




Introduction
Gastric cancer (GC) is the sixth most common cancer and the second most common cause of cancer-related death worldwide [1]. Radical gastrectomy with lymph node dissection remains the mainstay of GC treatment with curative intent. However, some GC patients were diagnosed at a late stage even stage IV disease, and most of them died within one year. The metastatic patterns of GC include peritoneal, hematogenous and distant lymphatic metastases. However, small metastatic peritoneal nodules without ascites formation, tiny liver metastases and borderline-sized paraaortic lymph nodes may be regarded as the absence of distant metastasis by abdominal computed tomography scan. In addition, tumor markers, carcinoembryonic antigen (CEA) and carbohydrate antigen 19-9 (CA 19-9), are even within the normal range in some stage IV GC patients.
Circulating cell-free DNA (cfDNA) can serve as a liquid biopsy and noninvasive method for cancer monitoring and management [2–4]. cfDNA levels in GC patients are more sensitive than CEA levels in the prediction of tumor recurrence [5, 6], and the cfDNA levels are significantly higher in stage IV GC [5].
Next-generation sequencing (NGS) has been used to investigate the concordance of genetic mutations between tumor tissue DNA and cfDNA [7]. Although high concordance of genetic mutation patterns was reported between tumor tissue DNA and cfDNA, some mutant variants were found in tumor tissue DNA only, and some were found in cfDNA only. The discrepancy in genetic mutations between primary tumor DNA and cfDNA was considered to be due to tumor heterogeneity. For stage I–III esophagus cancer and GC, a recent NGS study [8] with enrollment of 295 patients demonstrated that the cfDNA was detected in 96% preoperatively and 23.5% within 16 weeks after surgery. Their results showed that cfDNA detected at any time point after surgery was associated with shorter recurrence-free survival and poor prognosis. In addition, genetic methylation in cfDNA was reported to be a biomarker for predicting lymphatic metastasis, distant metastasis, peritoneal metastasis, and patient survival in GC [9–13]. However, the correlation between genetic mutations and metastatic patterns in tumor DNA and cfDNA in stage IV GC is still unknown.
In the current study, we used NGS and evaluated the concordance of genetic mutations between tumor DNA and cfDNA in stage IV GC patients. The relationship between the genetic mutations and metastatic patterns was analyzed.
Results
Clinicopathological characteristics
The clinicopathological features of the 56 stage IV GC patients are shown in Table 1. There were 29 males and 27 females. The mean age was 65.8 years old. The tumor was located mostly in the middle third of the stomach. Approximately 69.6% of the 56 patients had poorly differentiated tumors and 92.9% had lymphovascular invasion. The most common metastatic site was the peritoneum, followed by distant lymphatic and hematogenous metastases. Among the 56 stage IV GC patients, five patients had metastases in both the peritoneum and distant lymph nodes; one patient had metastases in both the peritoneum and hematogenous metastases.
Table 1. Clinicopathological features of 56 stage IV gastric cancer (GC) patients.
| Clinicopathological features | Stage IV GC (n = 56) (%) |
| Age | 65.8 ± 13.9 |
| Gender (M/F) | 29/27 |
| Tumor size (cm) | 7.8 ± 2.9 |
| Tumor location | |
| Upper third | 6 (10.7) |
| Middle third | 23 (41.1) |
| Lower third | 21 (37.5) |
| Whole stomach | 6 (10.7) |
| Cell differentiation | |
| Well/moderate | 17 (30.4) |
| Poor | 39 (69.6) |
| Lymphovascular invasion | 52 (92.9) |
| Lauren’s classification | |
| Intestinal type | 19 (33.9) |
| Diffuse type | 37 (66.1) |
| Metastatic site | |
| Hematogenous | 3 (5.4) |
| Peritoneum | 33 (58.9) |
| Distant lymph node | 26 (46.4) |
Analysis of the mutated genes in GC tumor samples and cfDNA
Among the 56 stage IV GC patients, genetic mutations were observed in both tumor DNA and cfDNA in 54 (96.4%) patients. Two patients had no genetic mutations detected in either tumor DNA or cfDNA.
We analyzed the genetic mutation according to the metastatic sites. As shown in Figure 1A, for patients with peritoneal metastasis, the most commonly mutated gene was KMT2D (45%), followed by TP53 (42%), KMT2C (42%), ARID1A (39%), MACF1 (27%), LRP1B (24%), FAT4 (21%), CDH1 (21%), APC (18%), and ABCA10 (18%). In Figure 1B, for patients with hematogenous metastasis, the most commonly mutated genes were LRP1B (67%) and KMT2C (67%), followed by TP53 (33%), PREX2 (33%), MACF1 (33%), KRAS (33%), KMT2D (33%), FAT4 (33%), CTNNB1 (33%), CREBBP (33%), CDH1 (33%), BNC2 (33%), BCOR (33%), and ARID1A (33%). In Figure 1C, for patients with distant lymphatic metastasis, the most commonly mutated gene was TP53 (46%), followed by FAT4 (38%), ARID1A (38%), KMT2C (35%), PLB1 (31%), MACF1 (23%), LRP1B (23%), MUC6 (23%), CREBBP (15%), CDH1 (15%), BCOR (15%), ERBB3 (15%), and BNC2 (15%). The most common mutated spot in both peritoneal and distant lymphatic metastases was KMT2C c.8390delA.
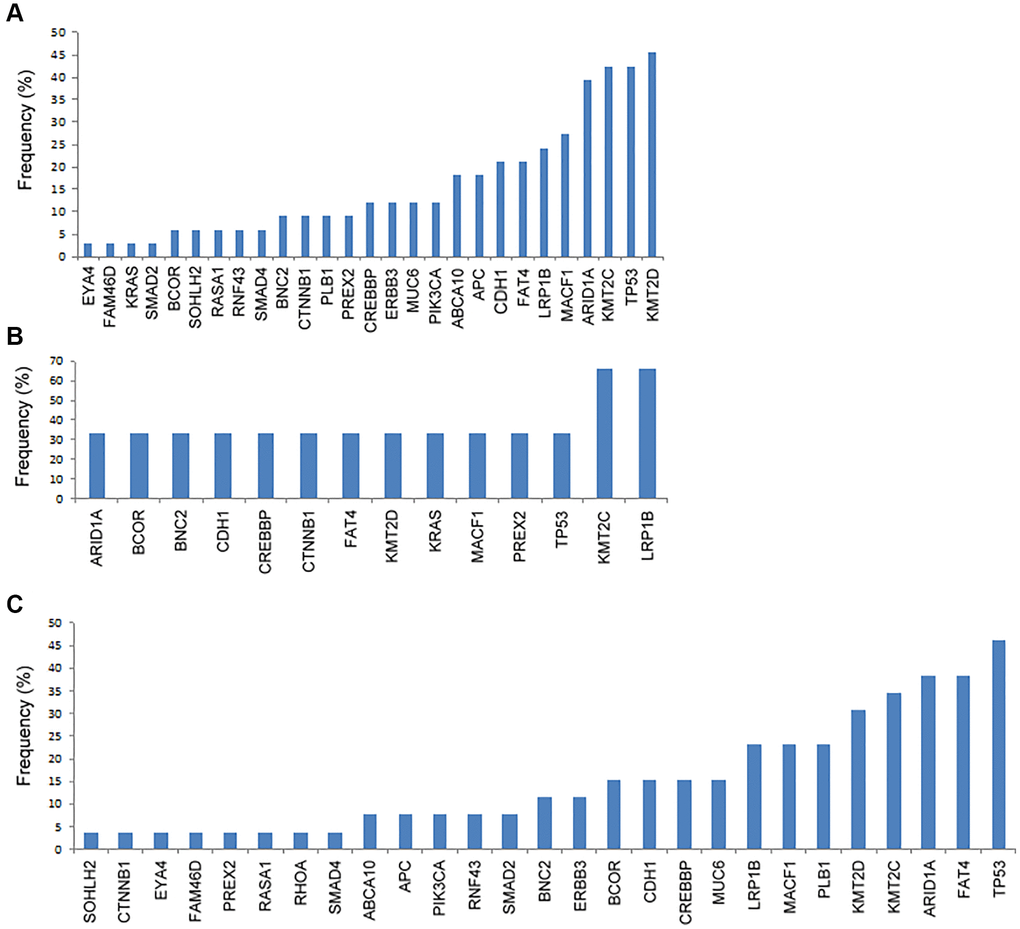
Figure 1. The frequency of genetic mutations according to the metastatic patterns. (A) Peritoneal metastasis, (B) hematogenous metastasis, and (C) distant lymphatic metastasis.
We further analyzed the immunohistochemical (IHC) stain for some common mutated genes in the tumor samples, including CDH1, MACF1, TP53, PLB1, ARID1A, KMT2C, FAT4, and KMT2D. As shown in Figure 2 and Table 2, tumors with CDH1 mutations were associated with significantly more negative expression of the correlated E-cadherin protein (P = 0.021), which was also observed in tumors with mutations in MACF1 (P < 0.001), TP53 (P < 0.001), PLB1 (P < 0.001), ARID1A (P < 0.001), KMT2C (P < 0.001), FAT4 (P < 0.001), and KMT2D (P < 0.001).
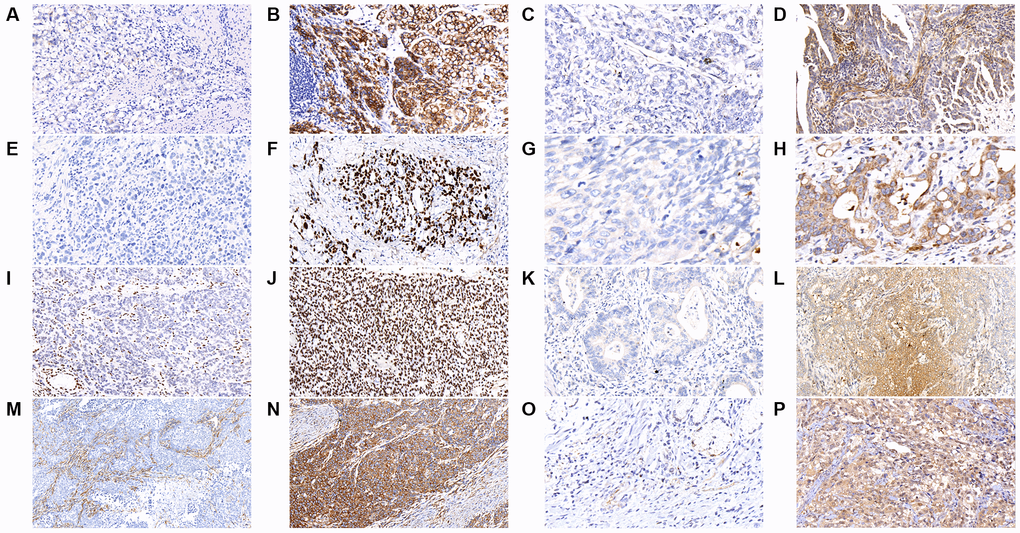
Figure 2. The results of immunohistochemical staining in gastric cancer tumor samples are shown as follows in the figures. (A) E-cadherin-negative (B) E-cadherin-positive (C) MACF1-negative (D) MACF1-positive (E) p53-negative (F) p53-positive (G) PLB1-negative (H) PLB1-positive (I) ARID1A-negative (J) ARID1A-positive (K) KMT2C-negative (L) KMT2C-positive (M) FAT4-negative (N) FAT4-positive (O) KMT2D-negative (P) KMT2D-positive. (x400).
Table 2. The correlation between the expression of IHC stain and genetic mutation.
| IHC stain | Genetic mutation | P value | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No n (%) | Yes n (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| E-cadherin (CDH1) | 0.021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 0 | 1 (11.1) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 47 (100) | 8 (88.9) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MACF1 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 0 | 7 (58.3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 44 (100) | 5 (41.7) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P53 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 3 (8.8) | 14 (63.6) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 31 (91.2) | 8 (36.4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PLB1 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 1 (2.1) | 8 (100) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 47 (97.9) | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ARID1A | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 1 (2.8) | 12 (60.0) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 35 (97.2) | 8 (40.0) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KMT2C | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 1 (2.4) | 11 (73.3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 40 (97.6) | 4 (26.7) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FAT4 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 2 (4.7) | 6 (46.2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 41 (95.3) | 7 (53.8) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KMT2D | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 0 | 8 (44.4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 38 (100) | 10 (55.6) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviation: IHC: immunohistochemical. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As shown in Figure 3, the most commonly mutated gene in cfDNA was MACF1 (19.3%), followed by KMT2D (17.0%), FAT4 (13.6%), KMT2C (13.6%), LRP1B (12.5%) and PLB1 (11.4%). The median number of mutated genes in the 56 GC cfDNA samples was 1 (range 0–5). Among them, twenty-six (46.4%) patients had more than one mutated gene.
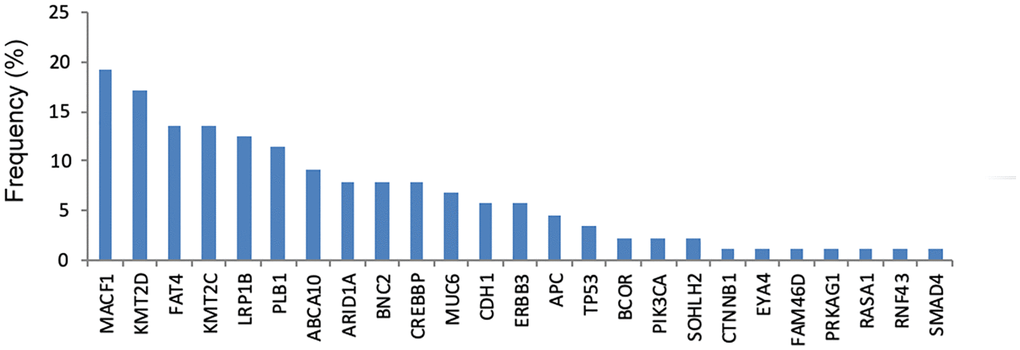
Figure 3. The frequency of genetic mutations in cfDNA.
As shown in Figure 4A, the frequency of genetic mutations (at least one gene mutation) in tumor DNA was highest in patients with hematogenous metastasis (100%), followed by peritoneal metastasis (97%) and distant lymphatic metastasis (96.2%). In Figure 4B, the frequency of genetic mutation in cfDNA was highest in patients with distant lymphatic metastasis (100%), followed by peritoneal metastasis (87.5%) and hematogenous metastasis (66.7%).
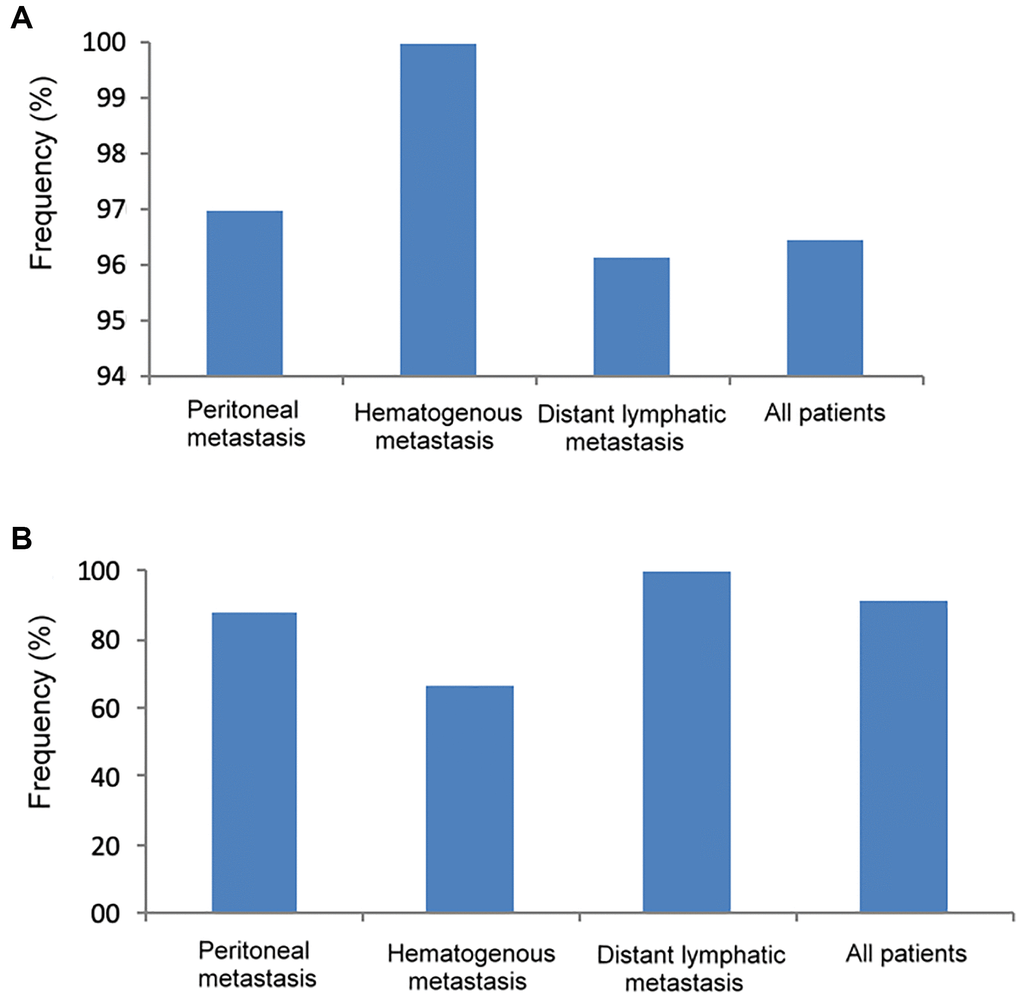
Figure 4. The frequency of genetic mutations in tumor DNA and cfDNA according to different metastatic patterns. (A) Tumor DNA, (B) cfDNA.
As shown in Table 3, comparing patients with peritoneal metastasis alone and distant lymphatic metastasis alone, there was no significant difference in the frequency of any of the genetic mutations between these two metastatic patterns in tumor DNA; however, in cfDNA, the frequency of PLB1 mutations was significantly higher in patients with distant lymphatic metastasis than in those with peritoneal metastasis (23.8% vs. 3.7%, P = 0.037).
Table 3. The correlation of tumor DNA mutation, cfDNA mutation, single peritoneal metastasis and single distant lymphatic metastasis.
| Genetic mutation | Tumor DNA | cfDNA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Peritoneum n = 27 n (%) | Distant LM n = 21 n (%) | P value | Peritoneum n = 27 n (%) | Distant LM n = 21 n (%) | P value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CREBBP | 2 (7.4) | 2 (9.5) | 0.792 | 1 (3.7) | 1 (4.8) | 0.856 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP53 | 11 (40.7) | 8 (38.1) | 0.853 | 3 (11.1) | 0 | 0.115 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ARID1A | 11 (40.7) | 7 (33.3) | 0.599 | 4 (14.8) | 3 (14.3) | 0.959 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BCOR | 1 (3.7) | 4 (19.0) | 0.084 | 0 | 1 (4.8) | 0.252 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTNNB1 | 1 (3.7) | 0 | 0.373 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KMT2D | 10 (37.0) | 6 (28.6) | 0.537 | 8 (29.6) | 2 (9.5) | 0.089 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RNF43 | 1 (3.7) | 1 (4.8) | 0.856 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| APC | 3 (11.1) | 0 | 0.115 | 2 (7.4) | 0 | 0.203 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FAT4 | 3 (11.1) | 7 (33.3) | 0.060 | 2 (7.4) | 6 (28.6) | 0.051 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KMT2C | 8 (29.6) | 6 (28.6) | 0.936 | 4 (14.8) | 2 (9.5) | 0.582 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ABCA10 | 5 (18.5) | 2 (9.5) | 0.381 | 5 (18.5) | 2 (9.5) | 0.381 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MUC6 | 2 (7.4) | 2 (9.5) | 0.792 | 1 (3.7) | 2 (9.5) | 0.409 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KRAS | 0 | 0 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SMAD4 | 1 (3.7) | 0 | 0.373 | 1 (3.7) | 0 | 0.373 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PIK3CA | 3 (11.1) | 1 (4.8) | 0.430 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BNC2 | 2 (7.4) | 2 (9.5) | 0.792 | 1 (3.7) | 2 (9.5) | 0.409 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PLB1 | 3 (11.1) | 5 (23.8) | 0.242 | 1 (3.7) | 5 (23.8) | 0.037 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MACF1 | 7 (25.9) | 4 (19.0) | 0.574 | 5 (18.5) | 3 (14.3) | 0.696 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ERBB3 | 4 (14.8) | 3 (14.3) | 0.959 | 2 (7.4) | 2 (9.5) | 0.792 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CDH1 | 4 (14.8) | 2 (9.5) | 0.582 | 3 (11.1) | 2 (9.5) | 0.858 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RASA1 | 2 (7.4) | 1 (4.8) | 0.707 | 1 (3.7) | 0 | 0.373 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PREX2 | 2 (7.4) | 1 (4.8) | 0.707 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EYA4 | 1 (3.7) | 1 (4.8) | 0.856 | 1 (3.7) | 0 | 0.373 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LRP18 | 6 (22.2) | 5 (23.8) | 0.897 | 3 (11.1) | 4 (19.0) | 0.440 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FAM46D | 1 (3.7) | 1 (4.8) | 0.856 | 0 | 1 (4.8) | 0.252 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SMAD2 | 1 (3.7) | 2 (9.5) | 0.409 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RHOA | 0 | 1 (4.8) | 0.252 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SOHLH2 | 2 (7.4) | 1 (4.8) | 0.707 | 1 (3.7) | 1 (4.8) | 0.856 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CNGA4 | 0 | 0 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: cfDNA: cell-free DNA; LM: lymphatic metastasis. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The specificity of all genetic mutations in cfDNA was 100% in predicting mutations in tumor DNA. As shown in Table 4, the sensitivity of the top nine mutated genes in tumor DNA and cfDNA was compared. Using the mutation pattern of cfDNA in the prediction of mutations in tumor DNA, the sensitivity was the highest in FAT4 (88.9%), followed by MACF1 (80%), CDH1 (75%), PLB1 (75%), KMT2D (72.7%), LRP1B (71.4%), KMT2C (40%), ARID1A (25%), and TP53 (13.6%).
Table 4. Sensitivity and specificity of nine top mutated genes between tumor DNA and cfDNA of stage IV GC patients.
| TP53 | ARID1A | KMT2C | KMT2D | FAT4 | LRP1B | MACF1 | CDH1 | PLB1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | Tumor DNA mutation | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yes | No | Yes | Yes | Yes | No | Yes | No | Yes | No | Yes | No | Yes | No | Yes | No | Yes | No | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cfDNA mutation | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yes | 3 | 0 | 5 | 0 | 6 | 0 | 8 | 0 | 8 | 0 | 5 | 0 | 4 | 0 | 3 | 0 | 3 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No | 19 | 34 | 15 | 36 | 9 | 41 | 3 | 45 | 1 | 47 | 2 | 49 | 1 | 51 | 1 | 52 | 1 | 52 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sensitivity | 13.6% | 25% | 40% | 72.7% | 88.9% | 71.4% | 80% | 75% | 75% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Specificity | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: cfDNA: cell-free DNA; GC: gastric cancer. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Concordance of genetic mutations between cfDNA and tumor samples
In total, 621 mutation spots among these 29 genes were found in tumor samples. Among these 56 patients, 360 mutation spots in tumor samples in 45 patients could not be detected in cfDNA. The concordance of mutation spots in these 29 genes was 42.0% (261/621) between cfDNA and tumor samples. Figure 5 shows the concordance of mutation patterns in these 29 genes between tumor samples (Figure 5A) and cfDNA (Figure 5B). All the genetic mutations in cfDNA could be identified in tumor DNA.
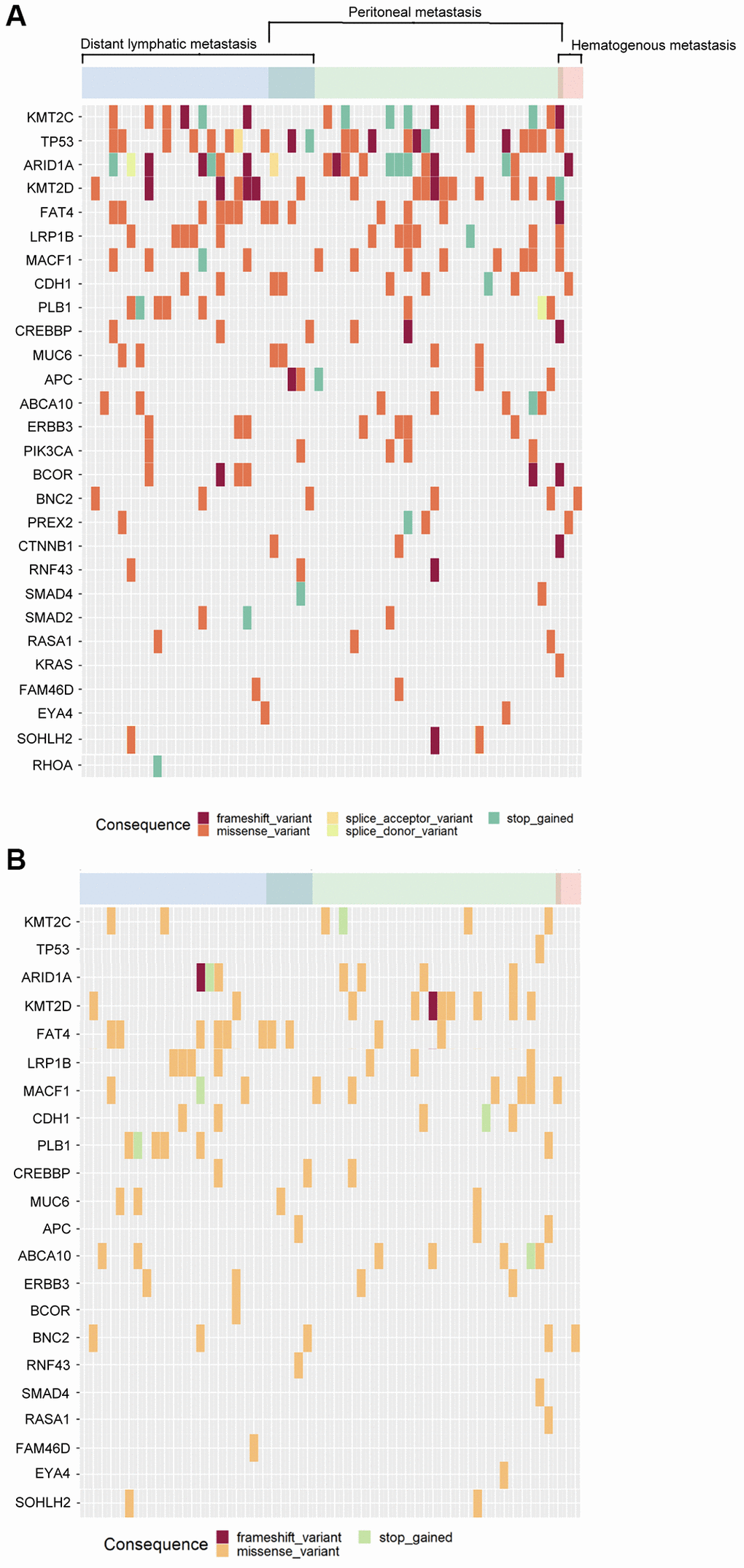
Figure 5. The mutational patterns of the tumor tissue and cfDNA. (A) Tumor tissue, (B) cfDNA.
The mutation patterns of tumor DNA and cfDNA in patients with more than one metastatic pattern
Among the 56 stage IV GC patients, one patient had both peritoneal and hematogenous metastases. The patient had ten mutated genes in the tumor DNA and a total of 60 mutated spots, and only one mutated gene, MACF1, was detected in both tumor DNA and cfDNA.
Among the 56 stage IV GC patients, five patients had both peritoneal and distant lymphatic metastases. All of them had at least two mutated genes in tumor DNA and at least one mutated gene in cfDNA. Regarding tumor DNA, there were two mutated genes in one patient (CDH1 and MUC6), five mutated genes in two patients (including BNC2, FAT4, APC, TP53, CREBBP), and ten mutated genes in two patients (CDH1, PIK3CA, SMAD4, MUC6, FAT4, APC, RNF43, KMT2D, CTNNB1, ARID1A). Regarding cfDNA, three patients had one mutated gene (including MUC6 mutation in one patient and FAT4 mutation in two patients), and two patients had two mutated genes (one patient with BNC2 and CREBBP mutations; another patient with RNF43 and APC mutations).
We further compared the number of mutated genes and mutated spots between single-site and multiple-site metastases in tumor DNA and cfDNA. Regarding tumor DNA, the number of mutated genes (3.5 ± 2.3 vs. 4.7 ± 2.9, P = 0.273) was not significantly different between single-site and multiple-site metastases, while patients with multiple-site metastases had significantly more mutated spots than patients with single-site metastases (23.7 ± 18.6 vs. 9.6 ± 8.6, P = 0.002). Regarding cfDNA, the number of mutated genes (1.8 ± 1.4 vs. 1.3 ± 0.5, P = 0.432) and mutated spots (5.0 ± 4.2 vs. 6.2 ± 2.3, P = 0.491) were not significantly different between single-site and multiple-site metastases.
Discussion
In the present study, NGS with a 29-gene panel was used to analyze cfDNA and tumor DNA in stage IV GC patients. The concordance of mutation patterns between cfDNA and tumor DNA was 42.0%. The specificity was 100% using the mutation status of cfDNA to predict mutation patterns in tumor samples. For cfDNA with PLB1 mutations, patients were more likely to develop distant lymphatic metastasis than peritoneal metastasis. Patients with multiple-site metastases had significantly more mutated spots than patients with single-site metastases. To the best of our knowledge, this is the first study to investigate the mutation patterns of different metastatic patterns between cfDNA and tumor DNA for stage IV GC.
Our results showed that the concordance rate between tumor DNA and cfDNA was 42%, which was similar to previous studies [14]. Varkalaite et al., [14] used whole-exome sequencing for mutation analysis; among the 152 mutations in the plasma samples, only 39 mutations were detected in the tissue samples, indicating that the specificity was 25.7%. In our study, the specificity was 100% for cfDNA to predict mutations in tumor samples. The difference between our study and a previous study [14] might be due to different technology in mutation detection, differences in tumor stage and races. The quantity of cfDNA was higher in stage IV GC than in stage I–III GC [5], which might increase the detection rate of genetic mutations in cfDNA and make our results more promising. A recent published study [15] demonstrated that by using a 605-gene sequencing panel to sequencing cfDNA and tumor DNA, the mutation concordance between plasma cfDNA and tumor DNA was higher in stage IV GC (70.6%) than stage III GC (30.2%). cfDNA is useful in molecular profiling of GC patient and may be helpful in the prediction of patient survival, clinical therapeutic response, and future development of personalized therapy regimens. It was reported that the quantity of tissue matching alterations and presence of any somatic mutation in cfDNA is significant for discrimination between M0 and M1 GC patients, indicating that quantitative and qualitative cfDNA mutational profile analysis is useful for the evaluation of GC disease status and patient prognosis [14].
Anti-PD-L1 is approved as a primary immunotherapy in stage IV GC. Higher mutational load in cfDNA was associated with a better response to pembrolizumab, and reduced cfDNA six weeks after therapy had a longer progression-free survival (PFS), indicating that cfDNA can serve as a predictor of treatment response and PFS for GC [16]. Jin et al., reported that mutation status of TGFBR2, RHOA, and PREX2 in cfDNA were associated with resistant to immunotherapy and a shorter PFS. Hence, cfDNA can serve as a biomarker in the response to immunotherapy in advanced GC [17]. It was reported that CCNE1 amplification in cfDNA was associated with resistant to HER2-targeted therapy, while HER2 amplification was sensitive to HER2-targeted therapy [18]. Consequently, cfDNA mutational profiles may serve as a potential biomarker in the prediction of immunotherapy and targeted therapy in GC. In addition, for stage I–III GC patients after curative surgery, monitoring cfDNA mutation patterns may serve as a liquid biopsy and a useful biomarker for predicting tumor recurrence, recurrence patterns and patient prognosis. For patients at a high risk of tumor recurrence according to the cfDNA mutation patterns, postoperative adjuvant therapy may be required for preventing tumor recurrence and improving patient survival. Our results may provide useful information regarding immunotherapy or targeted therapy for GC treatment in the future.
We also analyzed the correlation between metastatic pattern and genetic mutation pattern in tumor DNA and cfDNA in stage IV GC, which has not yet been reported. The four most commonly mutated genes among the three metastatic patterns were TP53, KMT2C, KMT2D and ARID1A, which were also among the top high-frequency mutations for GC reported by the Catalogue of Somatic Mutations in Cancer (COSMIC) database [19]. All four genes mentioned above could be considered tumor suppressor genes [20–23]. TP53 mutation was associated with lymph node metastasis and distant metastasis in GC, especially in the Asian population [20], which was similar to our results. ARID1A mutation was reported to be associated with distant metastasis in GC [22], and ARID1A mutation could serve as a biomarker for immunotherapy in GI tract cancer [23]. KMT2D and KMT2C mutations in GC were associated with the DNA repair process, and these two genetic mutations were considered targets for cancer treatment using poly ADP-ribose polymerase (PARP) inhibitors [22, 24]. According to our results, some common genetic mutations were identified in different metastatic patterns, and drug therapy might be suitable for stage IV GC treatment, such as PARP inhibitors for mutations of the KMT2 family and immunotherapy for ARID1A mutations.
Low expression of E-cadherin (CDH1), ARID1A, FAT4, and KMT2C were associated with a poor prognosis in GC [25–28], while high expression of p53 or KMT2D was reported to be associated with a poor survival [29, 30]. MACF1 mutations were associated with upregulation of the mTOR signaling pathway and had a worse prognosis in breast cancer [31]. PLB1 (phospholipase B1) is a secreted membrane-associated phospholipase that is involved in choline metabolism in tumors [32]. PLB1 mutation was reported to be associated with poor survival in non-small cell lung cancer and glioblastoma multiform [33, 34]. As shown in Table 2, our results demonstrated that the genetic mutations of these genes mentioned above (CDH1, ARID1A, FAT4, KMT2C, TP53, KMT2D, MACF1, and PLB1) were associated with the down regulation of the associated protein function, which may be involved in the up-regulation or down-regulation in the mechanism of cancer developing. In addition, our results showed that PLB1 mutation was more common in the cfDNA of GC patients with single distant lymphatic metastasis than in patients with peritoneal metastasis, which has not yet been reported yet. According to our results, PLB1 mutation in cfDNA may serve as a risk factor for distant lymphatic metastasis in GC.
Our results demonstrated that the sensitivity of 88.9% was the highest using cfDNA to predict the mutation patterns of tumor DNA with FAT4 mutation. FAT4 is involved in the Wnt pathway, which plays an important role in tumorigenesis, invasion, and vascularization [35]. FAT4 silencing enhanced proliferation, colony formation, invasion, and metastasis and reduced fibronectin adhesion [36]. According to our results, monitoring of FAT4 mutation in cfDNA may be a substitute for tumor biopsy in stage IV GC.
In colorectal cancer, higher cfDNA levels and increased number of genetic mutations in cfDNA were associated with a poor surgical and multiple-site metastasis [37], which has not yet been reported in GC. According to our results, GC patients with multiple-site metastases had at least two mutated genes in tumor DNA and at least one mutated gene in cfDNA. In tumor DNA, the number of mutated spots rather than the number of mutated genes was significantly higher in multiple-site metastases than in single-site metastases, which was not observed in cfDNA. It seems that more mutated spots in tumor DNA may diversify the mutation patterns and increase the possibility of multiple-site metastasis. Our findings may remind the physicians of the possibility of multiple-site metastasis when many mutated spots are found in tumor DNA.
There are limitations in the current study. First, this is a retrospective and single-center study. Second, the patient number was limited and selection bias may exist. More patients enrolled from different countries and races are required to verify our results.
Materials and Methods
Patients and sample collection
Tumor and preoperative serum samples were collected from 56 stage IV GC patients from Taipei Veterans General Hospital Biobank. Only stage IV GC patients with available tumor and preoperative serum samples in the biobank were enrolled in this study. The exclusion criteria included patients who had stage I–III GC, who received emergent surgery, or who did not have available tumor or preoperative serum samples in the biobank. The tumor tissues and normal gastric mucosa tissues were collected and stored in a biobank at our institution. Written informed consent before tumor tissue collection was obtained from all study participants. The study was approved by the Institutional Review Board of Taipei Veterans General Hospital (2021-07-022AC). The pathological staging of GC was performed according to the 8th American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) TNM classification system [38]. The metastatic patterns included peritoneal, hematogenous, and distant lymphatic metastases. Single-site metastasis was defined as a single metastatic pattern. Multiple-site metastasis was defined as a patient with more than one metastatic pattern.
cfDNA extraction from plasma
Whole blood (4~8 ml) was drawn from each clinical patient using disposable venous blood lancet and stored directly in cfDNA Blood Collection Tube (Streck). Blood samples were stored at room temperature and used for plasma separation extraction within three days. In plasma separation, tube containing whole blood was centrifuged at 3,000 × g for 10 minutes at room temperature, then the upper layer was transferred to the 1.5 ml micro-centrifuge tube. The transferred tubes were processed for the second centrifuge at 11,000 × g for 10 minutes, and finally transfer the supernatant to new sample tubes for cfDNA extraction. cfDNA was extracted from 1,000 μl of plasma by using the QIAamp MinElute ccfDNA Kits (Qiagen), and tumor DNA was extracted from tissue specimens by using a QIAamp DNA Tissue Kit (Qiagen, Valencia, CA, USA). After DNA extraction, DNA quantity was measured by using the Qubit dsDNA High-Sensitivity assay (Thermo Fisher Scientific).
Analysis of genetic alterations
A total of 250 ng DNA from each tumor tissue was used to construct an NGS library using an IDT Lotus Library Preparation Kit (IDT, USA). Each DNA sample was fragmented and then used to prepare a DNA library by performing end repair, a-overhang addition, adaptor ligation and size selection (250~350 bp). Fifteen microliters of each cfDNA sample was used to construct a library by using the xGen Prism DNA Library Prep Kit (IDT). Target DNA of exonic regions of 29 frequently-mutated genes in GC (ABCA10, APC, ARID1A, BCOR, BNC2, CDH1, CNGA4, CREBBP, CTNNB1, ERBB3, EYA4, FAM46D, FAT4, KMT2C, KMT2D, KRAS, LRP1B, MACF1, MUC6, PIK3CA, PLB1, PREX2, RASA1, RHOA, RNF43, SMAD2, SMAD4, SOHLH2 and TP53) was enriched using probe-based methods. The probes were synthesized by Integrated DNA Technologies (USA) according to our previously designed probe sequences, and the capture procedure was performed following the IDT guidelines. After probe-based enrichment, libraries of each pool were amplified with 14 cycles. The amplified libraries were quantified using an LC480 qPCR system (Roche) and pooled into a new 1.5-ml tube as a 10-nM pooled DNA library. The final pool was used for sequencing (Illumina NextSeq sequencer, 2 × 150 bp). The raw output of each tumor tissue was >1.5 Gb, and the average depth of target regions was >250X. The raw output of each cfDNA was >15 Gb, and the average depth of target regions was >2000X. We used the Illumina Basespace Dragen somatic mutation pipeline (https://www.illumina.com/products/by-type/informatics-products/basespace-sequence-hub/apps/edico-genome-inc-dragen-somatic-pipeline.html) to perform variant calling and annotated all variants by using Illumina variant interpreter (https://variantinterpreter.informatics.illumina.com/home). In this study, although adjacent-normal parts or white blood cells were not collected for genotyping and identifying germline variants of each case, single-nucleotide polymorphisms, defined as the minor allele frequency >1%, of the primary variants of tumor and cfDNA were filtered out based on the databases of Taiwan biobank, 1,000 Genome and GnomAD East Asian populations. Variants with >5% and >1% frequencies were retained for the following in tissue and cfDNA, respectively. Although some variants were found to have high confidences in variant calling with more than 2,000 depth, however, only >1% variants were considered in this study, which might reduce the sensitivity in this study.
Immunohistochemical (IHC) staining
IHC staining was performed according to the manufacturer’s recommendations, including E-cadherin (CDH1), MACF1, p53, PLB1, ARID1A, KMT2C, FAT4, and KMT2D. Antibodies utilized were: E-cadherin (Genemed #61-0192), MACF1 (Proteintech #13058-1-AP), p53 (Leica #P53-D07-L-CE), PLB1 (Invitrogen #PA5-119343), ARID1A (Sigma #HPA005456), KMT2C (Invitrogen #PA5-68419), FAT4 (Invitrogen #PA5-72970), KMT2D (Invitrogen #PA5-57490).
Immunoreactivity E-cadherin was localized in membranes; MACF1 and PLB1 were localized in the cytoplasm and membranes; p53 and ARID1A were localized in the nucleus; KMT2C and FAT4 were localized in the cytoplasm; KMT2D was localized in the cytoplasm and nucleus, and mainly in nucleus (Figure 2). Immunoreactivity of at least 10% of tumor cells was considered positive staining.
Statistical analysis
IBM SPSS Statistics 25.0 was used for statistical analyses. A χ2 test with Yates correction or Fisher’s exact test was used to compare the categorical data. The frequency of the specific gene mutation was calculated using the number of patients carrying the specific gene mutation divided by the total number of the patients and was presented as a percentage. A P value < 0.05 was defined as statistically significant.
Conclusions
Due to the high sensitivity and specificity of some genes in the prediction of mutation in tumor samples, monitoring the mutation pattern of cfDNA may be a substitute for tumor biopsy and may be applicable in the treatment of stage IV GC.
Abbreviations
AJCC: American Joint Committee on Cancer; CA 19-9: carbohydrate antigen 19-9; CEA: carcinoembryonic antigen; cfDNA: cell-free DNA; COSMIC: Catalogue of Somatic Mutations in Cancer; GC: Gastric cancer; NGS: Next-generation sequencing; PARP: poly ADP-ribose polymerase; PCR: Polymerase chain reaction; TCGA: The Cancer Genome Atlas; UICC: Union for International Cancer Control.
Author Contributions
Ching-Yun Kung: conceptualization and writing – original draft. Wen-Liang Fang: funding acquisition, conceptualization, formal analysis, data curation, writing – original draft, and writing – review and editing. Yi-Ping Hung: conceptualization and writing – review and editing. Kuo-Hung Huang: funding acquisition, conceptualization and writing – review and editing. Ming-Huang Chen: conceptualization and writing – review and editing. Yee Chao: conceptualization and writing – review and editing. Shih-Chieh Lin: conceptualization and writing – review and editing. Anna Fen-Yau Li: conceptualization and writing – review and editing. Su-Shun Lo: conceptualization and writing – review and editing. Chew-Wun Wu: conceptualization and writing – review and editing.
Acknowledgments
The authors thank Chien-Hsing Lin and Fang-Yu Chang for the molecular genetic analysis.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement and Consent
The study was approved by the Institutional Review Board of Taipei Veterans General Hospital (2021-07-022AC). Written informed consent before tumor tissue collection was obtained from all study participants.
Funding
This study was supported with funding from Melissa Lee Cancer Foundation (MLCF-V111_A11107, MLCF_V112_A11203, MLCF_V112_A11205) and Taipei Veterans General Hospital (V111C-184, V111C-222). The funding did not play a role in the study design, data collection, analysis, interpretation or manuscript writing.
References
- 1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, Znaor A, Bray F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019; 144:1941–53. https://doi.org/10.1002/ijc.31937 [PubMed]
- 2. Spindler KL, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012; 18:1177–85. https://doi.org/10.1158/1078-0432.CCR-11-0564 [PubMed]
- 3. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA
Jr . Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985–90. https://doi.org/10.1038/nm.1789 [PubMed] - 4. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108–12. https://doi.org/10.1038/nature12065 [PubMed]
- 5. Fang WL, Lan YT, Huang KH, Liu CA, Hung YP, Lin CH, Jhang FY, Chang SC, Chen MH, Chao Y, Lin WC, Lo SS, Fen-Yau Li A, et al. Clinical significance of circulating plasma DNA in gastric cancer. Int J Cancer. 2016; 138:2974–83. https://doi.org/10.1002/ijc.30018 [PubMed]
- 6. Kim YW, Kim YH, Song Y, Kim HS, Sim HW, Poojan S, Eom BW, Kook MC, Joo J, Hong KM. Monitoring circulating tumor DNA by analyzing personalized cancer-specific rearrangements to detect recurrence in gastric cancer. Exp Mol Med. 2019; 51:1–10. https://doi.org/10.1038/s12276-019-0292-5 [PubMed]
- 7. Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, Chen X, Sun J, Wang Z, Hong Z, Zhu L, Zhu C, Chen J, et al. Circulating Tumor DNA Mutation Profiling by Targeted Next Generation Sequencing Provides Guidance for Personalized Treatments in Multiple Cancer Types. Sci Rep. 2017; 7:583. https://doi.org/10.1038/s41598-017-00520-1 [PubMed]
- 8. Huffman BM, Aushev VN, Budde GL, Chao J, Dayyani F, Hanna D, Botta GP, Catenacci DVT, Maron SB, Krinshpun S, Sharma S, George GV, Malhotra M, et al. Analysis of Circulating Tumor DNA to Predict Risk of Recurrence in Patients With Esophageal and Gastric Cancers. JCO Precis Oncol. 2022; 6:e2200420. https://doi.org/10.1200/PO.22.00420 [PubMed]
- 9. Yan H, Chen W, Ge K, Mao X, Li X, Liu W, Wu J. Value of Plasma Methylated SFRP2 in Prognosis of Gastric Cancer. Dig Dis Sci. 2021; 66:3854–61. https://doi.org/10.1007/s10620-020-06710-8 [PubMed]
- 10. Balgkouranidou I, Matthaios D, Karayiannakis A, Bolanaki H, Michailidis P, Xenidis N, Amarantidis K, Chelis L, Trypsianis G, Chatzaki E, Lianidou ES, Kakolyris S. Prognostic role of APC and RASSF1A promoter methylation status in cell free circulating DNA of operable gastric cancer patients. Mutat Res. 2015; 778:46–51. https://doi.org/10.1016/j.mrfmmm.2015.05.002 [PubMed]
- 11. Balgkouranidou I, Karayiannakis A, Matthaios D, Bolanaki H, Tripsianis G, Tentes AA, Lianidou E, Chatzaki E, Fiska A, Lambropoulou M, Kolios G, Kakolyris S. Assessment of SOX17 DNA methylation in cell free DNA from patients with operable gastric cancer. Association with prognostic variables and survival. Clin Chem Lab Med. 2013; 51:1505–10. https://doi.org/10.1515/cclm-2012-0320 [PubMed]
- 12. Han J, Lv P, Yu JL, Wu YC, Zhu X, Hong LL, Zhu WY, Yu QM, Wang XB, Li P, Ling ZQ. Circulating methylated MINT2 promoter DNA is a potential poor prognostic factor in gastric cancer. Dig Dis Sci. 2014; 59:1160–8. https://doi.org/10.1007/s10620-013-3007-0 [PubMed]
- 13. Hu XY, Ling ZN, Hong LL, Yu QM, Li P, Ling ZQ. Circulating methylated THBS1 DNAs as a novel marker for predicting peritoneal dissemination in gastric cancer. J Clin Lab Anal. 2021; 35:e23936. https://doi.org/10.1002/jcla.23936 [PubMed]
- 14. Varkalaite G, Forster M, Franke A, Kupcinskas J, Skieceviciene J. Liquid Biopsy in Gastric Cancer: Analysis of Somatic Cancer Tissue Mutations in Plasma Cell-Free DNA for Predicting Disease State and Patient Survival. Clin Transl Gastroenterol. 2021; 12:e00403. https://doi.org/10.14309/ctg.0000000000000403 [PubMed]
- 15. He W, Yang J, Sun X, Jiang S, Jiang J, Liu M, Mu T, Li Y, Zhang X, Duan J, Xu R. Advantages and Limitations of Monitoring Circulating Tumor DNA Levels to Predict the Prognosis of Patients Diagnosed With Gastric Cancer. Biomark Insights. 2022; 17:11772719221141525. https://doi.org/10.1177/11772719221141525 [PubMed]
- 16. Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, Liu XQ, Sher X, Jung H, Lee M, Lee S, Park SH, Park JO, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018; 24:1449–58. https://doi.org/10.1038/s41591-018-0101-z [PubMed]
- 17. Jin Y, Chen DL, Wang F, Yang CP, Chen XX, You JQ, Huang JS, Shao Y, Zhu DQ, Ouyang YM, Luo HY, Wang ZQ, Wang FH, et al. The predicting role of circulating tumor DNA landscape in gastric cancer patients treated with immune checkpoint inhibitors. Mol Cancer. 2020; 19:154. https://doi.org/10.1186/s12943-020-01274-7 [PubMed]
- 18. Kim ST, Banks KC, Pectasides E, Kim SY, Kim K, Lanman RB, Talasaz A, An J, Choi MG, Lee JH, Sohn TS, Bae JM, Kim S, et al. Impact of genomic alterations on lapatinib treatment outcome and cell-free genomic landscape during HER2 therapy in HER2+ gastric cancer patients. Ann Oncol. 2018; 29:1037–48. https://doi.org/10.1093/annonc/mdy034 [PubMed]
- 19. Li Q, Wu R, Wu F, Chen Q. KMT2D promotes proliferation of gastric cancer cells: evidence from ctDNA sequencing. J Clin Lab Anal. 2021; 35:e23721. https://doi.org/10.1002/jcla.23721 [PubMed]
- 20. Wang J, Shao X, Liu Y, Shi R, Yang B, Xiao J, Liu Y, Qu X, Li Z. Mutations of key driver genes in gastric cancer metastasis risk: a systematic review and meta-analysis. Expert Rev Mol Diagn. 2021; 21:963–72. https://doi.org/10.1080/14737159.2021.1946394 [PubMed]
- 21. Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, Stock W, LeBeau MM, Shannon KM, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014; 25:652–65. https://doi.org/10.1016/j.ccr.2014.03.016 [PubMed]
- 22. Nemtsova MV, Kalinkin AI, Kuznetsova EB, Bure IV, Alekseeva EA, Bykov II, Khorobrykh TV, Mikhaylenko DS, Tanas AS, Strelnikov VV. Mutations in Epigenetic Regulation Genes in Gastric Cancer. Cancers (Basel). 2021; 13:4586. https://doi.org/10.3390/cancers13184586 [PubMed]
- 23. Li L, Li M, Jiang Z, Wang X. ARID1A Mutations Are Associated with Increased Immune Activity in Gastrointestinal Cancer. Cells. 2019; 8:678. https://doi.org/10.3390/cells8070678 [PubMed]
- 24. Rampias T, Karagiannis D, Avgeris M, Polyzos A, Kokkalis A, Kanaki Z, Kousidou E, Tzetis M, Kanavakis E, Stravodimos K, Manola KN, Pantelias GE, Scorilas A, Klinakis A. The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep. 2019; 20:e46821. https://doi.org/10.15252/embr.201846821 [PubMed]
- 25. Uchikado Y, Okumura H, Ishigami S, Setoyama T, Matsumoto M, Owaki T, Kita Y, Natsugoe S. Increased Slug and decreased E-cadherin expression is related to poor prognosis in patients with gastric cancer. Gastric Cancer. 2011; 14:41–9. https://doi.org/10.1007/s10120-011-0004-x [PubMed]
- 26. Zhu YP, Sheng LL, Wu J, Yang M, Cheng XF, Wu NN, Ye XB, Cai J, Wang L, Shen Q, Wu JQ. Loss of ARID1A expression is associated with poor prognosis in patients with gastric cancer. Hum Pathol. 2018; 78:28–35. https://doi.org/10.1016/j.humpath.2018.04.003 [PubMed]
- 27. Jiang X, Liu Z, Xia Y, Luo J, Xu J, He X, Tao H. Low FAT4 expression is associated with a poor prognosis in gastric cancer patients. Oncotarget. 2017; 9:5137–54. https://doi.org/10.18632/oncotarget.23702 [PubMed]
- 28. Cho SJ, Yoon C, Lee JH, Chang KK, Lin JX, Kim YH, Kook MC, Aksoy BA, Park DJ, Ashktorab H, Smoot DT, Schultz N, Yoon SS. KMT2C Mutations in Diffuse-Type Gastric Adenocarcinoma Promote Epithelial-to-Mesenchymal Transition. Clin Cancer Res. 2018; 24:6556–69. https://doi.org/10.1158/1078-0432.CCR-17-1679 [PubMed]
- 29. Zhang X, Yamamoto Y, Wang X, Sato M, Imanishi M, Sugaya A, Hirose M, Endo S, Moriwaki T, Yamato K, Hyodo I. MDM4 as a Prognostic Factor for Patients With Gastric Cancer With Low Expression of p53. Anticancer Res. 2021; 41:1475–83. https://doi.org/10.21873/anticanres.14906 [PubMed]
- 30. Xiong W, Deng Z, Tang Y, Deng Z, Li M. Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer. Biochem Biophys Res Commun. 2018; 504:129–36. https://doi.org/10.1016/j.bbrc.2018.08.143 [PubMed]
- 31. Tian Y, Zhu K, Li Y, Ren Z, Wang J. MACF1 mutations predict poor prognosis: a novel potential therapeutic target for breast cancer. Am J Transl Res. 2022; 14:7670–88. [PubMed]
- 32. Moestue SA, Borgan E, Huuse EM, Lindholm EM, Sitter B, Børresen-Dale AL, Engebraaten O, Maelandsmo GM, Gribbestad IS. Distinct choline metabolic profiles are associated with differences in gene expression for basal-like and luminal-like breast cancer xenograft models. BMC Cancer. 2010; 10:433. https://doi.org/10.1186/1471-2407-10-433 [PubMed]
- 33. Zhu M, Geng L, Shen W, Wang Y, Liu J, Cheng Y, Wang C, Dai J, Jin G, Hu Z, Ma H, Shen H. Exome-Wide Association Study Identifies Low-Frequency Coding Variants in 2p23.2 and 7p11.2 Associated with Survival of Non-Small Cell Lung Cancer Patients. J Thorac Oncol. 2017; 12:644–56. https://doi.org/10.1016/j.jtho.2016.12.025 [PubMed]
- 34. Lin H, Wang K, Xiong Y, Zhou L, Yang Y, Chen S, Xu P, Zhou Y, Mao R, Lv G, Wang P, Zhou D. Identification of Tumor Antigens and Immune Subtypes of Glioblastoma for mRNA Vaccine Development. Front Immunol. 2022; 13:773264. https://doi.org/10.3389/fimmu.2022.773264 [PubMed]
- 35. Wang Y. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther. 2009; 8:2103–9. https://doi.org/10.1158/1535-7163.MCT-09-0282 [PubMed]
- 36. Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012; 44:570–4. https://doi.org/10.1038/ng.2246 [PubMed]
- 37. Shahjehan F, Kamatham S, Kasi PM. Role of Circulating Tumor DNA in Gastrointestinal Cancers: Update From Abstracts and Sessions at ASCO 2018. Front Oncol. 2019; 9:358. https://doi.org/10.3389/fonc.2019.00358 [PubMed]
- 38. American Joint Committee on Cancer. AJCC cancer staging manual. 8th ed. New York: Springer; 2017.