



Introduction
Subarachnoid hemorrhage (SAH) is an acute cerebrovascular disease with high mortality and morbidity, mainly caused by the rupture of intracranial aneurysms [1]. Early brain injury (EBI) is one of the main factors for the poor prognosis of SAH patients. Much evidence indicates that neuroinflammation is the main pathophysiological mechanism of EBI. Inhibiting the inflammatory response after SAH can effectively alleviate EBI and significantly promote the recovery of neurological function after SAH [2]. The NLRP3 inflammasome is a multi-protein complex associated with various inflammatory diseases [3]. The NLRP3 protein consists of an amino-terminal pyrin domain (PYD), a NACHT domain, and a carboxy-terminal leucine-rich (LRR) repeat domain [4]. The PYD domain of NLRP3 binds to the PYD domain of ASC and then recruits inactive caspases-1 through the interaction of ASC with the common recruitment domain (CARD) to form the NLRP3 inflammasome. Activation of the NLRP3 inflammasome cleaves pro-caspase-1 into active caspase-1, which induces the pro-inflammatory cytokine IL-1β and IL-18 process and secretion, and causes inflammatory response.
Recent studies have been gradually revealing the effects of NLRP3 on the occurrence and development of the central nervous system. In the pathological process of Alzheimer's disease, abnormally activated NLRP3 inflammasome can lead to pathological damage of neurons and accelerate the deterioration of neural function [5]. Soares et al. have reported that in a mouse model of multiple sclerosis, pharmacological inhibition of NLRP3 activation can significantly reduce neuroinflammation and promote the improvement of motor function [6]. In addition to the above-mentioned chronic neurodegenerative diseases, studies have confirmed that NLRP3 inflammasome activated after SAH, and inhibition of NLRP3 inflammasome can alleviate EBI after SAH [7, 8]. However, the mechanism of the NLRP3 inflammasome activation is very complex, including abnormal distribution of intracellular ions (potassium, calcium, and chloride ions) [9–15], oxidized mitochondrial DNA (ox-mtDNA) [16], unstable soluble Enzymes [17] and reactive oxygen species (ROS) [18, 19]. The disruption of intracellular ion homeostasis has been considered to be one of the most important factors in inducing NLRP3 inflammasome activation. Recent studies have shown that calcium ions influx or migration plays an important role in the activation of the NLRP3 inflammasome [20–22], however, whether calcium ions are involved in NLRP3 inflammasome activation after SAH and the underlying mechanisms involved remains unclear.
TRPV1, also known as vanilloid receptor type 1 (VR1), is a member of the transient receptor potential channel protein family. Previous studies have focused on the role of TRPV1 in sensory transmission from nociceptive neurons in the peripheral nervous system [23]. Recent reports indicated that TRPV1 was widely distributed in the central nervous system, and it may involve regulating the neuroinflammatory processes [24]. Moreover, some studies have pointed out that TRPV1 regulated neuroinflammation by activating the NLRP3 inflammasome [25–27]. Herein, we speculate that TRPV1 and associated NLRP3 inflammasome might play an essential role in SAH-induced neuroinflammation.
Materials and Methods
Mouse models of SAH and drugs administration
C57BL/6 mice (male, 22–25 g) were purchased from Vital River Laboratory Animal Technology (Beijing, China). The procedures involved in mice were conformed to the guidelines of the National Institutes of Health on the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee of Shandong First Medical University.
SAH model was constructed via endovascular perforation as the previous study [28]. Briefly, the mice were anesthetized using 5% isoflurane, and then the anesthesia was maintained at 2% isoflurane during the entirety of the experiment, the nylon suture was passed through the external carotid artery to the bifurcation of the anterior and middle artery and ultimately punctured to cause blood to flow into the subarachnoid space.
Mice were injected with 30 mg/kg capsazepine (CPZ, MedChemExpress, Cat#HY-15640) subcutaneously or 10 mg/kg capsaicin (CAP, MedChemExpress, Cat# HY-10448) intraperitoneally post-modeling to inhibit or active TRPV1 [26, 29], respectively. MCC950 (MedChemExpress, Cat# HY-12815) (40 mg/kg) were intraperitoneally injected post-modeling and 12 h later [8]. The drugs above were all purchased from MedChemExpress (NJ, USA).
Magnetic-activated cell sorting (MACS) and qPCR analysis
Ipsilateral hemispheres were harvested at 24 h post-SAH, dissociated mechanically using a glass homogenizer, and filtered into a 70-μm strainer to obtain a single-cell suspension. The single-cell suspension was isolated with 30% Percoll (Cytiva, Cat#17089101) solutions, incubated with CD11b microbeads (Miltenyi Biotec, Germany, 130-126-725), and then divided into CD11b positive and negative cells.
Total mRNA of tissues and cells was extracted using TRIzol™ Reagent (Life Technologies, CA, USA), used to synthesize cDNA using Evo M-MLV Reverse Transcriptase (Takara, Japan, RR420A), and then performed the qPCR analysis using SYBR Premix Ex Taq™ Kit (Takara, Japan, RR036A) according to manufacturer’s protocol. The primers were listed in Supplementary Table 1.
Brain water content
Twenty-four hours post-operation, the mice were euthanized, and the left hemisphere were promptly excised and immediately weighed to determine its wet weight (WW). Subsequently, the left hemisphere was dehydrated in a dry bath at 105°C for 72 hours to ascertain their dry weight (DW). The water content for each brain segment was calculated using the formula: ((WW − DW)/WW) × 100%.
Neurobehavioral assessment
Neurobehavioral assessment containing modified Garcia and beam balance test was performed 24 h post-SAH to evaluate neurological function [30]. Modified Garcia and beam balance scores ranged from 3 to 18 and 0 to 4 respectively, with higher scores indicating better neurological function.
Histological staining analysis
Mice were anesthetized and perfused sufficiently with 4% paraformaldehyde (PFA) at 24 h after SAH to harvest the brain tissues. The brain tissues were fixed in 4% PFA for 24 h, dehydrated in 30% sucrose solution, embedded in OCT compound, and then sliced into 10 μm coronal sections for immunofluorescence and Nissl staining analysis.
For immunofluorescence staining analysis, mice brain sections were blocked and permeabilized with 5% bovine serum containing 0.3% Triton-X100 for 1 h at room temperature and incubated with primary antibodies overnight at 4°C followed by 1 hour incubation at room temperature with the corresponding secondary antibody. The primary antibodies included anti-Iba-1 (Abcam, Cambridge, UK, ab-5076), anti-TRPV1 (Abcam, Cambridge, UK, ab203103), and anti-CD16/CD32 antibody (Biolegend, CA, USA, 101329).
For Nissl staining, mice brain sections were stained with 0.5% cresyl violet for 0.5 h at room temperature and dehydrated with ethanol absolute [31]. The images were analyzed using Image J (NIH).
Western blot
Proteins of tissues and cells were extracted using RIPA lysis buffer (Beyotime, Shanghai, China, P0013B), separated by SDS-PAGE, transferred into PVDF membrane, blocked with 5% non-fat powdered milk, incubated with primary antibodies overnight at 4°C followed by 1 hour incubation at room temperature with the corresponding secondary antibody. Then, the PVDF membrane was visualized using the ECL Plus chemiluminescence reagent kit. The primary antibodies included anti-β-actin (Proteintech, Hubei, China, 60008-1-Ig), anti-ZO-1 (Santa Cruz, TX, USA, sc-33725), anti-claudin-5 (Santa Cruz, TX, USA, sc-28670), anti-occludin (Santa Cruz, TX, USA, sc-133256), anti-NLRP3(Abcam, Cambridge, UK, ab210491), anti-ASC (Santa Cruz, TX, USA, sc-271054), anti-caspase-1 p20 (Santa Cruz, TX, USA,sc-22165). The protein expression was quantified using Image J (NIH).
ELISA
The total protein content of tissues and cell culture supernatants was determined via BCA assay (Beyotime, China). The expression level of IL-1β was detected using ELISA kit (Boster, Wuhan, China, EK0394) according to the manufacturer’s protocol.
In vitro SAH model
Murine BV2 cells were stimulated by 200 μM hemin (Sigma-Aldrich, MO, USA, H9039) to establish the SAH model in vitro. Meanwhile, BV2 cells were treated with 10 μM CAP [32] and BAPTA-AM [26] to investigate the mechanism of the TRPV1-activated NLRP3 inflammasome.
Calcium concentration detection
Calcium concentration was detected as previously reported [33]. Briefly, BV2 cells were incubated with 4 μM Fura-2/AM (YEASEN, Shanghai, China, 40702ES50) for 30min, and captured the fluorescence intensities when excited at 340 and 380 nm and emitted at 510 nm. The calcium concentration was calculated as followed: the fluorescence intensities at 340 nm/ the fluorescence intensities at 380 nm.
Statistical analysis
The data were analyzed by SPSS 22.0 and GraphPad Prism 8.0 to test whether met the normal distribution and variance homogeneity by Shapiro-Wilk and Levene methods, respectively. When met, data were analyzed by Student’s t-test (two groups) or one-way analysis of variance (ANOVA) with Tukey’s post hoc contrasts (more than three groups); otherwise, data were analyzed by Mann–Whitney nonparametric test (two groups) and Kruskal-Wallis test with post hoc contrast by the Dunn-Bonferroni test (more than three groups).
Availability of data and materials
The datasets used during the present study are available from the corresponding author upon reasonable request.
Results
TRPV1 upregulated post-SAH
TRPV1 plays crucial roles in various physiological and pathological processes, including sensation of mechanical stimuli, voltage sensitivity, response to chemical substances, immune activation, modulation of temperature, gastrointestinal function regulation, and involvement in tumorigenesis. The results of qPCR showed that the mRNA level of Trpv1 was increased at 24 hours after SAH (Figure 1A). Using MACS to separate the brain cells into CD11b positive and negative cells, it was found that Trpv1 changed most significantly in the CD11b positive cells (Figure 1B). Consistently, immunofluorescence staining demonstrated that TRPV1 was mainly expressed in microglia/macrophages (Figure 1C).The expression status of TRPV1 in microglia and macrophages deserves further study.
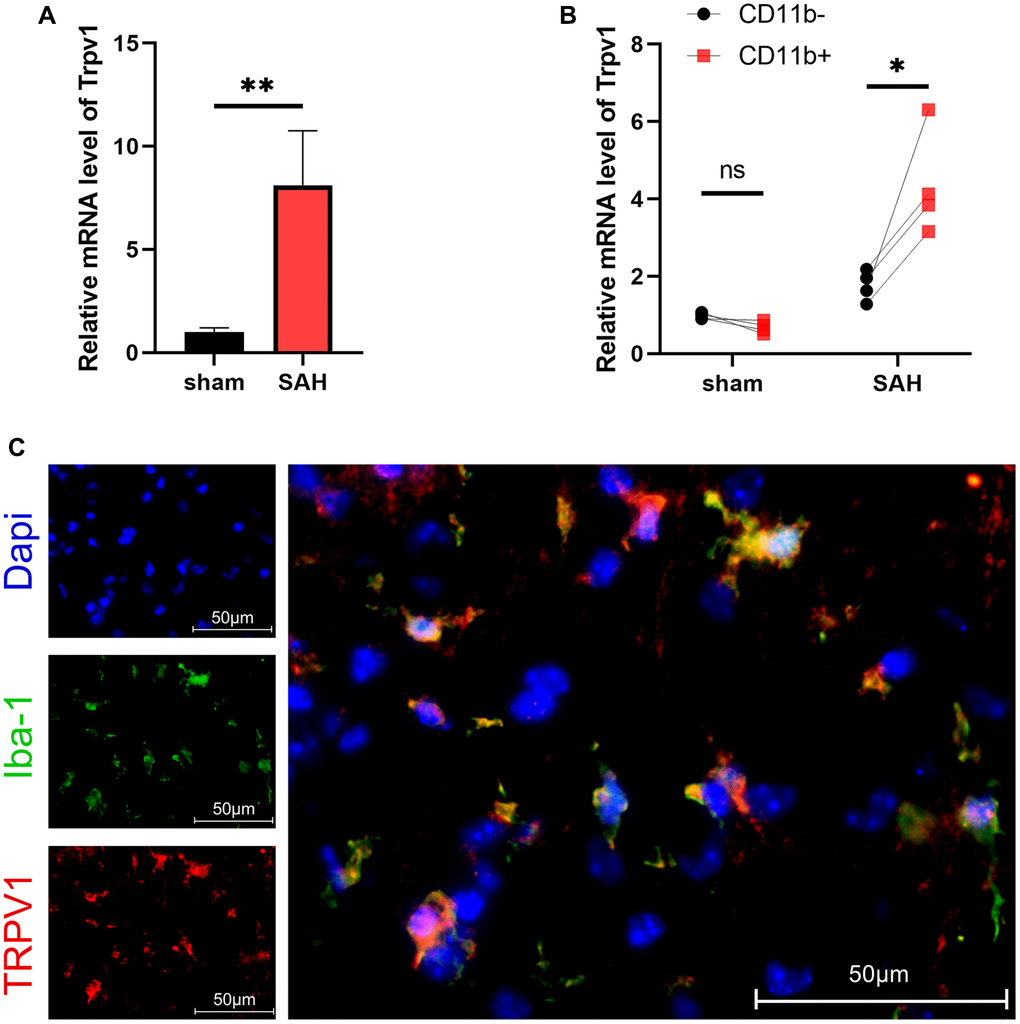
Figure 1. The expression and distribution of TRPV1. (A, B) The mRNA level of Trpv1 in brain tissues or CD11b positive/negative cells isolated via MACS was assessed by qPCR at 24 h post-SAH. (C) Representative immunofluorescence images showed the co-localization of TRPV1 with Iba-1. Abbreviation: Abbreviation: no significance; *p < 0.05 and **p < 0.01. Scale bar = 50 μm.
Antagonism of TRPV1 attenuated neurological dysfunction
We conducted follow-up studies to investigate the effect of TRPV1 on neurological impairment, CPZ, a selective TRPV1 antagonist, was introduced. Neurobehavioral scores including modified Garcia scores and beam scores showed severe neurological dysfunction after SAH when compared with the sham group. However, blockage of TRPV1 with CPZ improved the neurological dysfunction (Figure 2A, 2B). The administration of CPZ dramatically reduced the brain water content and upregulated the expression of tight junction proteins including ZO-1, occluding, and claudin-5 (Figure 2C–2G). Nissl staining also showed that CPZ increased the number of neurons (Figure 2H, 2I).
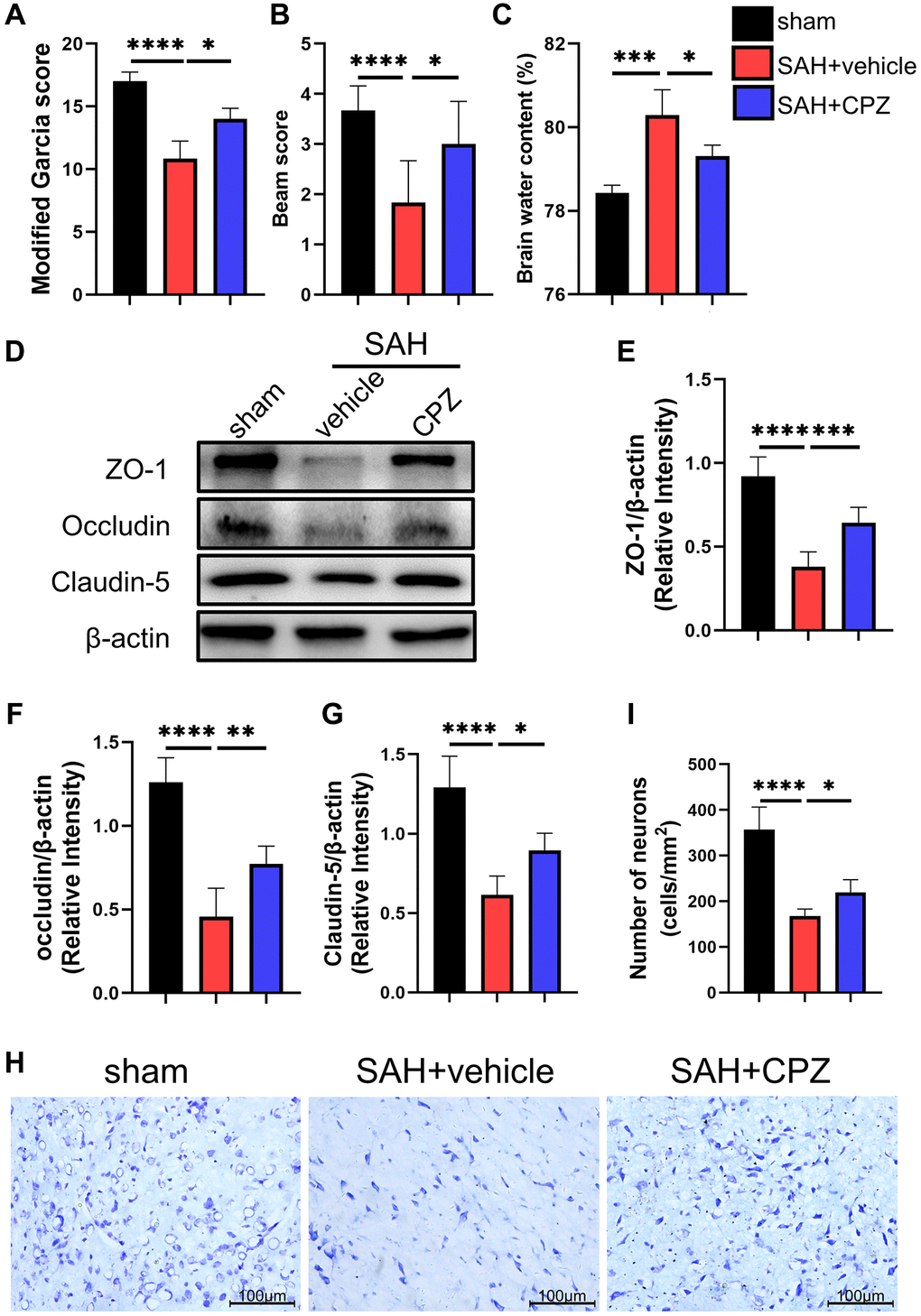
Figure 2. Effect of TRPV1 on neurological function, brain edema, and neuron injury. (A, B) Neurological function at 24 h post-SAH was evaluated via modified Garcia and beam score. (C) The water content of the left hemisphere at 24 h post-SAH. (D–G) The relative protein expression of ZO-1, Occludin, and Claudin-5 at 24 h post-SAH was assessed via western blot. (H, I) The neuron number at 24 h post-SAH was assessed via Nissl stain. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. Scale bar = 100 μm.
Antagonism of TRPV1 alleviated the neuroinflammation
We have learned that TRPV1 activation affects nerve function damage. Can the inhibition of TRPV1 alleviate nerve function damage? We have conducted further research on the possible mechanism of TRPV1 in alleviating nerve function damage. Immunofluorescence staining demonstrated that the number of CD16/32 positive microglia/macrophages increased after SAH and reduced after treatment with CPZ (Figure 3A). Consistent with immunofluorescence staining, the mRNA level of CD16, CD32, CD86, IL-1β, and TNF-α was elevated in the SAH+vehicle group, but was reduced in the SAH+CPZ group (Figure 3B–3F). Subsequently, we investigated whether TRPV1 was involved in the activation of the NLRP3 inflammasome. The western blot results showed that the expression of NLRP3, ASC, and caspase-1 p20 were increased in the SAH+vehicle group and reduced in the SAH+CPZ group (Figure 4A–4D). The level of IL-1β showed a similar change as determined via ELISA (Figure 4E).
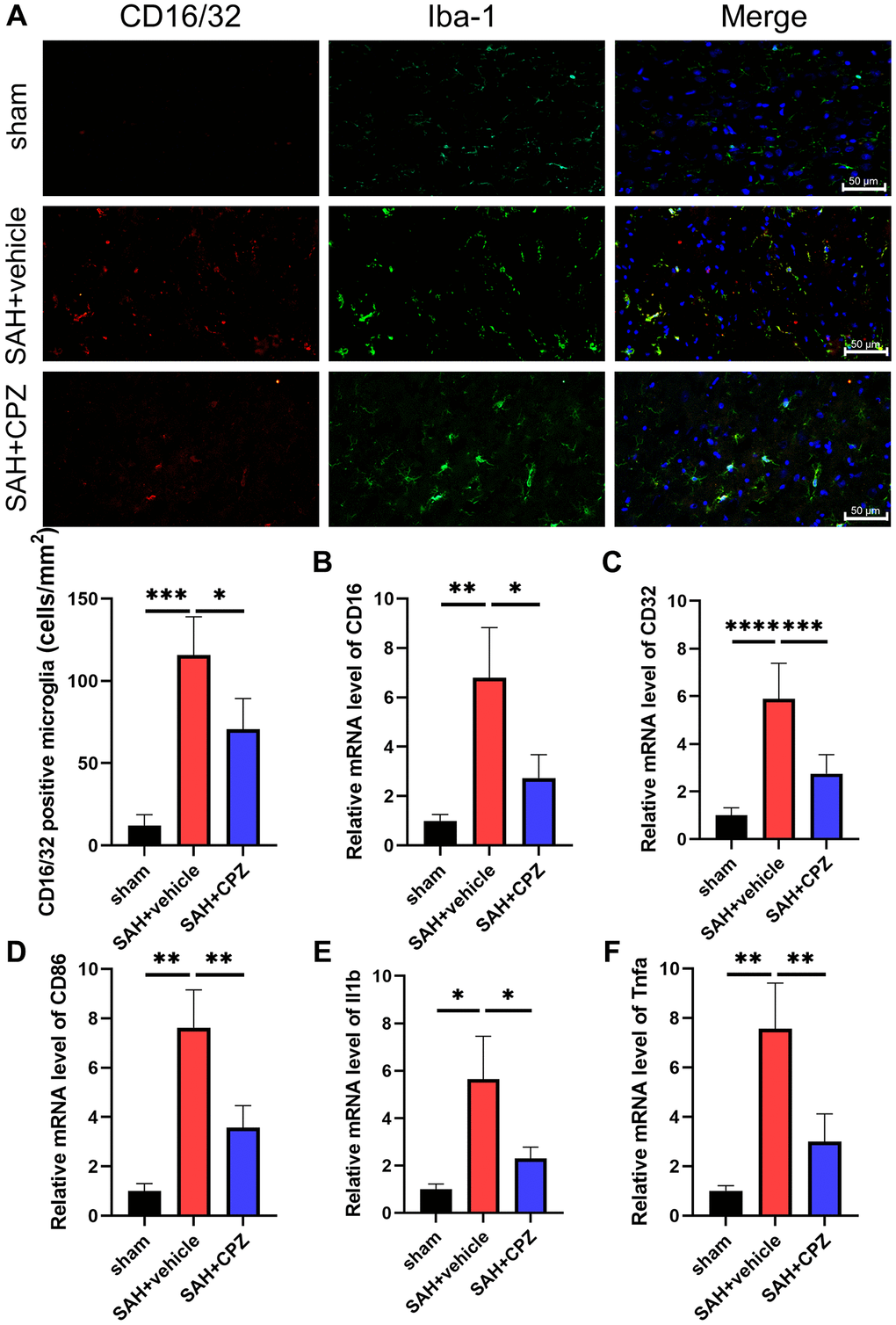
Figure 3. Effect of TRPV1 on inflammation response after SAH. (A) The immunofluorescence showed the number of CD16/32 positive microglia/macrophages at 24 h post-SAH. (B–F) The mRNA level of CD16, CD32, CD86, IL-1β, and TNF-α in brain tissues at 24 h post-SAH. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. Scale bar = 50 μm.
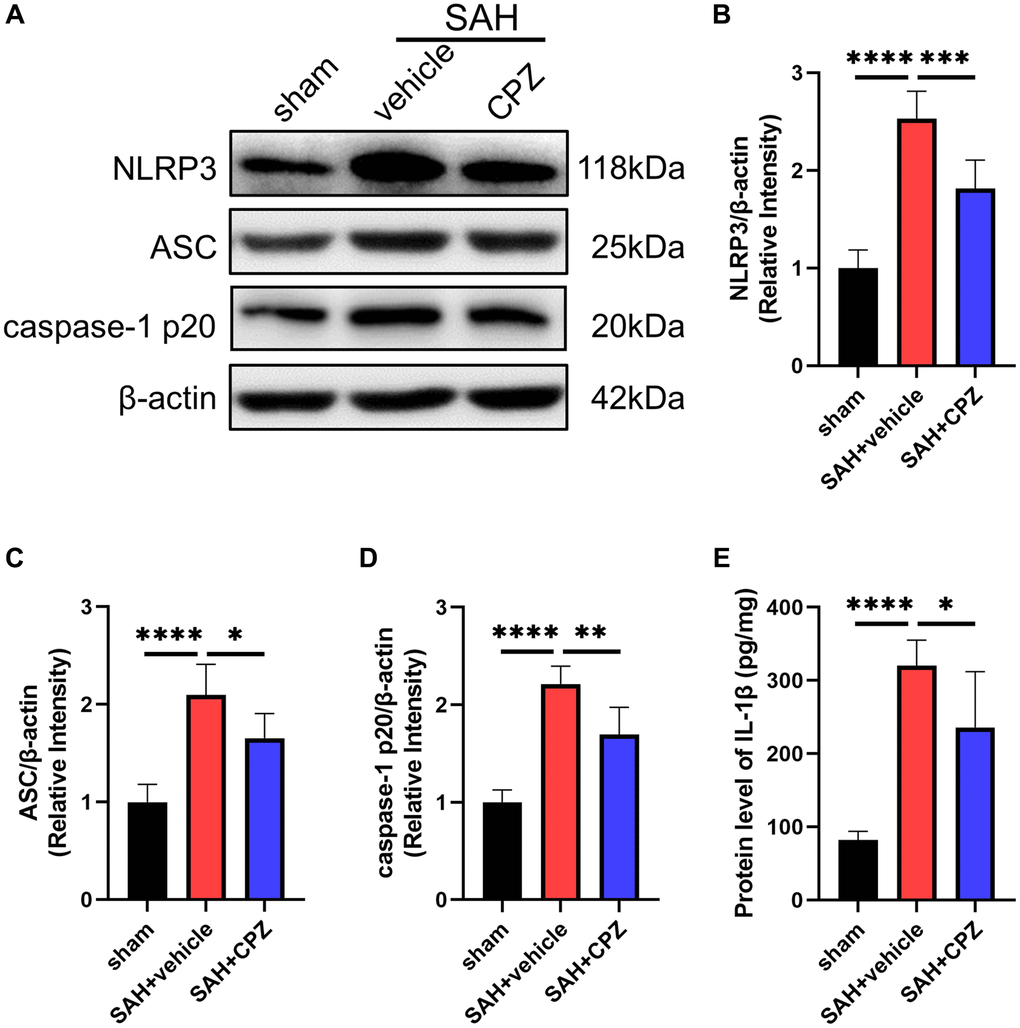
Figure 4. Effect of TRPV1 on NLRP3 inflammasome activation. (A–D) The relative protein expression of NLRP3, ASC, and caspase-1 p20 at 24 h post-SAH was assessed via western blot. (E) The protein level of IL-1β at 24 h post-SAH was assessed via ELISA. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
TRPV1 enhanced neuroinflammation via the NLRP3 inflammasome
To further investigate the effect of NLRP3 inflammasome on TRPV1-induced neuroinflammation, CAP, a selective TRPV1 agonist, and MCC950, a selective TRPV1 antagonist, were used to activate TRPV1 and inhibit NLRP3, respectively. Administration of CAP decreased the modified Garcia and beam scores and increased the brain water content compared with the SAH+vehicle group, while inhibition of NLRP3 with MCC950 reversed the aggravation of neurobehavioral assessment and brain water content (Figure 5A–5C). Data from ELISA and qPCR confirmed that MCC950 inhibited the elevation of the level of IL-1β, TNF-α, CD16, CD32, and CD86 induced by CAP (Figure 5D–5I).
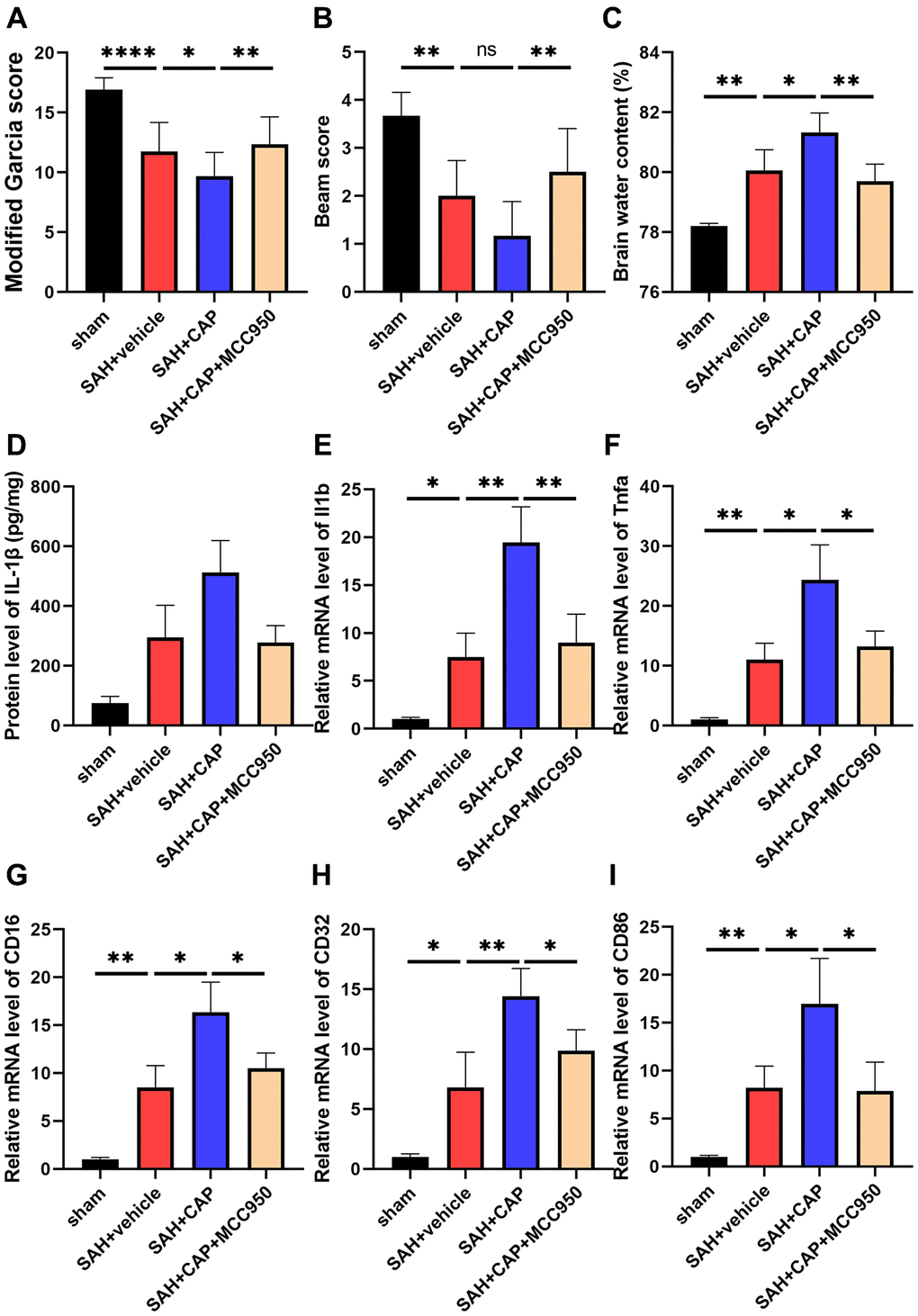
Figure 5. Effect of NLRP3 on TRPV1 mediated neurological dysfunction and inflammatory response at 24 h post SAH. (A, B) Neurological function was valued via modified Garcia and beam score. (C) The water content of the left hemisphere. (D) The protein level of IL-1β was assessed via ELISA. (E–I) The mRNA level of CD16, CD32, CD86, IL-1β, and TNF-α in brain tissues was assessed via qPCR. Abbreviation: no significance; *p < 0.05, **p < 0.01 and ****p < 0.0001.
TRPV1 activated NLRP3 inflammasome via calcium
Given TRPV1’s essential role in regulating calcium homeostasis and the involvement of calcium in NLRP3 inflammasome activation, calcium was introduced to investigate the underlying mechanism by which TRPV1 induced NLRP3 inflammasome activation. The calcium content of microglia was dramatically increased when stimulated with hemin, while it was reduced when treated with CPZ (Figure 6A). Chelation of calcium via BAPTA-AM reversed the increased levels of NLRP3, ASC, caspase-1 p20, and IL-1β induced by hemin (Figure 6B–6F).
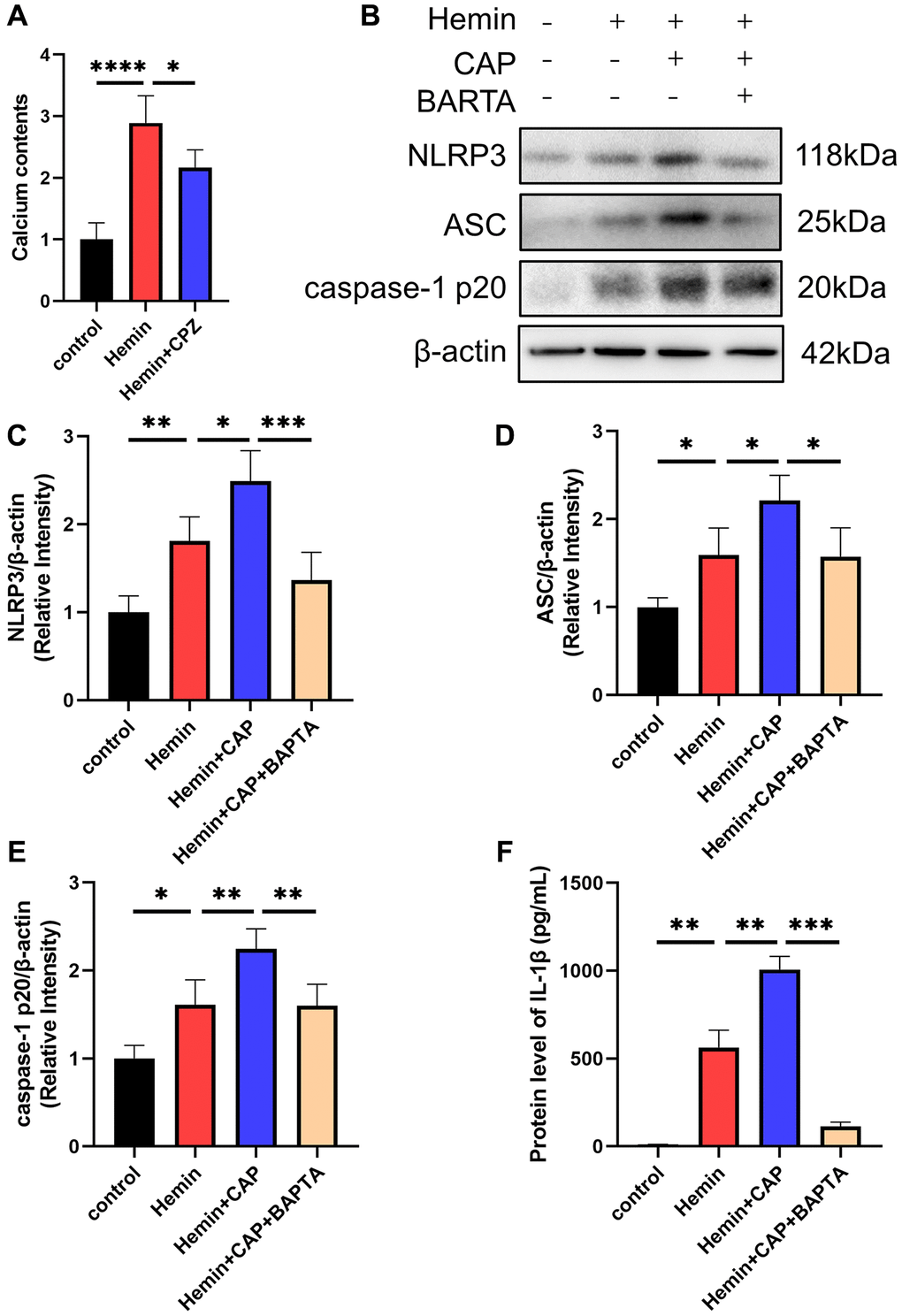
Figure 6. Effect of calcium on TRPV1 mediated NLRP3 inflammasome activation. (A) The calcium content of BV2 cells was assessed at 24 h after modeling. (B–E) The relative protein expression of NLRP3, ASC, and caspase-1 p20 in BV2 cells at 24 h post modeling was assessed via western blot. (F) The protein level of IL-1β in BV2 cells supernatant at 24 h post modeling was assessed via ELISA. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Discussion
As TRPV1 is not only expressed in peripheral sensory neurons but also widely expressed in the central nervous system, attention has gradually moved beyond its classical role as a pain sensor to focus on its role in the central nervous system [34–36]. Although many studies have pointed to the indispensability of TRPV1 in the central nervous system, particularly in the pathology of the inflammatory response, it is a puzzle to conclude exactly what role it plays, as it exhibits deteriorated or protective effects in the context of diseases [37, 38]. For deteriorated effects, the selective TRPV1 antagonists CPZ inhibited the morphine-induced increased expression of p38 MAPK and NF-κB [39]. Additionally, Miyanohara et al. confirmed that both TRPV1 knockout and CPZ showed an alleviated neurological function after stroke [40]. For the protective effects, TRPV1 suppressed the oxidative stress and enhanced the endogenous production of a neurotrophic factor in the neurodegeneration diseases including Parkinson’s disease [41], vascular dementia [42], and Huntington’s disease [43]. The mechanism was at least in part related to the anti-inflammatory effect [44–46]. Hence, it is essential to determine the specific effect of TRPV1 in the context of SAH. We found that CPZ inhibited TRPV1 alleviated the neurological function injury, BBB disruption and reduced the pro-inflammatory microglia, and was associated with the inflammatory response, indicating the deteriorated effects of TRPV1 after SAH.
Since TRPV1 was mainly expressed in microglia/macrophages and microglia/macrophages were thought to be one of the most important factors mediating the inflammatory response after SAH, we hypothesize that the brain injury effect of TRPV1 after SAH might be mainly due to its pro-inflammatory effect. The pro-inflammatory mechanism of TRPV1 remains unclear, however, there were some recent studies linking it to NLRP3 inflammasome [25–27, 47]. Moreover, growing evidence suggested that the NLRP3 inflammasome was involved in the inflammatory response after SAH, that inhibition of the NLRP3 inflammasome significantly alleviates neurological dysfunction, and that the NLRP3 inflammasome activation mechanisms after SAH related to SIRT1, NRF2, AMPK, etc., [8, 48–50]. However, the effect of TRPV1 on the NLRP3 inflammasome activation has not been determined in SAH. Our data confirmed that the expression of NLRP3 inflammasome-related proteins NLRP3, ASC, and caspase-1 p20 were down-regulated after pharmacological antagonism of TRPV1, and the level of inflammatory cytokine IL-1β was significantly decreased. Furthermore, inhibiting the NLRP3 inflammasome abolished the TRPV1 mediating the neurological dysfunction and inflammatory response. Since calcium ions can mediate NLRP3 inflammasome activation and TRPV1 can act as a non-selective calcium channel, we next explored the role of calcium ions on TRPV1-mediated NLRP3 inflammasome activation in vitro. The data showed that suppressing the TRPV1 reduced the calcium ion contents in BV2 cells. Moreover, chelated calcium ions reversed the TRPV1 mediating NLRP3 inflammasome activation. Previous studies have found that two mechanisms might be involved in calcium-mediated NLRP3 inflammasome activation: (1) promoting the interaction between NLRP3 and ASC to mediate the activation of NLRP3 inflammasome; (2) causing mitochondrial calcium overload that leads to mitochondrial damage, which in turn released ROS, ox-mtDNA to activate the NLRP3 inflammasome [51], but the mechanism under TRPV1-mediated calcium influx and NLRP3 activation remain to be defined. Zhang et al. revealed that calcium ion influx via TRPV1 directly bound to PP2A thus to active NLRP3 inflammasome in EAE [26], however, the specific mechanism in SAH needs further study.
In conclusion, our data showed that TRPV1 modulated NLRP3 inflammasome activation via calcium and that antagonizing TRPV1 significantly improves EBI after SAH (Figure 7). Targeting TRPV1 might be a promising therapeutic strategy for SAH.
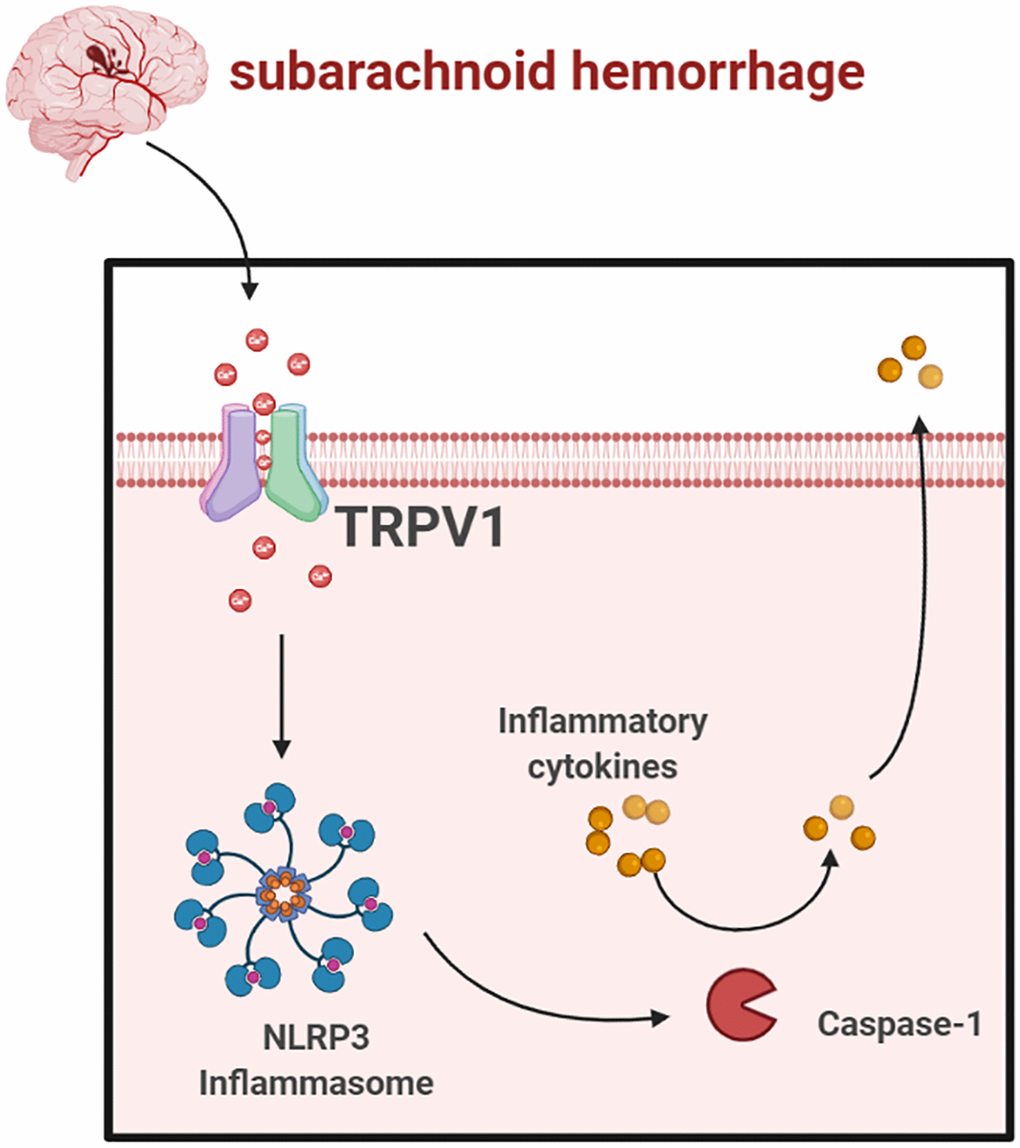
Figure 7. Schematic mechanism of TRPV1 activated NLRP3 inflammasome post-SAH (https://www.biorender.com/).
Supplementary Materials
Author Contributions
WS, ZZ, ZQ and KZ conceived and designed the research. WS, KZ, ZQ and JC constructed the SAH model, PCR, and immunofluorescence stain. WS, ZQ, JC and FW performed the experiment in vitro. XJ, JC and GG performed the western blot and Nissl staining. KZ and ZZ wrote and edited the manuscript. All authors read, corrected, and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
The experimental animal procedures were conformed to the guidelines of the National Institutes of Health on the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee of Shandong First Medical University (protocol No. 2021-109).
Funding
This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2021MH228).
References
- 1. Ji C, Chen G. Signaling Pathway in Early Brain Injury after Subarachnoid Hemorrhage: News Update. Acta Neurochir Suppl. 2016; 121:123–6. https://doi.org/10.1007/978-3-319-18497-5_21 [PubMed]
- 2. Xie Z, Huang L, Enkhjargal B, Reis C, Wan W, Tang J, Cheng Y, Zhang JH. Recombinant Netrin-1 binding UNC5B receptor attenuates neuroinflammation and brain injury via PPARγ/NFκB signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun. 2018; 69:190–202. https://doi.org/10.1016/j.bbi.2017.11.012 [PubMed]
- 3. Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015; 4:296–307. https://doi.org/10.1016/j.redox.2015.01.008 [PubMed]
- 4. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007; 7:31–40. https://doi.org/10.1038/nri1997 [PubMed]
- 5. Ren QG, Gong WG, Zhou H, Shu H, Wang YJ, Zhang ZJ. Spatial Training Ameliorates Long-Term Alzheimer's Disease-Like Pathological Deficits by Reducing NLRP3 Inflammasomes in PR5 Mice. Neurotherapeutics. 2019; 16:450–64. https://doi.org/10.1007/s13311-018-00698-w [PubMed]
- 6. Soares JL, Oliveira EM, Pontillo A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult Scler Relat Disord. 2019; 29:26–34. https://doi.org/10.1016/j.msard.2019.01.023 [PubMed]
- 7. Zhou K, Shi L, Wang Z, Zhou J, Manaenko A, Reis C, Chen S, Zhang J. RIP1-RIP3-DRP1 pathway regulates NLRP3 inflammasome activation following subarachnoid hemorrhage. Exp Neurol. 2017; 295:116–24. https://doi.org/10.1016/j.expneurol.2017.06.003 [PubMed]
- 8. Dodd WS, Noda I, Martinez M, Hosaka K, Hoh BL. NLRP3 inhibition attenuates early brain injury and delayed cerebral vasospasm after subarachnoid hemorrhage. J Neuroinflammation. 2021; 18:163. https://doi.org/10.1186/s12974-021-02207-x [PubMed]
- 9. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013; 38:1142–53. https://doi.org/10.1016/j.immuni.2013.05.016 [PubMed]
- 10. Green JP, Yu S, Martín-Sánchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, Brough D. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci U S A. 2018; 115:E9371–80. https://doi.org/10.1073/pnas.1812744115 [PubMed]
- 11. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012; 492:123–7. https://doi.org/10.1038/nature11588 [PubMed]
- 12. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012; 109:11282–7. https://doi.org/10.1073/pnas.1117765109 [PubMed]
- 13. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007; 14:1583–9. https://doi.org/10.1038/sj.cdd.4402195 [PubMed]
- 14. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, Cui J, Bai L, Wang J, Jiang W, Zhou R. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. 2017; 8:202. https://doi.org/10.1038/s41467-017-00227-x [PubMed]
- 15. Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, Unutmaz D, Feske S. Ca2+ Signaling but Not Store-Operated Ca2+ Entry Is Required for the Function of Macrophages and Dendritic Cells. J Immunol. 2015; 195:1202–17. https://doi.org/10.4049/jimmunol.1403013 [PubMed]
- 16. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, Yu JW, Alnemri ES. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013; 191:4358–66. https://doi.org/10.4049/jimmunol.1301170 [PubMed]
- 17. Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J Biol Chem. 2014; 289:32926–36. https://doi.org/10.1074/jbc.M114.579961 [PubMed]
- 18. Aminzadeh M, Roghani M, Sarfallah A, Riazi GH. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer's disease. Int Immunopharmacol. 2018; 54:78–85. https://doi.org/10.1016/j.intimp.2017.10.024 [PubMed]
- 19. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013; 4:1611. https://doi.org/10.1038/ncomms2608 [PubMed]
- 20. Tseng HH, Vong CT, Kwan YW, Lee SM, Hoi MP. TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci Rep. 2016; 6:35016. https://doi.org/10.1038/srep35016 [PubMed]
- 21. Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, Montero MT, Herranz AS, Bazán E, Reimers D, Mulero V, Pelegrín P. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity. 2012; 37:487–500. https://doi.org/10.1016/j.immuni.2012.06.013 [PubMed]
- 22. Wang M, Zhang Y, Xu M, Zhang H, Chen Y, Chung KF, Adcock IM, Li F. Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic Biol Med. 2019; 134:229–38. https://doi.org/10.1016/j.freeradbiomed.2019.01.004 [PubMed]
- 23. Ho KW, Ward NJ, Calkins DJ. TRPV1: a stress response protein in the central nervous system. Am J Neurodegener Dis. 2012; 1:1–14. [PubMed]
- 24. Martins D, Tavares I, Morgado C. "Hotheaded": the role OF TRPV1 in brain functions. Neuropharmacology. 2014; 85:151–7. https://doi.org/10.1016/j.neuropharm.2014.05.034 [PubMed]
- 25. Lin Y, Huang T, Shen W, Pang Q, Xie Q, Chen X, Tu F. TRPV1 Suppressed NLRP3 Through Regulating Autophagy in Microglia After Ischemia-Reperfusion Injury. J Mol Neurosci. 2022; 72:792–801. https://doi.org/10.1007/s12031-021-01935-2 [PubMed]
- 26. Zhang Y, Hou B, Liang P, Lu X, Wu Y, Zhang X, Fan Y, Liu Y, Chen T, Liu W, Peng B, Yin J, Han S, He X. TRPV1 channel mediates NLRP3 inflammasome-dependent neuroinflammation in microglia. Cell Death Dis. 2021; 12:1159. https://doi.org/10.1038/s41419-021-04450-9 [PubMed]
- 27. Luo J, Chen J, Yang C, Tan J, Zhao J, Jiang N, Zhao Y. 6-Gingerol protects against cerebral ischemia/ reperfusion injury by inhibiting NLRP3 inflammasome and apoptosis via TRPV1 / FAF1 complex dissociation-mediated autophagy. Int Immunopharmacol. 2021; 100:108146. https://doi.org/10.1016/j.intimp.2021.108146 [PubMed]
- 28. Zeng H, Chen H, Li M, Zhuang J, Peng Y, Zhou H, Xu C, Yu Q, Fu X, Cao S, Cai J, Yan F, Chen G. Autophagy protein NRBF2 attenuates endoplasmic reticulum stress-associated neuroinflammation and oxidative stress via promoting autophagosome maturation by interacting with Rab7 after SAH. J Neuroinflammation. 2021; 18:210. https://doi.org/10.1186/s12974-021-02270-4 [PubMed]
- 29. Anandakumar P, Kamaraj S, Jagan S, Ramakrishnan G, Devaki T. Capsaicin provokes apoptosis and restricts benzo(a)pyrene induced lung tumorigenesis in Swiss albino mice. Int Immunopharmacol. 2013; 17:254–9. https://doi.org/10.1016/j.intimp.2013.05.015 [PubMed]
- 30. Wu X, Zeng H, Xu C, Chen H, Fan L, Zhou H, Yu Q, Fu X, Peng Y, Yan F, Yu X, Chen G. TREM1 Regulates Neuroinflammatory Injury by Modulate Proinflammatory Subtype Transition of Microglia and Formation of Neutrophil Extracellular Traps via Interaction With SYK in Experimental Subarachnoid Hemorrhage. Front Immunol. 2021; 12:766178. https://doi.org/10.3389/fimmu.2021.766178 [PubMed]
- 31. Chen H, Yu X, Hu L, Peng Y, Yu Q, Zhou H, Xu C, Zeng H, Cao Y, Zhuang J, Fu X, Zhou G, Li J, et al. Activation of Nurr1 with Amodiaquine Protected Neuron and Alleviated Neuroinflammation after Subarachnoid Hemorrhage in Rats. Oxidative Medicine and Cellular Longevity. 2021; 2021. https://doi.org/10.1155/2021/6669787
- 32. Wang C, Huang W, Lu J, Chen H, Yu Z. TRPV1-Mediated Microglial Autophagy Attenuates Alzheimer's Disease-Associated Pathology and Cognitive Decline. Front Pharmacol. 2022; 12:763866. https://doi.org/10.3389/fphar.2021.763866 [PubMed]
- 33. Sha'fie MSA, Rathakrishnan S, Hazanol IN, Dali MHI, Khayat ME, Ahmad S, Hussin Y, Alitheen NB, Jiang LH, Syed Mortadza SA. Ethanol Induces Microglial Cell Death via the NOX/ROS/PARP/TRPM2 Signalling Pathway. Antioxidants (Basel). 2020; 9:1253. https://doi.org/10.3390/antiox9121253 [PubMed]
- 34. Tsuji F, Murai M, Oki K, Seki I, Ueda K, Inoue H, Nagelkerken L, Sasano M, Aono H. Transient receptor potential vanilloid 1 agonists as candidates for anti-inflammatory and immunomodulatory agents. Eur J Pharmacol. 2010; 627:332–9. https://doi.org/10.1016/j.ejphar.2009.10.044 [PubMed]
- 35. Balleza-Tapia H, Crux S, Andrade-Talavera Y, Dolz-Gaiton P, Papadia D, Chen G, Johansson J, Fisahn A. TrpV1 receptor activation rescues neuronal function and network gamma oscillations from Aβ-induced impairment in mouse hippocampus in vitro. Elife. 2018; 7:e37703. https://doi.org/10.7554/eLife.37703 [PubMed]
- 36. Marrone MC, Morabito A, Giustizieri M, Chiurchiù V, Leuti A, Mattioli M, Marinelli S, Riganti L, Lombardi M, Murana E, Totaro A, Piomelli D, Ragozzino D, et al. TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat Commun. 2017; 8:15292. https://doi.org/10.1038/ncomms15292 [PubMed]
- 37. Alawi K, Keeble J. The paradoxical role of the transient receptor potential vanilloid 1 receptor in inflammation. Pharmacol Ther. 2010; 125:181–95. https://doi.org/10.1016/j.pharmthera.2009.10.005 [PubMed]
- 38. Musumeci G, Grasselli G, Rossi S, De Chiara V, Musella A, Motta C, Studer V, Bernardi G, Haji N, Sepman H, Fresegna D, Maccarrone M, Mandolesi G, Centonze D. Transient receptor potential vanilloid 1 channels modulate the synaptic effects of TNF-α and of IL-1β in experimental autoimmune encephalomyelitis. Neurobiol Dis. 2011; 43:669–77. https://doi.org/10.1016/j.nbd.2011.05.018 [PubMed]
- 39. Nguyen TL, Kwon SH, Hong SI, Ma SX, Jung YH, Hwang JY, Kim HC, Lee SY, Jang CG. Transient receptor potential vanilloid type 1 channel may modulate opioid reward. Neuropsychopharmacology. 2014; 39:2414–22. https://doi.org/10.1038/npp.2014.90 [PubMed]
- 40. Miyanohara J, Shirakawa H, Sanpei K, Nakagawa T, Kaneko S. A pathophysiological role of TRPV1 in ischemic injury after transient focal cerebral ischemia in mice. Biochem Biophys Res Commun. 2015; 467:478–83. https://doi.org/10.1016/j.bbrc.2015.10.027 [PubMed]
- 41. Nam JH, Park ES, Won SY, Lee YA, Kim KI, Jeong JY, Baek JY, Cho EJ, Jin M, Chung YC, Lee BD, Kim SH, Kim EG, et al. TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson's disease via CNTF. Brain. 2015; 138:3610–22. https://doi.org/10.1093/brain/awv297 [PubMed]
- 42. Gupta S, Sharma B. Pharmacological modulation of I(1)-imidazoline and α2-adrenoceptors in sub acute brain ischemia induced vascular dementia. Eur J Pharmacol. 2014; 723:80–90. https://doi.org/10.1016/j.ejphar.2013.12.003 [PubMed]
- 43. Gupta S, Sharma B. Pharmacological benefits of agomelatine and vanillin in experimental model of Huntington's disease. Pharmacol Biochem Behav. 2014; 122:122–35. https://doi.org/10.1016/j.pbb.2014.03.022 [PubMed]
- 44. Chang YH, Lee ST, Lin WW. Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem. 2001; 81:715–23. https://doi.org/10.1002/jcb.1103 [PubMed]
- 45. Chen CW, Lee ST, Wu WT, Fu WM, Ho FM, Lin WW. Signal transduction for inhibition of inducible nitric oxide synthase and cyclooxygenase-2 induction by capsaicin and related analogs in macrophages. Br J Pharmacol. 2003; 140:1077–87. https://doi.org/10.1038/sj.bjp.0705533 [PubMed]
- 46. Kim CS, Kawada T, Kim BS, Han IS, Choe SY, Kurata T, Yu R. Capsaicin exhibits anti-inflammatory property by inhibiting IkB-a degradation in LPS-stimulated peritoneal macrophages. Cell Signal. 2003; 15:299–306. https://doi.org/10.1016/s0898-6568(02)00086-4 [PubMed]
- 47. Yin N, Gao Q, Tao W, Chen J, Bi J, Ding F, Wang Z. Paeoniflorin relieves LPS-induced inflammatory pain in mice by inhibiting NLRP3 inflammasome activation via transient receptor potential vanilloid 1. J Leukoc Biol. 2020; 108:229–41. https://doi.org/10.1002/JLB.3MA0220-355R [PubMed]
- 48. Jin L, Jin F, Guo S, Liu W, Wei B, Fan H, Li G, Zhang X, Su S, Li R, Fang D, Duan C, Li X. Metformin Inhibits NLR Family Pyrin Domain Containing 3 (NLRP)-Relevant Neuroinflammation via an Adenosine-5'-Monophosphate-Activated Protein Kinase (AMPK)-Dependent Pathway to Alleviate Early Brain Injury After Subarachnoid Hemorrhage in Mice. Front Pharmacol. 2022; 13:796616. https://doi.org/10.3389/fphar.2022.796616 [PubMed]
- 49. Xia DY, Yuan JL, Jiang XC, Qi M, Lai NS, Wu LY, Zhang XS. SIRT1 Promotes M2 Microglia Polarization via Reducing ROS-Mediated NLRP3 Inflammasome Signaling After Subarachnoid Hemorrhage. Front Immunol. 2021; 12:770744. https://doi.org/10.3389/fimmu.2021.770744 [PubMed]
- 50. Zhang ZH, Liu JQ, Hu CD, Zhao XT, Qin FY, Zhuang Z, Zhang XS. Luteolin Confers Cerebroprotection after Subarachnoid Hemorrhage by Suppression of NLPR3 Inflammasome Activation through Nrf2-Dependent Pathway. Oxid Med Cell Longev. 2021; 2021:5838101. https://doi.org/10.1155/2021/5838101 [PubMed]
- 51. Li C, Chen M, He X, Ouyang D. A mini-review on ion fluxes that regulate NLRP3 inflammasome activation. Acta Biochim Biophys Sin (Shanghai). 2021; 53:131–9. https://doi.org/10.1093/abbs/gmaa155 [PubMed]