



Introduction
As a provocative title has recently announced, “rapamycin fails to extend lifespan in DNA repair-deficient mice” [1]. The word “fails” implies bad news. Rapamycin tried but failed. Yet, it is expected that the anti-aging drug rapamycin should not restore lifespan of short-lived mice that fail to grow and die young from causes other than normal aging [2]. In such growth-retarded mice, rapamycin, an inhibitor of cell growth, further retards weight gain.
Similarly, rapamycin does not extend but even slightly shortens lifespan in telomerase-deficient mice, which die young from poor growth and intestinal atrophy caused by telomere shortening [3]. (As we will discuss, this is predictable by hyperfunction theory.) While shortening lifespan by 18% in unnatural telomerase-deficient mice, in the same study in natural mice, rapamycin increased lifespan by 39% and healthspan by 58% (measured as tumor-free survival) [3]. In dozens of independent studies, rapamycin has not failed to extend lifespan in normal mice [4]. However, while extending lifespan in normal mice, rapamycin may fail to save animals dying young from cellular growth retardation. But something important should not be overlooked. The failure of rapamycin to extend lifespan in these short-lived mice, dying from DNA damage, rules out the damage theory of aging. To understand this point, we must first discuss what limits animal lifespan.
Quasi-programmed (hyperfunctional) aging
In proliferating cells, growth-promoting pathways such as mTOR (Target of Rapamycin) and MAPK drive cellular growth, which is balanced by cell division. When the cell cycle is arrested, however, growth-promoting pathways drive cellular senescence, which is a continuation of cellular growth in the absence of cell division [5]. During geroconversion to senescence, cells become hypertrophic and hyperfunctional. One example of hyper-function is SASP or Senescence-Associated Secretory Phenotype [6]. Rapamycin can cause reversible cycle arrest but suppresses geroconversion, thus ensuring quiescence instead of senescence. (Note: Rapamycin does not prevent cell cycle arrest, it only prevents geroconversion that makes this arrest permanent [7]. This point is often miscited by others). Rapamycin slows down both growth and geroconversion, figuratively slowing down time [8]. Like cellular senescence is a continuation of growth, organismal aging is a continuation of growth too [9].
According to hyperfunction theory, aging is quasi-programmed, a continuation of developmental growth programs, driven in part by hyper-functional signaling pathways including the mTOR pathway [9]. Hyperfunction is an excessive normal function later in life. It’s not necessarily an increase of function; it may even be insufficient decrease of function. For example, protein synthesis is decreased in C elegans but is still too high: its further inhibition extends lifespan [10, 11].
Hyperfunction leads to age-related diseases, secondary organ damage and loss of function. For example, cellular hyperfunctions result in hypertension, culminating in stroke and damage of the brain. Aging is a sum of all age-related diseases [12, 13]. This theory was discussed in detail [9, 14–20] and has gained experimental support [11, 16, 21–26]. I will not discuss it here, just to mention that accumulation of molecular damage is not a driving force of development and therefore of aging. It is hyperfunctional signaling pathways such as mTOR (one of many) that drive both growth and aging, causing age-related diseases that in turn damage organs, leading to secondary loss of function.
Although molecular damage accumulates, this accumulation is not life-limiting because quasi-programmed aging terminates life first (Figure 1A). Quasi-programmed (hyperfunctional) aging is life-limiting, because it is favored by natural selection. Natural selection favors robust development and fitness early in life at the cost of aging. For example, growth hormone receptor-deficient mice (GHR-KO mice), with decreased mTORC1 activity, live longer but are small and weak early in life [27, 28]. In such mice mTORC1-driven aging is inhibited and mice live longer but would not survive in the wild and therefore do not exist in nature. As another example, knockout of PI3K, an activator of mTOR pathways, extends lifespan 10-fold in C. elegans [29]. The mutant worm undergoes prolonged developmental arrest, which would be lethal in the wild [29]. Therefore, natural selection favors hyperfunctional mTOR that is optimal for development but drives age-related diseases later in life.
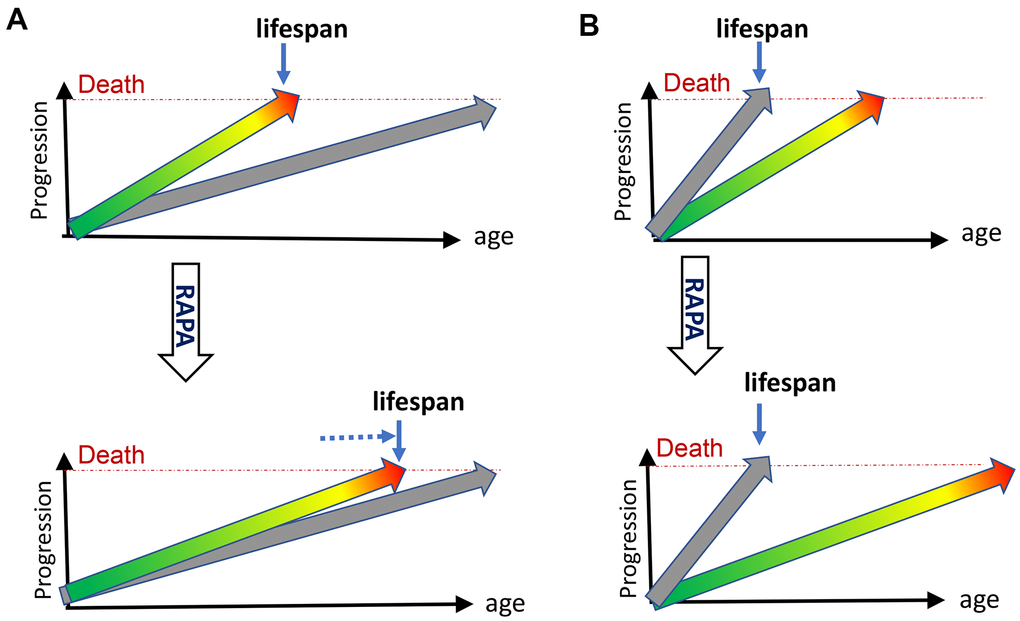
Figure 1. Rapamycin extends lifespan in natural but not progeroid mice. (A) Natural mice. Hyperfunctional aging (green/yellow/red arrow) progresses from development (green) to diseases (red), reaching death threshold and limiting lifespan. Accumulation of molecular damage (gray arrow) is slow and does not reach death threshold in animal lifetime. It would take longer to die from molecular damage. Treatment with rapamycin (RAPA) extends lifespan by slowing down mTOR-driven aging (B) Progeroid, telomerase- or DNA-repair-deficient mice. Accumulation of molecular damage (gray arrow) is artificially accelerated to become life-limiting. Treatment with rapamycin (RAPA) cannot extend lifespan.
According to damage theories, aging is functional decline caused by molecular damage. According to hyperfunction theory, quasi-programmed aging is not functional decline but a hyperfunction: cellular and systemic functions are higher than optimal for longevity. They are optimal for early life fitness and in part (only in part) mTOR-dependent.
In both molecular damage and hyperfunction theories, aging exists because late-life is shadowed from natural selection. But quasi-programmed aging is not simply shadowed from, it is promoted by natural selection, because accelerated aging is hardwired with fitness early in life. By selecting for fitness, nature indirectly selects for accelerated aging. This makes quasi-programmed aging life-limiting. One of predictions of hyperfunction theory is that rapamycin must extend lifespan in animals [9]. This prediction has been confirmed. In dozens of studies, rapamycin prolongs lifespan and healthspan in mice [3, 30–65]. Rapamycin extends lifespan in C elegans [66] and Drosophila [67–69]. Furthermore, rapamycin even extends life of the simplest animal, Hydra, which is thought to be immortal. Depending on conditions, Hydra can be either immortal or undergo aging. Rapamycin slows aging, stem cell exhaustion and extends life span in Hydra [70].
mTOR-driven aging is only one component of quasi-programmed (hyperfunction) aging. In addition, MEK/MAPK, NF-kB, p63, HIF-1 and many other signaling pathways are involved, interacting with the mTOR pathway and forming networks. Rapamycin cannot affect all of them. In theory, mTOR-independent quasi-programmed aging can be life-limiting in some conditions and diseases. I suggest that long-lived GHR-KO mice with low mTORC1 activity undergo partially mTORC1-independent quasi-programmed senescence, because rapamycin cannot prolong lifespan in these mice further, while prolonging lifespan in parental normal mice [71]. Discussion of mTOR-independent components of quasi-programmed aging is beyond the focus of this article. Let us return to stochastic accumulation of molecular damage.
How molecular damage can become life-limiting
Molecular damage can become life-limiting in two ways. First, hyper-functional aging should be eliminated or slowed down, so an organism lives long enough to die from accumulation of molecular damage. In this scenario, accumulation of molecular damage causes post-aging. Such examples are unknown, but it is a very intriguing possibility. Could a PI3K-null worm [29] with 10-fold longer lifespan die from molecular damage?
Second, accumulation of molecular damage can be greatly accelerated artificially by knockout of repair/maintenance enzymes (Figure 1B). Such animals do not exist in nature. But artificially created, they may provide a glimpse of how post-aging may look. Their pathology differs drastically from normal aging, for example, telomere shortening. Second-generation telomerase-deficient mice (G2 Terc−/−) with critically short telomeres fail to grow and die young from unfamiliar diseases such as intestinal atrophy due to failure of cell proliferation [3]. When telomeres reach critical length, it can cause DNA-damage response, leading to aplastic anemia, organ fibrosis, atrophy of the small intestine and the spleen, skin and hair lesions. In humans, diseases of short telomeres cause death from bone marrow failure and pulmonary fibrosis [72]. This does not resemble normal aging.
In humans, mice and C. elegans, telomere shortening is not life-limiting [73–75]. In mice lacking telomerase, even accelerated telomere shortening is still not life-limiting in the first generation [76]. It took several generations to achieve critically short telomeres, leading to syndromes strikingly different from normal aging. In humans, telomere length does not reach telomere threshold during life time [75, 77, 78]. Normal telomere shortening would cause telomere-driven pathologies, but normal animals do not live long enough to reach this threshold. Rapamycin prolongs life in normal mice, proving that telomere length does not constrain normal lifespan [3]. When artificially shortened, then telomeres become life-limiting and rapamycin cannot extend lifespan anymore [3].
Ercc1∆/− mutant mice are defective in DNA repair, such as transcription-coupled repair, global-genome nucleotide excision and crosslink repair [1, 2]. Therefore, multiple types of DNA damages accumulate. This leads to decreased cell proliferation, arrested development, poor growth, abnormal liver nuclei of liver and kidney, absence of subcutaneous fat, ferritin deposition, kidney malfunction and early death [2]. Unlike natural mice, short-lived Ercc1∆/− mice do not develop tumors, probably because they do not live long enough to suffer typical age-related diseases [1, 2]. In such mice, dying from molecular damage, rapamycin fails to extend lifespan [1].
Conclusions
Here I discussed new evidence that normal aging is not caused by accumulation of molecular damage or telomere shortening: while extending normal lifespan in mice, rapamycin failed to do so in mice dying from molecular damage (Figure 1).
Previously, several lines of evidence suggested that molecular damage does not cause normal aging. Their detailed discussion is beyond the focus of this article, so I will just mention some of them, without referencing them (I will reference these points in forthcoming review “When longevity drugs do not increase longevity: Unifying development-driven and damage-induced theories of aging”, In press). First, overexpression of enzymes that decrease damage does not extend lifespan in most studies. Similarly, antioxidants do not extend lifespan in animals and may increase mortality in humans. Furthermore, even data that support damage theory can be explained by other mechanisms. For example, N-Acetyl-L-Cysteine, a commonly used anti-oxidant, can inhibit mTOR. Second, according to calculations, molecular damage, especially mtDNA mutations and telomere shortening, cannot reach deadly threshold during animal lifetime. Third, genetic knockout of signaling pathways can extend lifespan without affecting molecular damage. Similarly, pharmacological interventions can extend life without affecting damage accumulation. Forth, dramatic intra- and inter-species differences in lifespan poorly correlate with the rate of molecular damage. Fifth, nuclear transfer and nuclear reprogramming both rule out DNA damage as a cause of aging. Following adult somatic cell nuclear transfer, cloned animals are healthy and have normal lifespan. Sixth, low levels of molecular damage may increase longevity. This phenomenon is known as hormesis. Regardless of mechanistic explanations, this indicates that molecular damage is not-life-limiting even when moderately increased. Finally, rapamycin increases lifespan in all normal animals tested, indicating that mTORC1-dependent quasi-program is life-limiting. The list can go on and on. Once again, damage accumulates and must cause death eventually, but quasi-programmed (hyperfunctional) aging terminates life first. Molecular damage can become life-limiting, when artificially accelerated or, potentially, when quasi-programmed aging is decelerated. Then interventions to repair molecular damage may increase life further.
Conflicts of Interest
The author declares that he has no conflicts of interest.
References
- 1. Birkisdóttir MB, Jaarsma D, Brandt RM, Barnhoorn S, van Vliet N, Imholz S, van Oostrom CT, Nagarajah B, Portilla Fernández E, Roks AJ, Elgersma Y, van Steeg H, Ferreira JA, et al. Unlike dietary restriction, rapamycin fails to extend lifespan and reduce transcription stress in progeroid DNA repair-deficient mice. Aging Cell. 2021. [Epub ahead of print]. https://doi.org/10.1111/acel.13302 [PubMed]
- 2. Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997; 7:427–39. https://doi.org/10.1016/s0960-9822(06)00190-4 [PubMed]
- 3. Ferrara-Romeo I, Martinez P, Saraswati S, Whittemore K, Graña-Castro O, Thelma Poluha L, Serrano R, Hernandez-Encinas E, Blanco-Aparicio C, Maria Flores J, Blasco MA. The mTOR pathway is necessary for survival of mice with short telomeres. Nat Commun. 2020; 11:1168. https://doi.org/10.1038/s41467-020-14962-1 [PubMed]
- 4. Blagosklonny MV. The goal of geroscience is life extension. Oncotarget. 2021; 12:131–44. https://doi.org/10.18632/oncotarget.27882
- 5. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7:3355–61. https://doi.org/10.4161/cc.7.21.6919 [PubMed]
- 6. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015; 17:1049–61. https://doi.org/10.1038/ncb3195 [PubMed]
- 7. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4:159–65. https://doi.org/10.18632/aging.100443 [PubMed]
- 8. Blagosklonny MV. Does rapamycin slow down time? Oncotarget. 2018; 9:30210–12. https://doi.org/10.18632/oncotarget.25788 [PubMed]
- 9. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5:2087–102. https://doi.org/10.4161/cc.5.18.3288 [PubMed]
- 10. Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in caenorhabditis elegans. Aging Cell. 2007; 6:111–19. https://doi.org/10.1111/j.1474-9726.2006.00266.x [PubMed]
- 11. Dhondt I, Petyuk VA, Cai H, Vandemeulebroucke L, Vierstraete A, Smith RD, Depuydt G, Braeckman BP. FOXO/DAF-16 activation slows down turnover of the majority of proteins in C. Elegans. Cell Rep. 2016; 16:3028–40. https://doi.org/10.1016/j.celrep.2016.07.088 [PubMed]
- 12. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany NY). 2009; 1:281–88. https://doi.org/10.18632/aging.100034 [PubMed]
- 13. Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012; 181:1142–46. https://doi.org/10.1016/j.ajpath.2012.06.024 [PubMed]
- 14. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7:3344–54. https://doi.org/10.4161/cc.7.21.6965 [PubMed]
- 15. Blagosklonny MV. Answering the ultimate question ”what is the proximal cause of aging?”. Aging (Albany NY). 2012; 4:861–77. https://doi.org/10.18632/aging.100525 [PubMed]
- 16. Gems D, de la Guardia Y. Alternative perspectives on aging in caenorhabditis elegans: reactive oxygen species or hyperfunction? Antioxid Redox Signal. 2013; 19:321–29. https://doi.org/10.1089/ars.2012.4840 [PubMed]
- 17. Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013; 75:621–44. https://doi.org/10.1146/annurev-physiol-030212-183712 [PubMed]
- 18. Wang H, Zhao Y, Ezcurra M, Benedetto A, Gilliat AF, Hellberg J, Ren Z, Galimov ER, Athigapanich T, Girstmair J, Telford MJ, Dolphin CT, Zhang Z, Gems D. A parthenogenetic quasi-program causes teratoma-like tumors during aging in wild-type C. Elegans. NPJ Aging Mech Dis. 2018; 4:6. https://doi.org/10.1038/s41514-018-0025-3 [PubMed]
- 19. Ezcurra M, Benedetto A, Sornda T, Gilliat AF, Au C, Zhang Q, van Schelt S, Petrache AL, Wang H, de la Guardia Y, Bar-Nun S, Tyler E, Wakelam MJ, Gems D. C. elegans Eats Its Own Intestine to Make Yolk Leading to Multiple Senescent Pathologies. Curr Biol. 2018; 28:2544–56.e5. https://doi.org/10.1016/j.cub.2018.06.035 [PubMed]
- 20. Xi J, Cai J, Cheng Y, Fu Y, Wei W, Zhang Z, Zhuang Z, Hao Y, Lilly MA, Wei Y. The TORC1 inhibitor Nprl2 protects age-related digestive function in Drosophila. Aging (Albany NY). 2019; 11:9811–28. https://doi.org/10.18632/aging.102428 [PubMed]
- 21. Scialò F, Sriram A, Naudí A, Ayala V, Jové M, Pamplona R, Sanz A. Target of rapamycin activation predicts lifespan in fruit flies. Cell Cycle. 2015; 14:2949–58. https://doi.org/10.1080/15384101.2015.1071745 [PubMed]
- 22. de la Guardia Y, Gilliat AF, Hellberg J, Rennert P, Cabreiro F, Gems D. Run-on of germline apoptosis promotes gonad senescence in C. Elegans. Oncotarget. 2016; 7:39082–96. https://doi.org/10.18632/oncotarget.9681 [PubMed]
- 23. Chen HY, Maklakov AA. The worm that lived: evolution of rapid aging under high extrinsic mortality revisited. Worm. 2013; 2:e23704. https://doi.org/10.4161/worm.23704 [PubMed]
- 24. Lind MI, Ravindran S, Sekajova Z, Carlsson H, Hinas A, Maklakov AA. Experimentally reduced insulin/IGF-1 signaling in adulthood extends lifespan of parents and improves darwinian fitness of their offspring. Evol Lett. 2019; 3:207–16. https://doi.org/10.1002/evl3.108 [PubMed]
- 25. Cheng Z, Ristow M. Mitochondria and metabolic homeostasis. Antioxid Redox Signal. 2013; 19:240–42. https://doi.org/10.1089/ars.2013.5255 [PubMed]
- 26. de Verges J, Nehring V. A critical look at proximate causes of social insect senescence: damage accumulation or hyperfunction? Curr Opin Insect Sci. 2016; 16:69–75. https://doi.org/10.1016/j.cois.2016.05.003 [PubMed]
- 27. Dominick G, Berryman DE, List EO, Kopchick JJ, Li X, Miller RA, Garcia GG. Regulation of mTOR activity in snell dwarf and GH receptor gene-disrupted mice. Endocrinology. 2015; 156:565–75. https://doi.org/10.1210/en.2014-1690 [PubMed]
- 28. Bartke A, Sun LY, Longo V. Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol Rev. 2013; 93:571–98. https://doi.org/10.1152/physrev.00006.2012 [PubMed]
- 29. Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ. Remarkable longevity and stress resistance of nematode PI3K-null mutants. Aging Cell. 2008; 7:13–22. https://doi.org/10.1111/j.1474-9726.2007.00348.x [PubMed]
- 30. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2:ra75. https://doi.org/10.1126/scisignal.2000559 [PubMed]
- 31. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392–95. https://doi.org/10.1038/nature08221 [PubMed]
- 32. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176:2092–97. https://doi.org/10.2353/ajpath.2010.091050 [PubMed]
- 33. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10:4230–36. https://doi.org/10.4161/cc.10.24.18486 [PubMed]
- 34. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66:191–201. https://doi.org/10.1093/gerona/glq178 [PubMed]
- 35. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4:715–22. https://doi.org/10.18632/aging.100496 [PubMed]
- 36. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY). 2012; 4:709–14. https://doi.org/10.18632/aging.100498 [PubMed]
- 37. Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012; 4:144ra103. https://doi.org/10.1126/scitranslmed.3003802 [PubMed]
- 38. Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012; 11:675–82. https://doi.org/10.1111/j.1474-9726.2012.00832.x [PubMed]
- 39. Fang Y, Westbrook R, Hill C, Boparai RK, Arum O, Spong A, Wang F, Javors MA, Chen J, Sun LY, Bartke A. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. 2013; 17:456–62. https://doi.org/10.1016/j.cmet.2013.02.008 [PubMed]
- 40. Flynn JM, O’Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, Zykovich A, Mooney SD, Strong R, Rosen CJ, Kapahi P, Nelson MD, Kennedy BK, Melov S. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013; 12:851–62. https://doi.org/10.1111/acel.12109 [PubMed]
- 41. Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science. 2013; 342:1524–28. https://doi.org/10.1126/science.1244360 [PubMed]
- 42. Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, Strong R, Bokov A, Javors MA, Ikeno Y, Hubbard G, Hasty P, Sharp ZD. Rapamycin extends life span of Rb1+/- mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013; 5:100–10. https://doi.org/10.18632/aging.100533 [PubMed]
- 43. Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schröder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, Hans W, Hettich MM, Holtmeier R, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013; 123:3272–91. https://doi.org/10.1172/JCI67674 [PubMed]
- 44. Ye L, Widlund AL, Sims CA, Lamming DW, Guan Y, Davis JG, Sabatini DM, Harrison DE, Vang O, Baur JA. Rapamycin doses sufficient to extend lifespan do not compromise muscle mitochondrial content or endurance. Aging (Albany NY). 2013; 5:539–50. https://doi.org/10.18632/aging.100576 [PubMed]
- 45. Fok WC, Chen Y, Bokov A, Zhang Y, Salmon AB, Diaz V, Javors M, Wood WH 3rd, Zhang Y, Becker KG, Pérez VI, Richardson A. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One. 2014; 9:e83988. https://doi.org/10.1371/journal.pone.0083988 [PubMed]
- 46. Hasty P, Livi CB, Dodds SG, Jones D, Strong R, Javors M, Fischer KE, Sloane L, Murthy K, Hubbard G, Sun L, Hurez V, Curiel TJ, Sharp ZD. eRapa restores a normal life span in a FAP mouse model. Cancer Prev Res (Phila). 2014; 7:169–78. https://doi.org/10.1158/1940-6207.CAPR-13-0299 [PubMed]
- 47. Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP, Kondratov RV. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014; 6:48–57. https://doi.org/10.18632/aging.100633 [PubMed]
- 48. Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Weekly administration of rapamycin improves survival and biomarkers in obese male mice on high-fat diet. Aging Cell. 2014; 13:616–22. https://doi.org/10.1111/acel.12211 [PubMed]
- 49. Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, Javors MA, Li X, Nadon NL, Nelson JF, Pletcher S, Salmon AB, Sharp ZD, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014; 13:468–77. https://doi.org/10.1111/acel.12194 [PubMed]
- 50. Popovich IG, Anisimov VN, Zabezhinski MA, Semenchenko AV, Tyndyk ML, Yurova MN, Blagosklonny MV. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol Ther. 2014; 15:586–92. https://doi.org/10.4161/cbt.28164 [PubMed]
- 51. Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, Diaz V, Sloane L, Maslin K, Treaster S, Réndon S, van Remmen H, Ward W, et al. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci. 2014; 69:119–30. https://doi.org/10.1093/gerona/glt056 [PubMed]
- 52. Hurez V, Dao V, Liu A, Pandeswara S, Gelfond J, Sun L, Bergman M, Orihuela CJ, Galvan V, Padrón Á, Drerup J, Liu Y, Hasty P, et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell. 2015; 14:945–56. https://doi.org/10.1111/acel.12380 [PubMed]
- 53. Johnson SC, Yanos ME, Bitto A, Castanza A, Gagnidze A, Gonzalez B, Gupta K, Hui J, Jarvie C, Johnson BM, Letexier N, McCanta L, Sangesland M, et al. Dose-dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet. 2015; 6:247. https://doi.org/10.3389/fgene.2015.00247 [PubMed]
- 54. Karunadharma PP, Basisty N, Dai DF, Chiao YA, Quarles EK, Hsieh EJ, Crispin D, Bielas JH, Ericson NG, Beyer RP, MacKay VL, MacCoss MJ, Rabinovitch PS. Subacute calorie restriction and rapamycin discordantly alter mouse liver proteome homeostasis and reverse aging effects. Aging Cell. 2015; 14:547–57. https://doi.org/10.1111/acel.12317 [PubMed]
- 55. Arriola Apelo SI, Pumper CP, Baar EL, Cummings NE, Lamming DW. Intermittent administration of rapamycin extends the life span of female C57BL/6J mice. J Gerontol A Biol Sci Med Sci. 2016; 71:876–81. https://doi.org/10.1093/gerona/glw064 [PubMed]
- 56. Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E, Tung H, Vizzini N, Chen B, Smith K, Meza D, Yajima M, Beyer RP, et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife. 2016; 5:e16351. https://doi.org/10.7554/eLife.16351 [PubMed]
- 57. Liao CY, Anderson SS, Chicoine NH, Mayfield JR, Academia EC, Wilson JA, Pongkietisak C, Thompson MA, Lagmay EP, Miller DM, Hsu YM, McCormick MA, O’Leary MN, Kennedy BK. Rapamycin reverses metabolic deficits in lamin A/C-deficient mice. Cell Rep. 2016; 17:2542–52. https://doi.org/10.1016/j.celrep.2016.10.040 [PubMed]
- 58. Felici R, Buonvicino D, Muzzi M, Cavone L, Guasti D, Lapucci A, Pratesi S, De Cesaris F, Luceri F, Chiarugi A. Post onset, oral rapamycin treatment delays development of mitochondrial encephalopathy only at supramaximal doses. Neuropharmacology. 2017; 117:74–84. https://doi.org/10.1016/j.neuropharm.2017.01.039 [PubMed]
- 59. Siegmund SE, Yang H, Sharma R, Javors M, Skinner O, Mootha V, Hirano M, Schon EA. Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum Mol Genet. 2017; 26:4588–605. https://doi.org/10.1093/hmg/ddx341 [PubMed]
- 60. Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, Ideker T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017; 18:57. https://doi.org/10.1186/s13059-017-1186-2 [PubMed]
- 61. Bielas J, Herbst A, Widjaja K, Hui J, Aiken JM, McKenzie D, Miller RA, Brooks SV, Wanagat J. Long term rapamycin treatment improves mitochondrial DNA quality in aging mice. Exp Gerontol. 2018; 106:125–31. https://doi.org/10.1016/j.exger.2018.02.021 [PubMed]
- 62. Weiss R, Fernandez E, Liu Y, Strong R, Salmon AB. Metformin reduces glucose intolerance caused by rapamycin treatment in genetically heterogeneous female mice. Aging (Albany NY). 2018; 10:386–401. https://doi.org/10.18632/aging.101401 [PubMed]
- 63. Parihar M, Dodds SG, Hubbard G, Javors MA, Strong R, Hasty P, Sharp ZD. Rapamycin extends life span in ApcMin/+ colon cancer FAP model. Clin Colorectal Cancer. 2020; S1533-0028:30123–27. https://doi.org/10.1016/j.clcc.2020.08.006 [PubMed]
- 64. Strong R, Miller RA, Bogue M, Fernandez E, Javors MA, Libert S, Marinez PA, Murphy MP, Musi N, Nelson JF, Petrascheck M, Reifsnyder P, Richardson A, et al. Rapamycin-mediated mouse lifespan extension: late-life dosage regimes with sex-specific effects. Aging Cell. 2020; 19:e13269. https://doi.org/10.1111/acel.13269 [PubMed]
- 65. Christy B, Demaria M, Campisi J, Huang J, Jones D, Dodds SG, Williams C, Hubbard G, Livi CB, Gao X, Weintraub S, Curiel T, Sharp ZD, Hasty P. P53 and rapamycin are additive. Oncotarget. 2015; 6:15802–13. https://doi.org/10.18632/oncotarget.4602 [PubMed]
- 66. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15:713–24. https://doi.org/10.1016/j.cmet.2012.04.007 [PubMed]
- 67. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly drosophila melanogaster. Cell Metab. 2010; 11:35–46. https://doi.org/10.1016/j.cmet.2009.11.010 [PubMed]
- 68. Wang A, Mouser J, Pitt J, Promislow D, Kaeberlein M. Rapamycin enhances survival in a drosophila model of mitochondrial disease. Oncotarget. 2016; 7:80131–39. https://doi.org/10.18632/oncotarget.12560 [PubMed]
- 69. Castillo-Quan JI, Tain LS, Kinghorn KJ, Li L, Grönke S, Hinze Y, Blackwell TK, Bjedov I, Partridge L. A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc Natl Acad Sci USA. 2019; 116:20817–19. https://doi.org/10.1073/pnas.1913212116 [PubMed]
- 70. Tomczyk S, Suknovic N, Schenkelaars Q, Wenger Y, Ekundayo K, Buzgariu W, Bauer C, Fischer K, Austad S, Galliot B. Deficient autophagy in epithelial stem cells drives aging in the freshwater cnidarian Hydra. Development. 2020; 147:dev177840. https://doi.org/10.1242/dev.177840 [PubMed]
- 71. Fang Y, Hill CM, Darcy J, Reyes-Ordoñez A, Arauz E, McFadden S, Zhang C, Osland J, Gao J, Zhang T, Frank SJ, Javors MA, Yuan R, et al. Effects of rapamycin on growth hormone receptor knockout mice. Proc Natl Acad Sci USA. 2018; 115:E1495–503. https://doi.org/10.1073/pnas.1717065115 [PubMed]
- 72. Gramatges MM, Bertuch AA. Short telomeres: from dyskeratosis congenita to sporadic aplastic anemia and Malignancy. Transl Res. 2013; 162:353–63. https://doi.org/10.1016/j.trsl.2013.05.003 [PubMed]
- 73. Raices M, Maruyama H, Dillin A, Karlseder J. Uncoupling of longevity and telomere length in C. Elegans. PLoS Genet. 2005; 1:e30. https://doi.org/10.1371/journal.pgen.0010030 [PubMed]
- 74. Simons MJ. Questioning causal involvement of telomeres in aging. Ageing Res Rev. 2015; 24:191–96. https://doi.org/10.1016/j.arr.2015.08.002 [PubMed]
- 75. Steenstrup T, Kark JD, Verhulst S, Thinggaard M, Hjelmborg JV, Dalgård C, Kyvik KO, Christiansen L, Mangino M, Spector TD, Petersen I, Kimura M, Benetos A, et al. Telomeres and the natural lifespan limit in humans. Aging (Albany NY). 2017; 9:1130–42. https://doi.org/10.18632/aging.101216 [PubMed]
- 76. Herrera E, Samper E, Martín-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999; 18:2950–60. https://doi.org/10.1093/emboj/18.11.2950 [PubMed]
- 77. Bischoff C, Petersen HC, Graakjaer J, Andersen-Ranberg K, Vaupel JW, Bohr VA, Kølvraa S, Christensen K. No association between telomere length and survival among the elderly and oldest old. Epidemiology. 2006; 17:190–94. https://doi.org/10.1097/01.ede.0000199436.55248.10 [PubMed]
- 78. Martin-Ruiz CM, Gussekloo J, van Heemst D, von Zglinicki T, Westendorp RG. Telomere length in white blood cells is not associated with morbidity or mortality in the oldest old: a population-based study. Aging Cell. 2005; 4:287–90. https://doi.org/10.1111/j.1474-9726.2005.00171.x [PubMed]