Sci. 2020; 24:4263–70. https://doi.org/10.26355/eurrev_202004_21006 [PubMed]
- 47. Ding Y, Qi N, Wang K, Huang Y, Liao J, Wang H, Tan A, Liu L, Zhang Z, Li J, Kong J, Qin S, Jiang Y. FTO facilitates lung adenocarcinoma cell progression by activating cell migration through mRNA demethylation. Onco Targets Ther. 2020; 13:1461–70. https://doi.org/10.2147/OTT.S231914 [PubMed]