Discussion
In this study, we examined the levels of Humanin and inflammation proteins in plasma from AMD vs. normal groups and investigated the effects of Humanin G on inflammatory proteins in normal and AMD RPE transmitochondrial cybrid cells. We demonstrated significantly reduced Humanin protein levels in the plasma of AMD patients compared to that in the plasma of age-matched normal subjects that served as controls. In addition, we reported activation of inflammation markers in AMD plasma and AMD RPE cybrid cells compared to their normal/control counterparts. Furthermore, we demonstrated that treatment with exogenous Humanin G can revert the abnormal levels of inflammation proteins found in AMD samples to close to normal ranges. These findings are novel and significant as ocular inflammation contributes to retinal degeneration and is a crucial player in the etiology and pathogenesis of AMD [24].
The strong potential of the most conserved mitochondrial-derived peptide, Humanin and its more potent variant Humanin G in protecting damaged mitochondria and RPE cells is well-established in retinal diseases including AMD [17, 25, 26]. Our previous study with Humanin G showed that mitochondria from AMD patients are severely damaged and highlighted the protective role of Humanin G against mitochondria-mediated and amyloid-β-induced cell death in AMD transmitochondrial ARPE-19 cybrids. One mechanism by which Humanin G protected AMD cybrid cells from death was through stabilization of mitochondria and prevention of mitochondrial death. In that study, real-time PCR with primers spanning 503–2484 bps mtDNA regions showed increased numbers of mtDNA lesions in this region of the AMD mitochondrial genome [16].
In the current study, we found that the plasma levels of endogenous Humanin protein were significantly lower by 36.58 % in AMD patients compared to that in age-matched normal subjects. To our knowledge, this is the first study that has reported notably reduced Humanin protein levels in AMD patients, thereby corroborating the pivotal role of Humanin in maintaining tissue homeostasis and normal functioning in the eye. Our study is consistent with previous studies showing that aging is accompanied by markedly reduced Humanin levels and adequate Humanin levels are proportional to increased lifespan and better health, since reduced Humanin levels are observed in aging-related illnesses such as Alzheimer’s disease and diabetes [27, 28]. Along similar lines, Humanin protein levels are remarkably higher in the plasma and skeletal muscle of humans following high-intensity exercise and resistance training, indicating a role of Humanin in regulating glucose metabolism as well [29, 30]. With age, the amount of baseline Humanin decreases in the hypothalamus, cortex, and skeletal muscle [31]. Moreover, treatment with exogenous Humanin G is known to reduce the expression of markers associated with aging-related disorders [32]. Therefore, we tested the effects of Humanin G on inflammatory markers in this study.
During the course of inflammation, endothelial cell activation leads to the expression of cell adhesion molecules, which mediate the trafficking of inflammatory and immune cells toward the sites of inflammation and promote their interaction with the activated endothelium [33]. Selectins/CD62 are cell adhesion molecules that are cell surface C-type lectins and soluble transmembrane glycoproteins involved in acute and chronic inflammation processes and facilitate the adhesive process in inflammation by enabling leukocyte rolling on vascular surfaces [34–37]. E-Selectin i.e., CD62E/ ELAM-1 (Endothelial-Leukocyte Adhesion Molecule 1), is a 115 kDa endothelial cell surface specific adhesion molecule that is expressed by the SELE (Selectin E) gene in humans. It is composed of an N-terminal C-type lectin domain, an EGF (Epidermal-Growth-Factor)-like domain, a conserved protein domain of six short consensus repeats of cysteine residues, a transmembrane domain, and a cytoplasmic tail. The expression of E-Selectin on endothelial cells is stimulated by proinflammatory cytokines, and results in adhesion of endothelial cells to the vascular lining and accumulation of blood leukocytes at the sites of inflammation [38]. In our study, we found that compared to their normal counterparts, AMD plasma and AMD RPE cells expressed significantly elevated protein levels of E-Selectin (CD62E) by 77.1 % and 158.5 % respectively, indicating the involvement of E-Selectin in AMD-related inflammation. Next, we demonstrated notably higher P-Selectin (CD62P) protein levels in the AMD plasma and AMD RPE cybrid cells by 75 % and 198.9 % respectively, indicating the significant contribution of P-Selectin in ocular inflammation. Consistent with our results, a previous study reported elevated levels of P-Selectin are found in non-arteritic anterior ischemic optic neuropathy, indicating that increased P-Selectin is a pathological marker associated with this ophthalmologic disease [39]. P-Selectin was also elevated in diseases with an inflammatory component such as rheumatoid arthritis [40]. P-Selectin i.e., CD62P is encoded by the SELP gene and is localized to the α-granules of platelets and Weibel-Palade bodies of endothelial cells. In response to stimulation by thrombin or other agonists such as histamine or collagen, P-Selectin is translocated to the surface of activated platelets (thrombocytes) and activated endothelial cells, where it plays a critical role in the recruitment of leukocytes to the site of injury during inflammation. P-Selectin is composed of an N-terminal C-type lectin domain, an EGF (Epidermal-Growth-Factor)-like domain, a conserved complement regulatory domain consisting of nine short consensus repeats of cysteine residues, a transmembrane domain, and a cytoplasmic tail region [41–44]. A recent study that examined the expression of Selectins in the retina and choroid of AMD patients and compared the allele and genotype frequencies between AMD patients and controls, demonstrated that a single SNP (Single Nucleotide Polymorphism) located within an intron of SELP gene (rs3917751) is statistically associated with dry AMD in their study cohort. This was the only ancestral risk allele for dry AMD that was found in the P-Selectin gene [45]. Our current study revealed that treatment with HNG decreased the protein levels of both E-Selectin and P-Selectin by 64.62 % and 60.99 % in AMD RPE cybrid cells compared to normal, thereby indicating that Humanin G suppresses inflammation by downregulating E- and P-Selectins. This is an important finding since inhibition of E-Selectin using microRNAs suppresses endothelial cell inflammation [46].
Apart from Selectins, we also examined another cell adhesion molecule – ICAM-1 (Intercellular adhesion molecule-1). ICAM-1 i.e., CD54 (Cluster of Differentiation 54) encoded by the ICAM-1 gene is a transmembrane glycoprotein that is typically expressed on endothelial cells and facilitates the binding of leukocytes to vascular endothelium [47]. In the current study, we found significantly increased protein levels of ICAM-1 in AMD plasma and AMD RPE cybrid cells by 83.6 % and 47.5 % respectively, and treatment with HNG reduced the ICAM-1 protein by 30.82 % in AMD RPE cybrid cells. Similar to our study, a previous study demonstrated that subfoveal choroidal neovascular membranes (CNVMs) surgically excised from AMD patients have higher protein levels of ICAM-1 compared with those in the normal eye [48]. Increased ICAM-1 protein levels were also found in the choroid and sclera of hypercholesterolemic New Zealand rabbits [49]. Our results are consistent with previous studies showing enhanced ICAM-1 protein levels in the aqueous humor samples of wet AMD patients and notable upregulation of retinal ICAM-1 in a VEGF (Vascular Endothelial Growth Factor)-induced retinal leukostasis rat model. Inhibition of ICAM-1 bioactivity significantly reduced retinal leukostasis and vascular permeability, thereby indicating that ICAM-1 plays a key role in inflammation and VEGF-induced retinal leukostasis [50, 51]. Retinal leukostasis, a manifestation of retinal inflammation, is induced by pro-inflammatory cytokines and VEGF, and is characterized by an increase in the number of static leukocytes in the retina, enhanced adhesion of leukocytes and monocytes to retinal endothelial cells, reduced retinal blood flow, luminal narrowing of retinal capillaries, and decreased perfusion pressure [52–54]. Humanin G’s ability to reduce ICAM-1 protein is remarkable because targeted homozygous knockout of ICAM-1 gene in a laser injury-induced choroidal neovascularization (CNV) mouse model notably inhibited CNV as evidenced by substantially diminished volume of CNV lesions and decreased fluorescein leakage in ICAM-1 deficient mice compared to wild-type mice after laser photocoagulation injury [55]. In addition to the retina, ICAM-1 is involved in corneal inflammatory processes as well. ICAM-1-deficient mice show significant reduced neovascularization compared to controls, thereby indicating that ICAM-1 is a mediator of inflammation-associated and VEGF-induced corneal neovascularization [56]. Our results underscore the importance of cell adhesion molecules in AMD pathology and highlight the vital role of HNG in alleviating retinal inflammation in AMD.
Next, we tested the protein levels of cytokines and chemokines and investigated the effects of Humanin G treatment on the expression of the pro-inflammatory markers. TNF-α (Tumor Necrosis Factor alpha) is a key player in the pathogenesis of AMD as reduced TNF-α levels in the serum are associated with higher visual acuity score in AMD patients [57]. The transcription of TNF-α is genetically regulated and in the promoter region of the TNF-α gene, three SNPs were detected: TNF-α-863 (rs1800630), TNF-α-308 (rs1800629) and TNF-α-238 (rs361525). Promoter polymorphisms at -238, -308, and -863 SNP positions could potentially regulate TNF-α production, and these polymorphisms may have implications for AMD pathogenesis due to an imbalance in inflammatory processes caused by dysregulation of TNF-α production [58–60]. TNF-863CC/TNF-308GA and TNF-308GA/TNF-238GG SNP genotypes combinations are associated with increased risk of AMD [61]. Only -308 G/A TNF-α gene polymorphism is associated with AMD. The TNF-α -1031 T/C polymorphism is significantly associated with wet AMD in the Taiwan Chinese population [62]. We report a 98.4 % increase in TNF-α protein levels in the plasma of AMD patients and a 111.3 % elevated TNF-α protein in the AMD RPE cybrid cells compared to their normal counterparts. This suggests the pivotal role of TNF-α in retinal inflammation in AMD. Consistent with our results, TNF-α protein levels were found to be significantly higher in the vitreous of AMD patients compared to that in normal individuals [63]. Significantly increased serum levels of TNF-α protein and associated retinal ischemia were found in Eales’ disease which is an idiopathic inflammatory retinal vasculopathy [64]. Moreover, TNF-α protein levels were markedly increased in the retina, RPE and choroid of Cxcr5 receptor-deficient mice [65]. Chronic exposure to TNF-α alters RPE morphology by interfering with RPE cell differentiation, development of transepithelial potential, and RPE tight junction formation, thereby resulting in RPE cells that resemble aged and diseased RPE cells found in AMD [66]. Higher levels of soluble TNF-receptor II protein have been found in the plasma of both early dry AMD and wet AMD patients, indicating systemic inflammation [67]. In our study, we discovered that addition of exogenous Humanin G to AMD RPE cybrid cells reduces the protein levels of TNF-α by 46.09 % compared to their untreated counterparts, thereby indicating that Humanin G could be used an effective inhibitor of TNF-α-induced ocular inflammation and might therefore alleviate AMD pathology. Although the effects of Humanin G on TNF-α in AMD have not been demonstrated before, along similar lines, it has been shown that administration of TNF-α inhibitors such as Adalimumab, a subcutaneous anti-TNF-α drug, in combination with anti-VEGF therapy improves visual acuity [68]. Intravitreal and intraperitoneal injections of Adalimumab prevent retinal degeneration and photoreceptor cell death by preventing TNF-α upregulation and reducing inflammation, oxidative stress, and apoptosis [69, 70].
Retinal inflammation, a major contributory factor in AMD pathogenesis, is characterized by chemokine-mediated infiltration of macrophages and microglia from the inner retina into the subretinal space. We found that the Monocyte Chemoattractant Protein-1 (MCP-1)/ CCL2 protein levels were significantly upregulated by 113.5 % in the plasma of AMD patients compared to normal plasma. This finding is important as MCP-1 is known to be secreted by RPE cells in response to oxidative stress damage and facilitates the migration and infiltration of macrophages and monocytes [71]. We next investigated the role of MIP-1α (Macrophage Inflammatory Protein-1α) i.e., CCL3 (C-C Motif Chemokine Ligand 3), a CC chemokine that is secreted by macrophages and binds to CCR1 and CCR5 chemokine receptors. MIP-1α mediates the recruitment of leukocytes and inflammatory cells to the site of inflammation in response to tissue injury. MIP-1α mediates wound healing and is involved in both cell-mediated immunity and systemic and mucosal humoral immune responses [72, 73]. We found a significant increase in MIP-1α protein levels by 185.2 % and 212.2 % in the AMD plasma as well as in the AMD RPE cybrid cells respectively, compared to that in the normal counterparts. Our results are consistent with previous studies that showed MIP-1α gene expression is notably upregulated and its function is nonredundant in retinal degeneration [74]. MIP-1α protein levels were remarkably elevated in aqueous humor samples obtained from the eyes of patients with exudative AMD and polypoidal choroidal vasculopathy, compared to that in control eyes. In addition, MIP-1α gene expression and protein levels were significantly upregulated due to post-ischemic pro-inflammatory response, in a mouse model of ischemic retinopathy [75, 76]. Humanin G-treated AMD RPE cybrid cells showed 61.98 % decreased MIP-1α protein levels compared with the untreated AMD RPE cybrid cells. Humanin G-induced suppression of MIP-1α might prevent AMD-associated ocular inflammation since previous studies have shown that inhibition of MIP-1α activity using neutralizing antibodies caused partial suppression of inflammation-induced retinal neovascularization, thereby indicating the critical contribution of MIP-1α in inflammation-induced retinal neovascularization [77]. MIP-1α plays a vital nonredundant role in chronic and acute inflammation-mediated retinal degeneration, and MIP-1α-deficient mice show reduced damage to the blood-retinal barrier compared with controls [65].
We demonstrated that AMD plasma and AMD RPE cybrid cells had 186.1 % and 137.3 % higher protein levels of IFN-γ (Interferon-gamma), which is secreted by the T Helper Type 1 (Th1) cells and its activation is a hallmark of adaptive and innate immune responses [78, 79], IFN-γ causes RPE cell death by increasing intracellular iron concentration, oxidative stress, lipid peroxidation, glutathione depletion, and activation of the JAK-STAT signaling pathway [80]. IFN-γ along with other cytokines induces the secretion of IL-6 from RPE cells [81]. In our study, addition of Humanin G reduced the IFN-γ protein by 62.86 % in AMD RPE cybrid cells compared to untreated AMD RPE cybrid cells. This highlights the potential of Humanin G to alleviate IFN-γ-mediated inflammatory responses in AMD, and it is important since IFN-γ plays a crucial role in the pathogenesis of AMD by: a) inducing the expression of VEGF in human RPE cells via the Phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling pathway; b) enhancing mitochondria-generated ROS in human RPE cells; c) regulating the activation of the complement cascade; d) reducing complement inhibition; e) macrophage polarization; and f) diminishing the deposition of amyloid-β plaques in neuroinflammation [82–87]. Furthermore, Interferon gamma-inducible protein-10 (IP-10) i.e., C-X-C motif chemokine ligand 10 (CXCL10) is an α-chemokine that plays a key role in T cell adhesion to endothelial cells and as a chemoattractant to lymphocytes and monocytes [88]. We observed 193.5 % higher IP-10 protein levels in AMD plasma vs. normal plasma samples. This is consistent with recent studies which reported markedly elevated IP-10 levels in the postmortem eyes from dry AMD, geographic atrophy i.e., advanced AMD, and neovascular AMD patients [89]. Moreover, in our study, we found notably enhanced Interferon-alpha (IFN-α) protein levels by 133.3 % in AMD plasma samples compared to normal plasma. This finding suggests the involvement of IFN-α in AMD pathology since IFN-α causes retinopathy by inducing retinal vein occlusion, retinal ischemia, leukocyte infiltration, capillary non-perfusion, cotton wool spot formation, and accumulation of immune cells in the retinal vasculature [90, 91].
The IL-1β cytokine binds to the IL-1RI receptor and is an established biomarker for retinal diseases. It mediates the degeneration of rods by impairment of retinal glutamate homeostasis and severe loss of cone segments causing subsequent loss of visual acuity in AMD [92, 93]. IL-1β protein was notably increased by 330.6 % in the plasma and by 224.5 % in the cell lysates of AMD samples vs. controls, showing similar degree of involvement of IL-1β in both the AMD patients as well as AMD RPE cybrid cells. Our results are consistent with a previous study that demonstrated significant increase in the IL-1β protein levels in the serum of AMD patients [94]. Previous studies reported that both the ~31 kDa inactive pro-form and the cleaved ~17 kDa active form of IL-1β protein were significantly elevated in the vitreous samples obtained from patients with neovascular AMD and polypoidal choroidal vasculopathy, compared to controls [95–98]. Furthermore, IL-1β gene was markedly upregulated in the retinal vessels of diabetic retinopathy rats, and IL-1β protein levels were also elevated in the serum, vitreous fluid, and aqueous humor samples of diabetic retinopathy patients. These studies demonstrated a correlation between IL-1β and cell death in diabetic retinopathy [99–106]. Higher IL-1β protein levels are also associated with photoreceptor cell death and diminished visual fields in retinitis pigmentosa [107, 108]. Remarkably increased gene and protein levels of IL-1β were found in the blood and aqueous humor of glaucoma patients, establishing IL-1β as a risk factor in glaucoma pathogenesis [109]. In the retina and vitreous of patients with retinal detachments, higher IL-1β levels were reported [110, 111]. Macular edema also led to an increase in the IL-1β protein levels in the aqueous humor and vitreous samples [112, 113]. Elevated IL-1β levels were demonstrated in the aqueous humor and supernatants of posterior eye cups in an in vivo experimental autoimmune uveoretinitis (EAU) model, and administration of anti-IL-1β antibody remarkably decreased the EAU scores [114, 115]. Intravitreal injections of amyloid-beta, a component of the drusen in dry AMD, caused a remarkable increase in IL-1β protein in the vitreous of the treated rats compared with controls [116]. Moreover, in the in vivo, ex vivo, and in vitro models of retinopathy of prematurity, IL-1β was notably increased in the RPE/ choroid and caused choroidal involution, loss of RPE and photoreceptors, retinal degeneration, and visual deterioration. In addition, we found that exogenous Humanin G caused significant decrease in the IL-1β protein levels by 68.31 % in AMD RPE cybrid cells vs. untreated controls. Our results are novel as no previous study has demonstrated the Humanin G-mediated suppression of IL-1β protein in transmitochondrial AMD RPE cybrid cells. This finding is significant because inhibition of IL-1β using IL-1R receptor antagonist and recombinant IL-1Ra alleviates the damaging effects of IL-1β, markedly decreases subretinal neovascularization after laser injury, and substantially reduces photoreceptor cell apoptosis in AMD [117–120]. In addition, the expression of IL-1β gene and protein is upregulated several fold in rodent retinas following photo-oxidative damage and inhibition of IL-1β using siRNAs or neutralizing antibodies suppressed chemokine-induced inflammation and retinal degeneration in AMD [121, 122].
IL-13 is a 13 kDa cytokine secreted by T helper type 2 (Th2) cells, CD4 cells, natural killer T cell, basophils, and eosinophils among other cell types, and regulates allergic inflammation and IgE synthesis. In the current study, plasma from AMD patients had 336.5 % higher IL-13 protein levels whereas AMD RPE cybrid cells showed 177.6 % increase in IL-13 protein. Along similar lines, IL-13 protein levels were remarkably higher in the aqueous humor of AMD patients vs. controls, and IL-13 inhibited RPE cell proliferation [123]. Moreover, treatment of AMD RPE cybrid cells with Humanin G reduced IL-13 protein levels by 57.63 % compared to their untreated counterparts. This suggests Humanin G’s potential in suppressing IL-13-induced ocular inflammatory responses and is significant since inhibition of IL-13 reduces intraocular inflammation in acute-inflammation-induced retinal pathology [124].
IL-17 is a key cytokine produced by the T helper 17 (Th17) cells, which are derived from CD4+ cells. IL-17 is also secreted by CD8+ T cells, NK cells, and other immune cells and is involved in the neovascularization and pathogenesis of ocular diseases such as AMD, diabetic retinopathy, retinal vein occlusion, and retinopathy of prematurity. IL-17 causes ocular neovascularization by cytoskeleton remodeling, regulation of VEGF, and activation of complement components [125]. IL-17 mediates VEGF-induced angiogenesis by promoting the mitogenic activity of VEGF and VEGF-induced growth of vascular endothelial cells and triggers the secretion of IL-1β from RPE cells via activation of NLRP3 inflammasome [126, 127]. Similar to previous studies that have demonstrated notably elevated IL-17 protein levels in the serum of AMD patients and in RPE cells which constitutively expressed IL-17 receptors i.e., IL17 RA, IL-17RC, and ACT1 [128], we found that IL-17 protein levels were increased by 145.7 % in AMD plasma and were 101.4 % higher in AMD RPE cybrid cells compared to controls. Our findings corroborate the important role of IL-17 in AMD-associated inflammation and pathology since the expression of IL-17A and its receptor IL-17RC is markedly upregulated in the macular lesions of advanced-stage AMD donors compared to normal tissue [129]. IL-17A activates Caspase-3 and Caspase-9 pro-apoptotic proteins, thereby causing cytotoxicity in RPE cells. The expression of IL-17 and its receptor IL-17RC is upregulated in AMD eyes compared to controls [130]. IL-17A mediates blood-retinal barrier damage by activating the JAK1 signaling cascade, thereby leading to the disruption of tight junctions in RPE cells and endothelial cells [131]. IL-17 promotes the proliferation, migration, and tube formation in human choroidal endothelial cells by activating CCL2 and CXCL8 in RPE cells, thereby contributing to choroidal endothelial cell angiogenesis [132]. Next, we investigated the effects of Humanin G and discovered that treatment with Humanin G reduced IL-17A protein levels by 48.31 % in AMD RPE cybrid cells, thereby highlighting the ability of Humanin G to suppress IL-17-mediated retinal inflammation. This finding is important since it has been shown that knockdown of IL-17RC using siRNA reduces IL-17-mediated pathology in RPE cells [124].
IL-4 is a cytokine that mediates the differentiation and activation of T helper 2 cells and is a key regulator of humoral immune responses [133]. Polymorphisms of IL-4 -590 T/T or T/C genotypes is a potential genetic marker for the development of AMD pathology, and IL-4 promotes pathological angiogenesis and choroidal neovascularization, thereby leading to retinal degeneration [134, 135]. The 118.5 % upregulation of IL-4 protein in AMD plasma vs. normal plasma highlights its vital role in AMD-related inflammation.
IL-10 regulates macrophage functioning in the eye and promotes choroidal neovascularization in AMD. IL-10 deficiency alleviates choroidal neovascularization-induced damage and favors retinal health. [136] IL-10 protein was higher by 189.7 % in AMD plasma vs. normal plasma, consistent with previous studies and underlining its involvement in ocular inflammatory response in AMD.
IL-12p70 is primarily secreted by macrophages and dendritic cells and is a heterodimer comprised of p35 and p40 subunits. IL-12p70 which mediates the expression of IFN-γ and contributes to innate and adaptive immune responses, was found to have 107.7 % elevated protein levels in AMD plasma compared to normal plasma in our study. This underlines the crucial role of IL-12p70 in AMD pathology as it has been previously confirmed that IL-12p70 is upregulated in response to inflammation and oxidative stress in ARPE-19 cells [137].
To our knowledge, this is the first report that confirms the protective role of Humanin G against inflammation in AMD RPE transmitochondrial cybrid cells and it is significant because reducing ocular inflammation could alleviate its damaging effects observed in the RPE cells that eventually lead to retinal degeneration in AMD pathogenesis.
In conclusion, our study reports novel findings that: A) demonstrate a decline in endogenous Humanin protein levels in plasma from AMD patients compared to normal subjects; B) underscore the role of cell adhesion molecules, cytokines, and chemokines in AMD-associated inflammation (Figure 6) and subsequent cellular damage in AMD RPE cybrid cells that has been observed in our previous studies; and C) establish the positive effects of Humanin G in reducing the expression of inflammatory markers in AMD RPE transmitochondrial cybrid cells and therefore demonstrate the potential of Humanin G in reducing AMD-associated inflammation in vitro (Figure 7). Since we found elevated inflammatory proteins in the AMD plasma, which suggests that inflammation might be more systemic and not just confined to the retina, we speculate that a baseline systemic inflammation may make the retinal cells more susceptible to damage, thereby leading to AMD pathology.
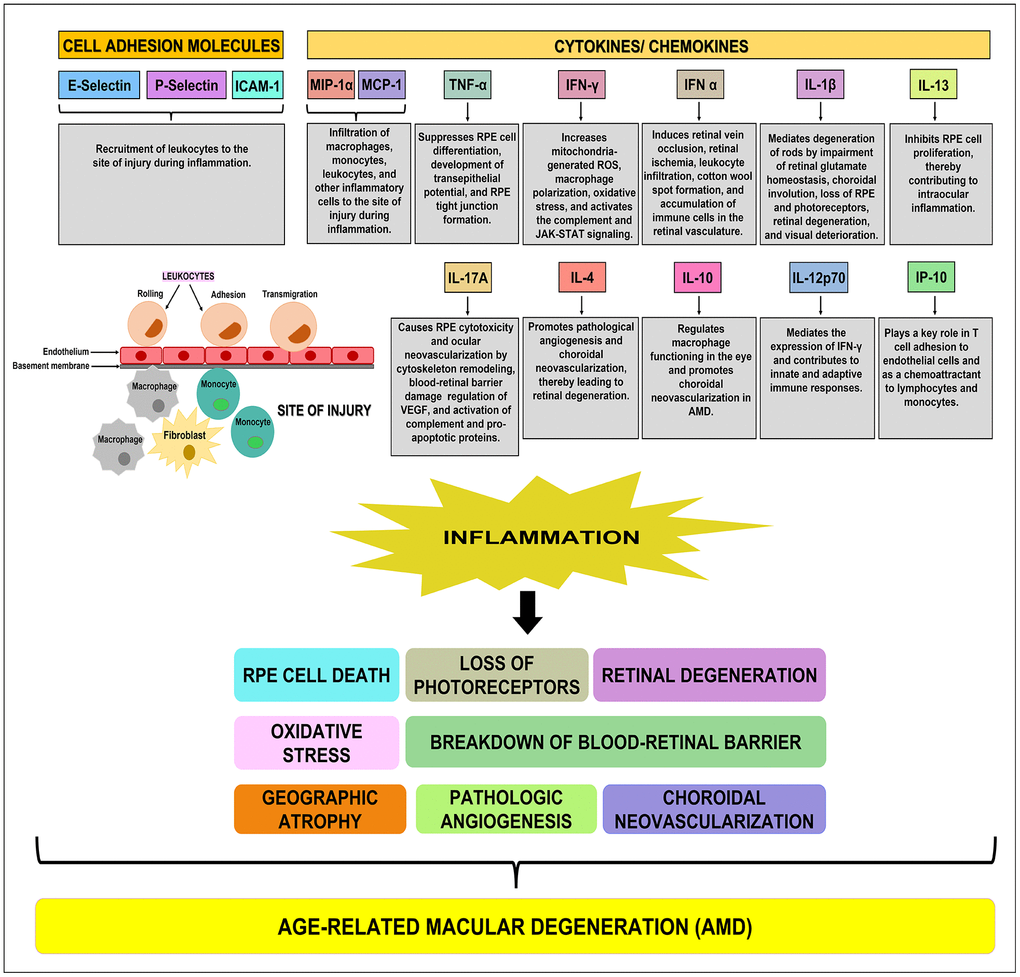
Figure 6. Schematic showing the function(s) of cell adhesion molecules, cytokines, and chemokines. E-Selectin, P-Selectin, and ICAM-1 are cell adhesion molecules that are involved in the recruitment of leukocytes to the site of injury during inflammation. MIP-1α, MCP-1, TNF-α, IFN-γ, IFN-α, IL-1β, IL-13, IL-17A, IL-4, IL-10, IL-12p70, and IP-10 are pro-inflammatory cytokines/ chemokines that promote RPE cell death, loss of photoreceptors, oxidative stress, retinal degeneration, breakdown of blood-retinal barrier, geographic atrophy, pathologic angiogenesis, and choroidal neovascularization, subsequently leading to the development of AMD.
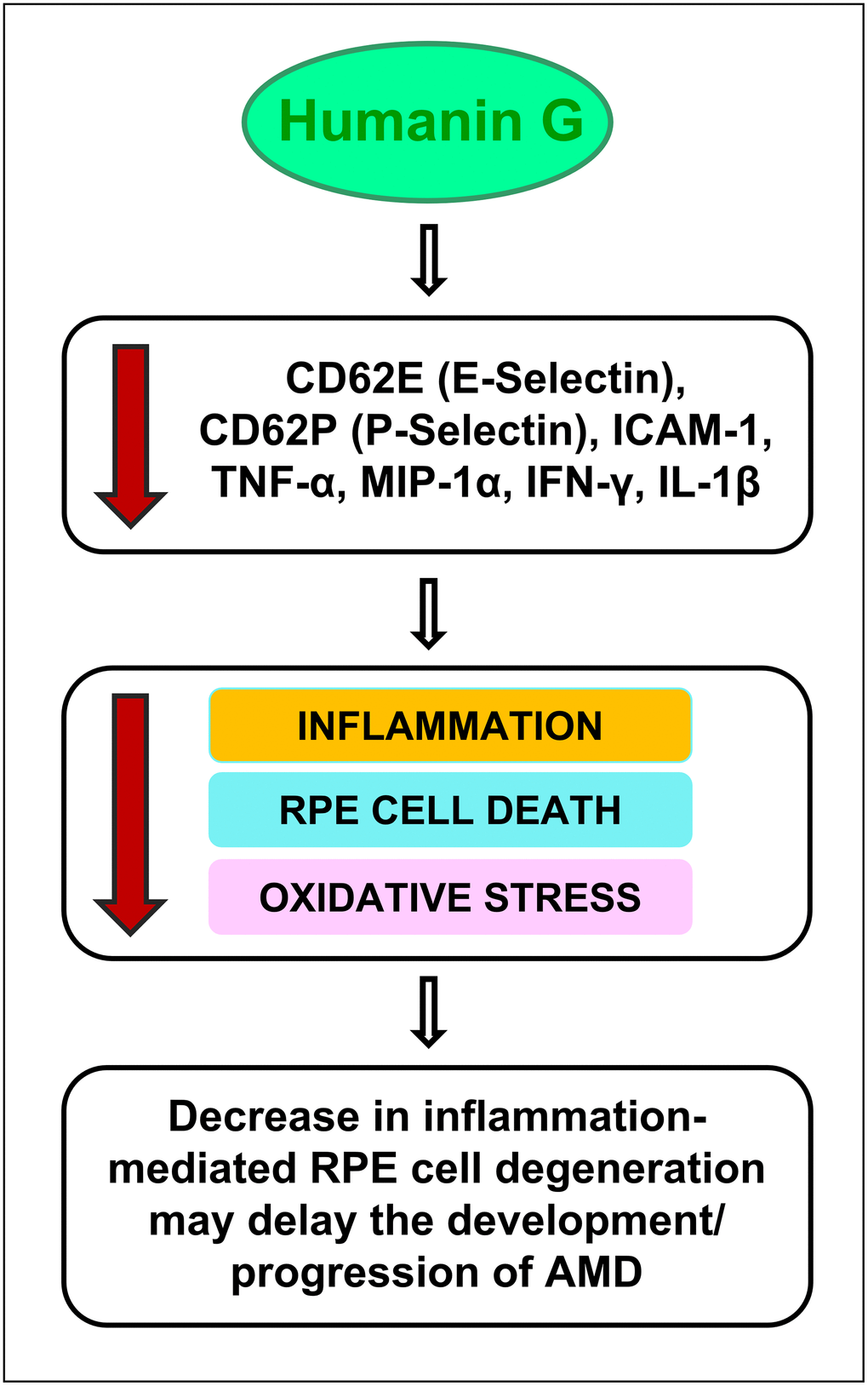
Figure 7. Schematic showing the potential action of Humanin G. Treatment with Humanin G reduces the levels of inflammation-associated proteins namely CD62E (E-Selectin), CD62P (P-Selectin), ICAM-1, TNF-α, MIP-1α, IFN-γ, and IL-1β. This is turn might decrease retinal inflammation, reduce RPE cell death and oxidative stress, thereby preventing retinal degeneration. This may delay the development/ progression of AMD.
It is important to note that in cells that are stressed/ damaged, Humanin G acts to suppress the inflammatory proteins that may contribute to the development of AMD. It is noteworthy that in the cells that are healthy and have a normal homeostasis, Humanin G does not exert any negative impact. This is significant because to be used successfully to treat diseases, you want the drug to be targeting the diseased cells but have no negative impact on the healthy cells.
Further studies are required to gain an in-depth understanding of the mechanisms underlying Humanin G-mediated suppression of inflammation in AMD and to establish Humanin G’s therapeutic potential as an inhibitor of AMD-associated inflammation. Furthermore, in addition to administration of Humanin G, knockdown or knock-out of the studied inflammatory markers using siRNA or CRISPR editing, may present a new line of treatment for AMD.
S.N.: Designed the study and performed the experiments; acquired, analyzed, and interpreted data; wrote and edited the manuscript. J.W.: Humanin ELISA experiment; M.C.K. and P.C.: Provided resources; M.C.K.: Corresponding author.
The authors declare that they have no conflicts of interest.
This research was funded by Arnold and Mabel Beckman foundation, Discovery Eye Foundation, Polly and Michael Smith, Edith and Roy Carver, Iris and B. Gerald Cantor Foundation, Unrestricted Departmental Grant from Research to Prevent Blindness and NEI R01 EY027363, UCI School of Medicine, and support of the Institute for Clinical and Translational Science (ICTS) at University of California Irvine. S.N. is a recipient of the 2017 Genentech/ ARVO AMD Translational Research Fellowship and the 2016 RPB pilot research grant.