



Introduction
Sleep is a basic condition essential for human health [1]. However, owing to changes in anatomy and hormone levels, as well as because of psychological factors, pregnant women are more susceptible to sleep dysfunction [2], especially during late pregnancy [3]. Sleep inadequate during late pregnancy not only increases the risk of hypertension and postpartum depression in pregnant women but also has a negative effect on the development of offspring [4]. Several basic studies have documented that sleep deprivation (SD) during pregnancy in rodents can affect the neurodevelopment of the offspring, manifesting as cognitive impairment, increase in anxiety-like behaviors, and immature sleep patterns [5, 6].
Maternal sleep deprivation (MSD) can lead to the reprogramming of the immune cells of the brain in utero, disrupt neurogenesis, impair synaptic plasticity, and activate microglia in later life, subsequently increasing the risk of cognitive impairment in the offspring during adolescence or adulthood [6–8]. However, relatively few studies have explored the effects of MSD on cognitive function in aging offspring, including the underlying mechanisms. We previously provided preliminary evidence indicating that the adverse effects of late-pregnancy SD on cognitive function in offspring can persist into old age, with mechanisms involving neuroinflammation [9]. However, the mechanisms responsible for cognition impairment in aged offspring induced by SD during late pregnancy remain largely unknown.
The maintenance and regulation of cognitive function largely rely on two critical brain regions, namely, the prefrontal cortex and the hippocampus [10]. The brain is susceptible to oxidative stress owing to its high oxygen consumption, high polyunsaturated fatty acid content, and low antioxidant defense capacity [11]. Growing evidence suggests that cognitive dysfunction is closely associated with oxidative stress in the brain. Indeed, neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease, and amyotrophic lateral sclerosis are accompanied by a significant increase in oxidative stress, as showed by the elevated expression levels of reactive oxygen species (ROS) and malondialdehyde (MDA) and the decrease in the contents of antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase [12, 13]. Inflammation also plays an important role in cognitive disorders. Preclinical and clinical studies have both shown that cognitive impairment induced by AD is accompanied by elevated levels of pro-inflammatory cytokines including interleukin-1 beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) [14, 15]. Together, these evidences suggest that oxidative stress and inflammation may be important mechanisms leading to cognitive impairment.
Mitochondrial biogenesis disruption is closely related to oxidative stress and neuroinflammation [16]. Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) is a transcription factor involved in the regulation of mitochondrial biogenesis and antioxidant damage in the nervous system, thus contributing to the regulation of cognitive impairment induced by various neurodegenerative diseases [17]. The transcription factor Sirtuin1 (Sirt1) can activate PGC-1α by deacetylation, resulting in increased synthesis and secretion of synaptic proteins in the brain, and thereby mitigating cognitive decline [18, 19]. The activated Sirt1/PGC-1α pathway can improve cognitive deficits induced by chronic cerebral hypoperfusion, scopolamine, or a high-fat diet by modulating oxidative stress and neuroinflammation [18–20]. This suggests that the Sirt1/PGC-1α pathway has potential as a therapeutic target for improving cognitive impairment induced by SD in late pregnancy. Treatment for cognitive impairment in older offspring is critical to the health of older patients. However, no drugs with a clear therapeutic target have been identified, and therefore, non-pharmacological treatments have the potential to be a promising therapeutic modality.
Environmental enrichment (EE) is an effective and simple non-pharmacological intervention for improving cognitive dysfunction by providing increased opportunities for social interaction, exercise, and somatosensory stimulation [21]. EE can not only alleviate or even prevent cognitive dysfunction in normal aging [22] but can also counteract cognitive deficits caused by early-life stress [23]. The mechanisms by which EE ameliorates cognitive deficits, at least in part, were through improvements in neuroinflammation, oxidative stress, and mitochondrial function, as well as increases in hippocampal synaptic plasticity [24–26]. Our preliminary results suggested that cognitive impairment in aging offspring resulting from MSD can be alleviated by long-term EE [9]. However, how EE affects cognitive impairment in aging offspring resulting from gestational exposure to SD is unclear, as are the putative accompanying changes in mitochondrial function, oxidative stress, and the Sirt1/PGC-1α pathway.
In this study, we explored whether SD during late pregnancy accelerates cognitive decline in aging offspring mice and, if so, whether long-term EE can ameliorate cognitive impairment through improving oxidative stress and neuroinflammation in a Sirt1/PGC-1α pathway-dependent manner.
Results
EE improved MSD-induced learning and memory decline
In the acquisition phase of the MWM test, the results showed no sex differences in escape latency and swim distance for the control and other treatment groups (Figure 1A, 1B and Supplementary Figures 1A, 1B, 2A, 2B). However, marked differences in latency and swim distance were observed between treatments in all three groups (latency: male: F(2, 21) = 11.46, P < 0.01; female: F(2, 21) = 13.68, P < 0.01; distance: male: F(2, 21) = 10.18; female: F(2, 21) = 18.94, P < 0.01; Figure 1D, 1E, 1G, 1H). Post hoc comparisons showed that the escape latency and swim distance were increased in the MSD group when compared with the Control group (Ps < 0.05). However, the escape latency and swim distance were shorter in the MSD+EE group than in the MSD group (Ps < 0.05). No difference in swimming velocity among the groups whether controlling for sex or treatment (Figure 1C, 1F, 1I and Supplementary Figures 1C, 2C).
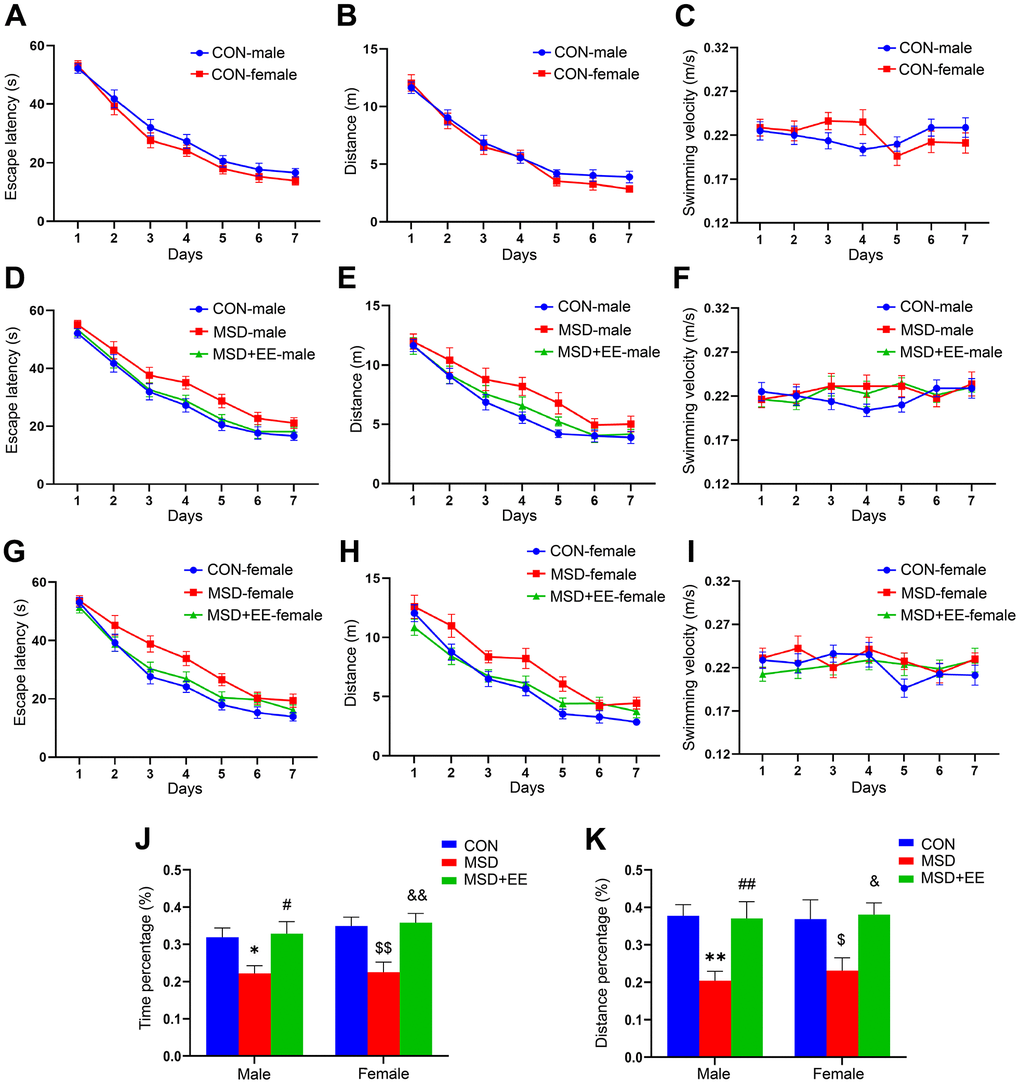
Figure 1. The effects of EE on MSD-induced changes in cognitive performance (n = 8). Escape latency, swim distance and velocity of control (CON) group (A–C), males (D–F), and females (G–I). (J) Percent time spent in the target quadrant. (K) Percent distance swam in the target quadrant. *P < 0.05 and **P< 0.01 vs. male mice in the CON group; #P < 0.05 and ##P < 0.01 vs. male mice in the MSD group; $P < 0.05 and $$P < 0.01 vs. female mice in the CON group; &P < 0.05 and &&P < 0.01 vs. female mice in the MSD group.
In the memory phase of the MWM test, the percentage of time spent and the distance swam in the target quadrant differed in the three groups (precent of time: F(2, 42) = 13.23, P < 0.01; percent of distance: F(2, 42) = 11.80, P < 0.01; Figure 1J, 1K). Furthermore, the time and distance percentages were clearly lower in the MSD group than in controls (Ps < 0.05) but markedly higher in the MSD+EE group than in the MSD group (Ps < 0.05).
EE improved MSD-induced oxidative stress
In the hippocampus and prefrontal cortex, the levels of SOD, MDA, and ROS were obviously different in all groups (hippocampus: SOD: F(2, 42) = 28.73, P < 0.01; MDA: F(2, 42) = 63.83, P < 0.01; ROS: F(2, 30) = 17.16, P < 0.01; prefrontal cortex: SOD: F(2, 42) = 48.86, P < 0.01; MDA: F(2, 42) = 55.12, P < 0.01; ROS: F(2, 30) = 24.45, P < 0.01; Figure 2A–2F). Compared with the control group, MDA and ROS levels in both brain regions were significantly increased in the MSD group, whereas SOD levels were decreased (Ps < 0.05). However, MDA and ROS levels were lower in the MSD+EE group than in the MSD group in both brain regions, whereas the opposite was observed for SOD content (Ps < 0.05).
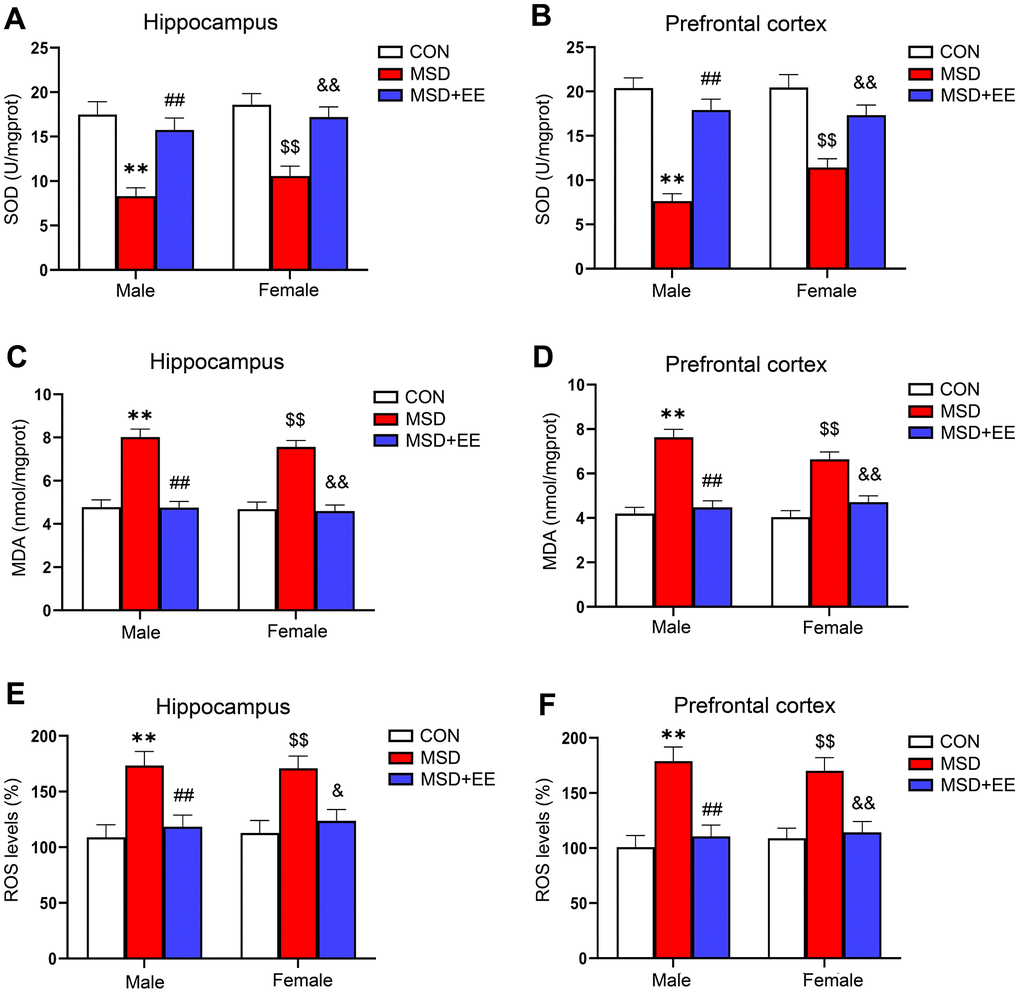
Figure 2. The effect of EE on change in MSD-induced oxidative stress levels (n = 8). The level of SOD in the hippocampus (A) and prefrontal cortex (B). The level of MDA in the hippocampus (C) and prefrontal cortex (D). The level of ROS in the hippocampus (E) and prefrontal cortex (F). **P < 0.01 vs. males in the control (CON) group; ##P < 0.01 vs. males in the MSD group; $$P < 0.01 vs. females in the CON group; &P < 0.05 and &&P < 0.01 vs. females in the MSD group.
EE alleviated MSD-induced inflammation
In the hippocampus and prefrontal cortex, the IL-1β, IL-6, and TNF-α levels differed among the groups (hippocampus: IL-1β: F(2, 42) = 48.44, P < 0.01; IL-6: F(2, 42) = 57.38, P < 0.01; TNF-α: F(2, 42) = 12.52, P < 0.01; prefrontal cortex: IL-1β: F(2, 42) = 20.42, P < 0.01; IL-6: F(2, 42) = 51.13, P < 0.01; TNF-α: F(2, 42) = 13.67, P < 0.01; Figure 3A–3F). Additionally, the IL-1β, IL-6, and TNF-α levels were increased in the MSD group relative to Control group (Ps < 0.05). Nevertheless, EE significantly mitigated these increases (Ps < 0.05).
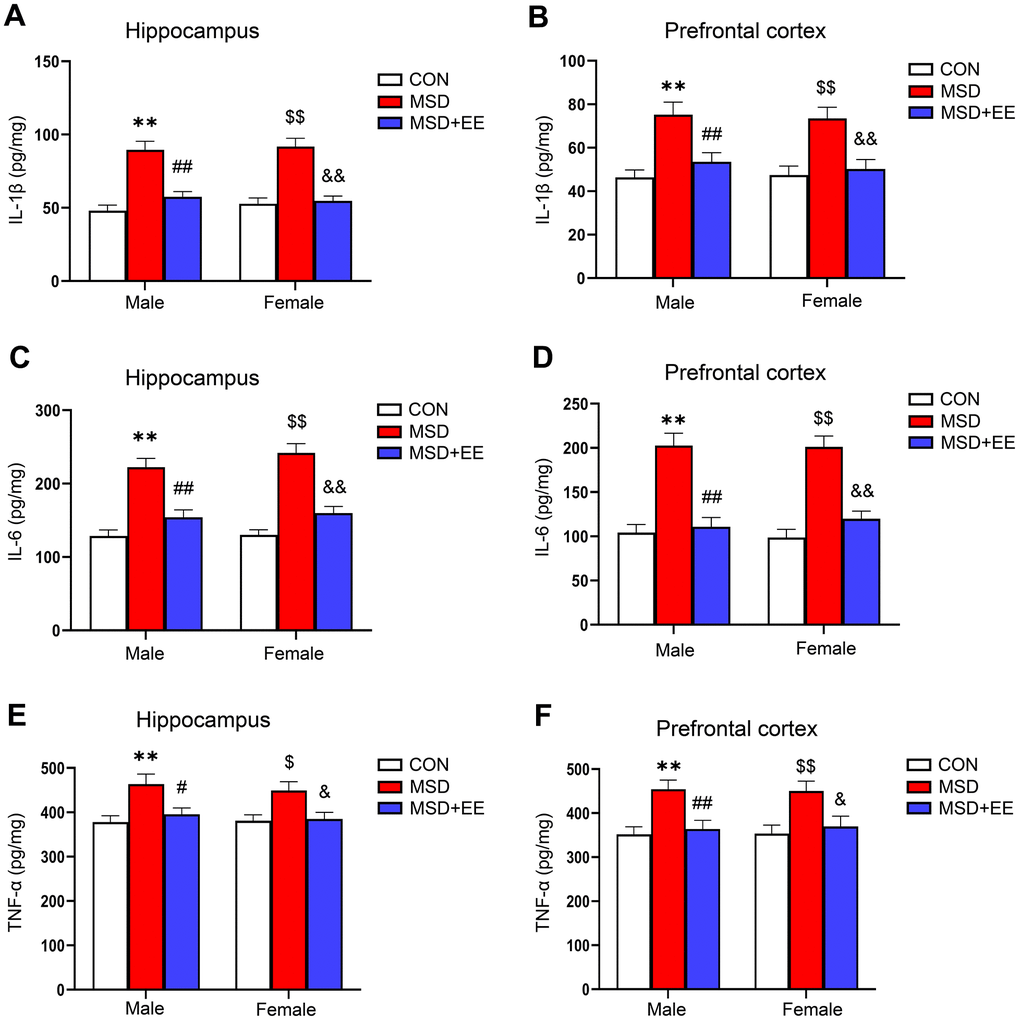
Figure 3. The effect of EE on the increased levels of proinflammation cytokines of aging mice induced by MSD (n = 8). The level of IL-1β in the hippocampus (A) and prefrontal cortex (B). The level of IL-6 in the hippocampus (C) and prefrontal cortex (D). The level of TNF-α in the hippocampus (E) and prefrontal cortex (F). **P < 0.01 vs. males in the control (CON) group; #P < 0.05 and ##P < 0.01 vs. males in the MSD group; $P < 0.05 and $$P < 0.01 vs. females in the CON group; &P < 0.05 and &&P < 0.01 vs. females in the MSD group.
EE decreased the mRNA level of Iba-1 and increased those of SYN, PSD-95, PGC-1α, and Sirt1 in the hippocampus
There were remarkable differences in hippocampal ionized calcium-binding adapter molecule 1 (Iba-1), synaptophysin (SYN), postsynaptic density protein-95 (PSD-95), PGC-1α, and Sirt1 mRNA levels between the three groups (Iba-1: F(2, 42) = 44.09, P < 0.01; SYN: F(2, 42) = 29.66, P < 0.01; PSD-95: F(2, 42) = 23.27, P < 0.01; PGC-1α: F(2, 42) = 26.91, P < 0.01; Sirt1: F(2, 42) = 29.17, P < 0.01; Figure 4A–4E). Compared with the Control group, the hippocampal mRNA levels of SYN, PSD-95, PGC-1α, and Sirt1 were significantly decreased, whereas that of Iba-1 was increased (Ps < 0.05). Furthermore, the mRNA levels of SYN, PSD-95, PGC-1α, and Sirt1 were higher and that of Iba-1 was lower in the MSD+EE group compared to the MSD group (Ps < 0.05).
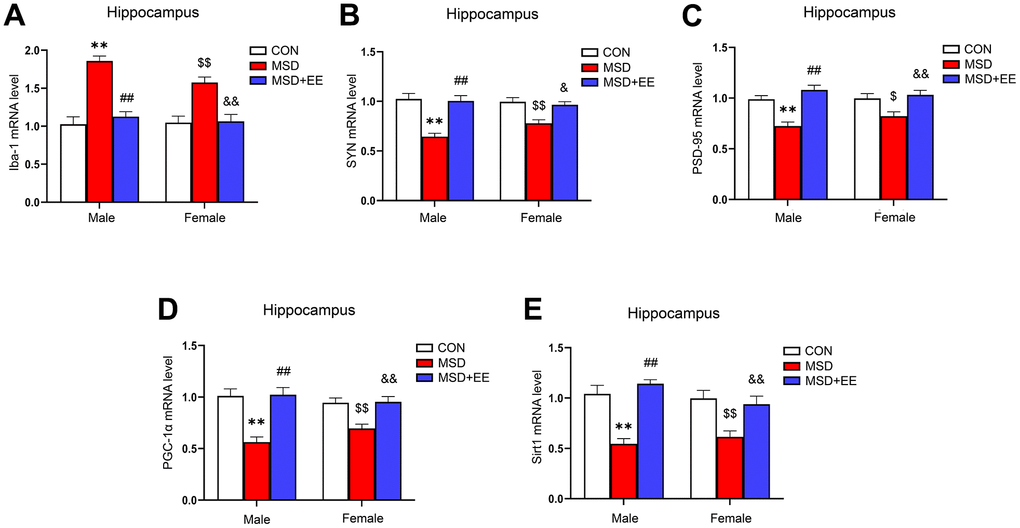
Figure 4. The effects of EE on MSD-induced alterations in Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 levels in the hippocampus of aging mice (n = 8). The Iba-1 (A), SYN (B), PSD-95 (C), PGC-1α (D), and Sirt1 (E) mRNA levels in the hippocampus. **P < 0.01 vs. males in the control (CON) group; ##P < 0.01 vs. males in the MSD group; $P < 0.05 and $$P < 0.01 vs. females in the CON group; &P < 0.05 and &&P < 0.01 vs. females in the MSD group.
EE decreased the mRNA level of Iba-1 and increased those of SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex
The mRNA levels of Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex differed significantly in all groups (Iba-1: F(2, 42) = 39.98, P < 0.01; SYN: F(2, 42) = 28.68, P < 0.01; PSD-95: F(2, 42) = 31.89, P < 0.01; PGC-1α: F(2, 42) = 24.17; P < 0.01; Sirt1: F(2, 42) = 37.12, P < 0.01; Figure 5A–5E). The Tukey test indicated that, compared with the controls, the mRNA levels of SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex were significantly decreased, whereas that of Iba-1 was increased in mice of the MSD group (Ps < 0.05). Furthermore, the prefrontal cortical levels of SYN, PSD-95, PGC-1α, and Sirt1 were higher, and that of Iba-1 was lower in the MSD+EE group relative to MSD group (Ps < 0.05).
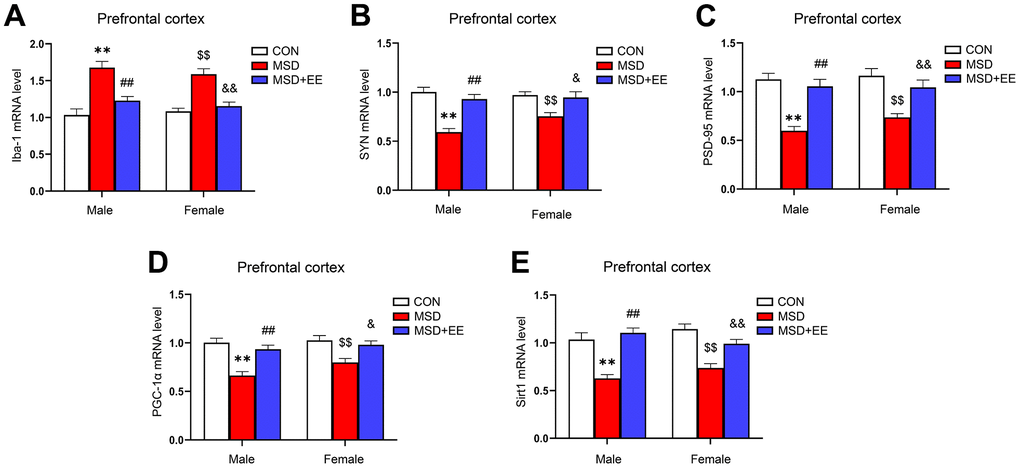
Figure 5. The effects of EE on MSD-induced alterations in Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 levels in the prefrontal cortex of aging mice (n = 8). The mRNA levels of Iba-1 (A), SYN (B), PSD-95 (C), PGC-1α (D), and Sirt1 (E) in the prefrontal cortex. **P < 0.01 vs. males in the control (CON) group; ##P < 0.01 vs. males in the MSD group; $$P < 0.01 vs. females in the CON group; &P < 0.05 and &&P < 0.01 vs. females in the MSD group.
EE decreased the protein level of Iba-1 and increased those of SYN, PSD-95, PGC-1α, and Sirt1 in the hippocampus
There were differences in protein levels of Iba-1, SYN, PSD-95, PGC-1α and Sirt1 in the hippocampus of the three groups (Iba-1: F(2, 30) = 48.42, P < 0.01; SYN: F(2, 30) = 59.18, P < 0.01; PSD-95: F(2, 30) = 39.13, P < 0.01; PGC-1α: F(2, 30) = 28.45, P < 0.01; Sirt1: F(2, 30) = 43.24, P < 0.01; Figure 6A–6F). Compared with control animals, the hippocampal protein levels of SYN, PSD-95, PGC-1α, and Sirt1 were significantly decreased in aging MSD-treated mice, whereas that of Iba-1 was increased (Ps < 0.05). However, these MSD-induced effects were reversed when mice were exposed to EE (Ps < 0.05).
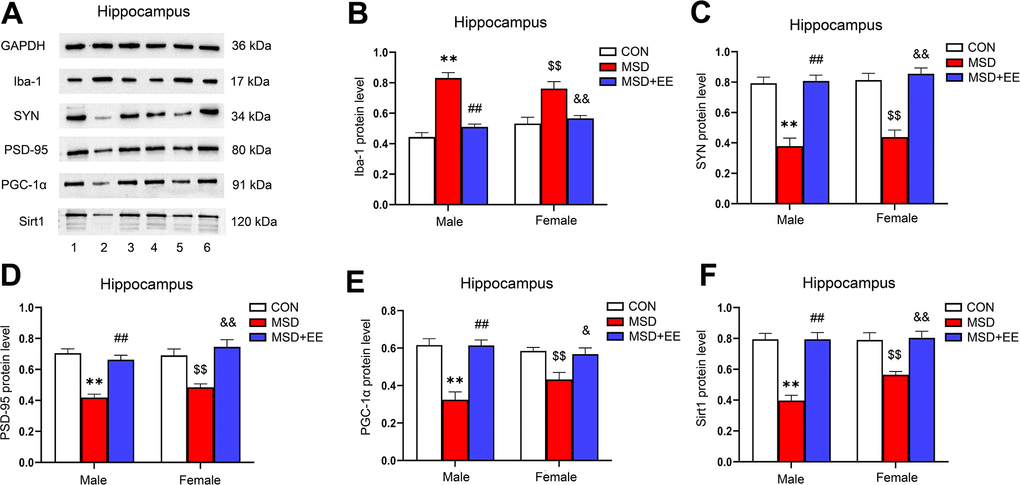
Figure 6. The effect of EE on MSD-induced changes in hippocampal Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 protein levels (n = 6). (A) The hippocampal expression levels of Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 measured by western blotting. 1: males in the control (CON) group; 2: males in the MSD group; 3: males in the MSD+EE group; 4: females in the CON group; 5: females in the MSD group; 6: females in the MSD+EE group. (B–F) Protein quantification results. (B) Iba-1, (C) SYN, (D) PSD-95, (E) PGC-1α, and (F) Sirt1. **P < 0.01 vs. males in the CON group; ##P < 0.01 vs. males in the MSD group; $$P < 0.01 vs. females in the CON group; &P < 0.05 and &&P < 0.01 vs. females in the MSD group.
EE decreased the protein level of Iba-1 and increased those of SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex
The expression levels of Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex were different in all groups (Iba-1: F(2, 30) = 52.42, P < 0.01; SYN: F(2, 30) = 50.46, P < 0.01; PSD-95: F(2, 30) = 44.23, P < 0.01; PGC-1α: F(2, 30) = 52.62, P < 0.01; Sirt1: F(2, 30) = 48.86, P < 0.01; Figure 7A–7F). Comparison results show that the protein levels of SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex were significantly decreased in the MSD group compared with the Control group, whereas that of Iba-1 was increased (Ps < 0.05). However, long-term EE reversed these MSD-mediated changes in the levels of the above proteins (Ps < 0.05).
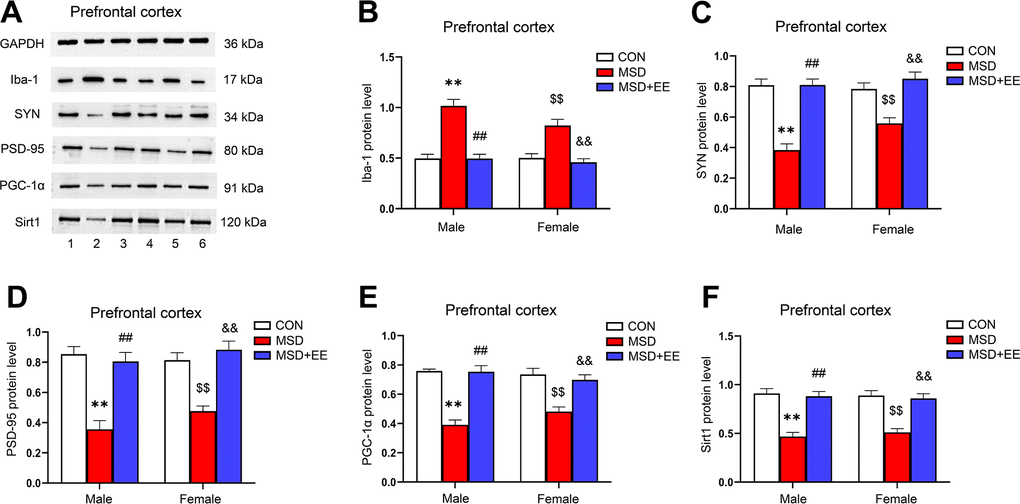
Figure 7. The effect of EE on MSD-induced changes in prefrontal cortical Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 protein levels (n = 6). (A) The expression levels of Iba-1, SYN, PSD-95, PGC-1α, and Sirt1 in the prefrontal cortex measured by western blotting. 1: males in the control (CON) group; 2: males in the MSD group; 3: males in the MSD+EE group; 4: females in the CON group; 5: females in the MSD group; 6: females in the MSD+EE group. (B–F) Protein quantification results. (B) Iba-1, (C) SYN, (D) PSD-95, (E) PGC-1α, and (F) Sirt1. **P < 0.01 vs. males in the CON group; ##P < 0.01 vs. males in the MSD group; $$P < 0.01 vs. females in the CON group; &&P < 0.01 vs. females in the MSD group.
Discussion
An increasing number of studies have shown that MSD exerts adverse effects on the nervous system of offspring rodents, including immature sleep patterns, an increase in anxiety-like behaviors, and a decrease in cognitive abilities [5, 27, 28]. In this study, we showed that MSD significantly impaired the spatial learning and memory abilities of aged offspring CD-1 mice, increased the levels of Iba-1, pro-inflammatory factors (IL-1β, IL-6, TNF-α), ROS, and MDA, decreased the expression levels of SOD and synaptic proteins (PSD-95, SYN), and inhibited the Sirt1/PGC-1α pathway in the hippocampus and prefrontal cortex. Importantly, EE reversed these adverse effects caused by MSD during late pregnancy by upregulating the Sirt1/PGC-1α pathway, thereby promoting mitochondrial biogenesis.
EE reversed MSD-induced cognitive impairment
The environment experienced early in life can have lifelong effects on physical health, referred to as the “fetal origins of adult diseases” hypothesis. Specifically, fetal or childhood exposure to adverse stimuli can lead to changes in immune function and epigenetics, as well as the reprogramming of the hypothalamic–pituitary–adrenal axis. These changes can trigger structural or functional alterations in relevant brain regions, leading to depression and cognitive impairment later in life [29, 30]. We have previously revealed that maternal immune stress in late pregnancy exacerbates age-related cognitive impairment in offspring mice [31]. Here, we demonstrated that MSD during late pregnancy leads to cognitive impairment in older offspring mice. EE can improve cognitive function by increasing social, sensory, and spatial complexity [32]. The results of the MWM test exhibited that EE reversed the MSD-induced deficits in cognitive performance in aged offspring, as evidenced by the shorter swim distance and escape latency in the training phase of the test and the increase in the percent distance and time spent in the target quadrant in the memory phase of the MWM test by mice in the MSD+EE group. These findings are in accordance with our previous studies showing that EE improved the cognitive performance in aging mice of spatial learning and memory impairment induced by MSD [9].
EE relieved oxidative stress and neuroinflammation caused by MSD
Oxidative stress and inflammation are strongly related to cognitive dysfunction [33]. Numerous neurodegenerative diseases are accompanied by elevated oxidative stress [34]. Exposure to harmful stimuli can result in excessive ROS production in the brain, which will lead not only to a decrease in the activity of antioxidant enzymes, such as SOD but also to an increase in the contents of endogenous oxidative damage-related products, such as MDA, which is a product of lipid peroxidation [35]. Excessive MDA production in the brain will further reduce SOD levels, resulting in a vicious cycle of oxidative stress in the central nervous system. If excess ROS cannot be removed from the brain, microglia will be activated and release pro-inflammatory factors including IL-1β, IL-6, and TNF-α through downstream signaling pathways, thereby triggering neuroinflammation and eventually causing brain tissue damage and impairing cognitive function [36, 37]. Lead exposure in rats early in life leads to cognitive decline, accompanied by upregulated oxidative stress and inflammatory responses in the brain [38]. Consistent with this observation, we found that SD during late pregnancy resulted in oxidative stress and neuroinflammation in the hippocampus and prefrontal cortex in aged offspring, as evidenced by the increased levels of ROS and MDA, decreased SOD activity, and upregulation of the levels of Iba-1 and IL-1β, IL-6, and TNF-α. EE could improve cognitive impairment by inhibiting neuroinflammation and oxidative stress [39]. For example, an enriched environment was reported to improve middle cerebral artery occlusion-induced cognition impairment in Sprague–Dawley rats by suppressing neuroinflammation and oxidative stress and increasing the expression levels of synaptic plasticity-related proteins in the hippocampus [25]. Similarly, our current results showed that long-term exposure to EE suppressed oxidative stress and neuroinflammation in the hippocampus and prefrontal cortex of aged offspring induced by SD during late pregnancy.
EE reversed the MSD-induced decline in synaptic protein levels
The synaptic plasticity in the hippocampus and prefrontal cortex plays a critical role in cognitive function [40]. PSD-95 is a post-synaptic protein involved in long-term potentiation as well as synaptic connectivity and maintenance [41]. SYN is a specific calcium-binding glycoprotein widely existed in the pre-synaptic vesicle membrane and plays a role in synaptic plasticity by regulating synaptic structure and function in addition to neurotransmitter release [42]. Basic studies have shown that PSD-95 and SYN knockout mice exhibit impaired synaptic morphology and function accompanied with learning and memory deficits [43, 44]. In clinical studies, the levels of PSD-95 and SYN were significantly decreased in postmortem brain tissue of AD patients compared with those of controls [45]. This clearly indicates that PSD-95 and SYN are critical for synaptic plasticity, synaptic integrity, and cognitive function [46, 47]. In the present study, the expression levels of PSD-95 and SYN were decreased in the hippocampus and prefrontal cortex of offspring mice in the MSD group when compared to those in Control animals, suggesting that exposure to MSD can lead to deficits in synaptic plasticity in aged offspring. Furthermore, EE can ameliorate impaired synaptic plasticity caused by exposure to adverse environments early in life. For example, an enriched environment reportedly increased the levels of synaptic plasticity-associated proteins such as BDNF, PSD-95, and SYN in elderly Sprague–Dawley rats subjected to prenatal mobile phone exposure [48]. Similarly, our results showed that long-term EE reversed the downregulated levels of PSD-95 and SYN in aged offspring CD-1 mice of dams that underwent SD during late pregnancy, suggesting that EE may protect cognitive function by improving synaptic plasticity.
EE attenuated mitochondrial biogenesis dysfunction by activating the Sirt1/PGC-1α pathway
Impaired mitochondrial biogenesis is associated with neuroinflammation, oxidative stress, and synaptic dysfunction [49]. PGC-1α is thought to be a central regulator of mitochondrial biogenesis [50]. The overexpression of PGC-1α can suppress neuroinflammation by regulating microglial polarization [51]; reduce oxidative stress by regulating the redox balance, such as by scavenging ROS and increasing the activities of antioxidant enzymes such as glutathione (GSH) and SOD [52]; and regulate the expression levels of synapse-associated proteins [53]. Furthermore, Sirt1, a histone deacetylase, can modulate mitochondrial biogenesis by modulating PGC-1α [54]. Chronic cerebral hypoperfusion can induce cognitive dysfunction in Sprague–Dawley rats, accompanied by the downregulation of the Sirt1/PGC-1α pathway, impairment of mitochondrial biogenesis, neuroinflammation, and oxidative stress [18]. Similarly, in the present study, we found that MSD led to a decrease in Sirt1 and PGC-1α levels in the hippocampus and prefrontal cortex of older offspring CD-1 mice. Interestingly, one of the mechanisms by which EE improves neurological function in the brain may be by enhancing or stimulating mitochondrial biogenesis [55]. We have previously demonstrated that EE counteracts alterations in mitochondrial quality control patterns, such as mitochondrial biogenesis, mitophagy, and dynamics, resulting from prenatal inflammatory stimulation in older offspring [23]. Consistent with previous results, we showed here that EE can reverse the decline in the levels of Sirt1 and PGC-1α in the hippocampus and prefrontal cortex of older offspring mice induced by MSD during late pregnancy. These results suggest that EE can ameliorate MSD-mediated mitochondrial biogenesis dysfunction in aged offspring mice by activating the Sirt1/PGC-1α pathway.
Despite the importance of our findings, our study had some limitations. First, we did not examine changes in mitochondrial morphology and other functions such as mitochondrial dynamics and autophagy; secondly, we did not use pharmacological modalities, such as Sirt1 blockers, to inhibit the Sirt1/PGC-1α pathway to observe the effects of EE; thirdly, a control+EE group was did not set for further comparison; thirdly, we did not set up a control group of young mice to observe the effects of sleep deprivation in late pregnancy on aging-related cognitive dysfunction and the putative ameliorative effect of an enriched environment in this process; finally, we did not use immunohistochemistry to quantitatively analyze the effect of EE on the expression levels of synaptic proteins and Iba-1 in different subregions of the hippocampus.
Conclusions
We found that long-term EE ameliorated late-pregnancy SD-induced cognitive impairment, attenuated neuroinflammation and oxidative stress, and upregulated synaptic protein levels in aged offspring mice. These effectively beneficial effects were associated with the attenuation of mitochondrial biogenesis dysfunction via the activation of the Sirt1/PGC-1α pathway (Figure 8). These findings suggest that long-term EE may be an effective non-pharmacological intervention for early-life stress-induced cognitive dysfunction, and the Sirt1/PGC-1α signaling pathway may be a promising therapeutic target for this condition.
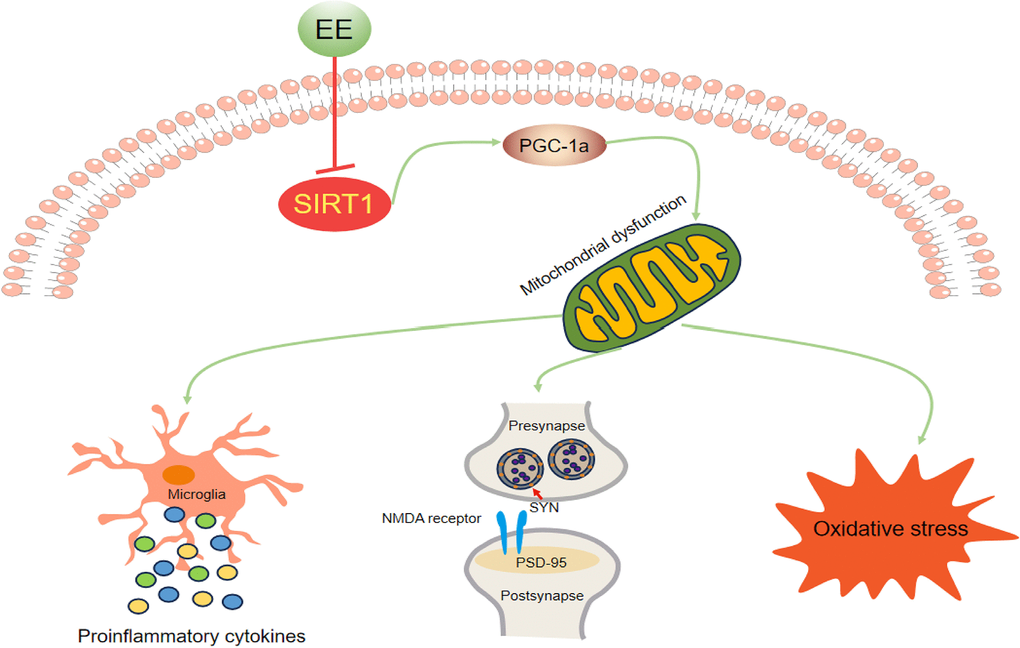
Figure 8. Schematic representation of the mechanisms underlying the inhibitory effect of EE on MSD-induced cognitive impairment in aging CD-1 mice.
Materials and Methods
Animals
2-month-old CD-1 mice were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). After two weeks of environmental adaptation, the mice were mated at a male: female ratio of 1:2. Pregnant female mice were randomized into a control group and a SD group. Female mice in the SD group were subjected to SD during late pregnancy, whereas no treatment was administered to animals in the control group. At 21 days of age, the male and female offspring were divided into the following three groups respectively: control group, MSD group, and MSD+EE group. Mice in the MSD+EE group were given an enriched environment from 21 days to 18 months after birth. During the experiments, all animals had access to water and food ad libitum and were reared at a temperature of 22-25° C and a humidity of 50 ± 5%. In addition, a 12-hour/12-hour light/dark cycle was provided with lights on at 8:00 am (Figure 9).
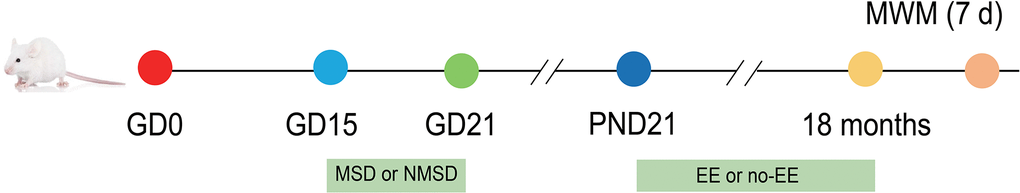
Figure 9. Experimental procedure and treatment schedule. GD: gestational day; PND: postnatal day; MWM: Morris water maze test; MSD: maternal sleep deprivation; NMSD: no MSD; EE: environmental enrichment.
Exposure to sleep deprivation
Female CD-1 mice were performed to SD from 12:00 to 18:00 h daily between day 15 and day 21 of pregnancy. SD was administered using a SD apparatus (BW-NSD404, China). The apparatus worked uninterruptedly at a speed of 0.5 m/min to keep mice awake during the deprivation period as previously reported [9].
Exposure to an enriched environment
Offspring CD-1 mice were subjected to long-term EE from 21 days to 18 months of age. Seven or eight mice were placed in a large cage (52 × 40 × 20 cm3) containing colored toys such as plastic tunnels, stairs, running wheels, and wooden houses. The environmental enrichment cages provided more opportunities for social interaction and space for somatosensory and cognitive stimulation as well as physical activity [56]. In contrast, the standard cages (36 × 18 × 14 cm3) did not contain any objects, and each housed three mice.
Morris water maze test
The Morris water maze test (MWM) was used to assess cognitive performance in mice [31]. The equipment mainly consists of a circular pool containing 22° C water (radius: 75 cm, height: 30 cm) and a target platform of cylindrical shape (radius: 5 cm, height: 24 cm). The training phase included four training sessions per day for a total of seven consecutive days. During this period, there is a target platform 1 cm underwater in the target quadrant, and the mice were placed in the water facing the wall and allowed to freely explore for 60 s. When mice found the platform, it was allowed to stay there for 30 s. Each mouse was trained four times a day from different quadrants in order. Mice without finding the target platform within 60 s were led to the platform and also allowed to rest there for 30 s. A video tracking system (ANY-Maze, Stoeling, USA) was applied to record the escape latency (time taken to locate the target platform), the swimming distance, and the swimming velocity of mice. A spatial probe task was conducted 2 h after the last trial of the training phase. For the probe task, removed the target platform, the animals were entered into the water maze from the opposite quadrant of the target quadrant and were given free exploration of 60 s. The percent of time and distance swam in the target quadrant were calculated.
Assays for ROS, MDA, and SOD contents
Hippocampal and prefrontal cortical tissues were homogenized on ice. After centrifugation (12,000 rpm for 20 min), store supernatant at –80° C. MDA and SOD contents were assayed by the respective kits (Nanjing Jiancheng, China) following the manual. A ROS assay kit (Beyotime Biotechnology, China) with fluorescent probe (DCFH-DA) was employed to determine ROS content. Absorbance was measured in a spectrofluorometer (emission 525 nm, excitation 488 nm).
Measurement of proinflammatory cytokine levels
The levels of IL-1β, IL-6, and TNF-α in the hippocampus and prefrontal cortex were assessed by the ELISA kits (MyBioscience, Inc., USA) following the operation instructions, respectively.
Real-time fluorescent quantitative PCR (qPCR)
Total RNA was obtained from hippocampal or prefrontal cortical tissues using TRIzol reagent (Life Technologies, USA). Then the PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara Bio Inc., Japan) was utilized to reverse transcribe RNA into cDNA. The qPCR mixture contained 2× SYBR Green Mix, forward and reverse primers, cDNA, and RNase-free water. A Real-Time PCR System (Thermo Fisher Scientific, USA) was employed for fluorescent quantitative PCR reactions. The analytical method used in this experiment was relative quantification study, calculated as 2-ΔΔCt. The primers used for qPCR are described below (Table 1).
Table 1. The primers used for qPCR.
| Gene | Forward primer (5’→3’) | Reverse primer (5’→3’) |
| β-actin | AGTGTGACGTTGACATCCGT | TGCTAGGAGCCAGAGCAGTA |
| Psd95 | GCTCCCTGGAGAATGTGCTA | TGAGAAGCACTCCGTGAACT |
| Syn | GCCTACCTTCTCCACCCTTT | GCACTACCAACGTCACAGAC |
| Sirt1 | TAATGTGAGGAGTCAGCACC | GCCTGTTTGGACATTACCAC |
| Pgc-1α | TGTGACTGGGGACTGTAGTA | AGAGCAGCACACTCTATGTC |
| Iba-1 | ATTCCTCGATGATCCCAAAT | CCAAGTTTCTCCAGCATTCG |
Western blotting
In the hippocampus and prefrontal cortex, western blot was examined for the protein expression levels of PSD-95, SYN, Iba-1, PGC-1α, and Sirt1. Briefly, hippocampal or prefrontal cortical tissues were lysed in RIPA lysis buffer (Beyotime Biotechnology, China) and then collect the supernatant after centrifugation. Protein samples were treated with SDS–PAGE and transferred to PVDF membranes (Millipore, USA), then blocked in 5% skimmed milk for 2 h at room temperature. The incubation was first carried out with the primary antibody against Iba-1 (1:200, Santa Cruz, USA), SYN (1:2,000, Bioss, China), PSD-95 (1:1,000, Abcam, UK), PGC-1α (1:1,000, Abcam, UK), and Sirt1 (1:500, Santa Cruz, USA) overnight at 4° C. Then incubated with the horseradish peroxidase-conjugated secondary antibodies (anti-rabbit/mouse IgG; 1:20,000; Zsbio, China) for 1.2 h, respectively. Protein band intensities were quantified using ImageJ (Media Cybernetics, USA).
Statistical analysis
Data were presented as means ± standard error of the mean (SEM) and conformed to the chi-square test for normality. Differences among groups were assessed by repeated-measures analysis of variance (ANOVA) or two-way ANOVA with Tukey’s post hoc test. All data were analyzed in GraphPad Prism 8.0 and the statistical significance level was set at P-value < 0.05.
Supplementary Materials
Author Contributions
Gui-Hai Chen and Xue-Chun Liu designed the study and revised the manuscript. Ru-Meng Wei, Yue-Ming Zhang, and Kai-Xuan Zhang performed the behavioral tests, and drafted the manuscript. Gao-Xia Liu, Xue-Yan Li, Jing-Ya Zhang, and Wei-Zhong Lun were responsible for the western blotting, RT-qPCR and ELISA. Ru-Meng Wei, Yue-Ming Zhang, and Kai-Xuan Zhang analyzed the data and constructed the graphs. All authors read and approved the final version of manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Statement
All animal experiments were approved by the guidelines for the Center for Laboratory Animal Science and the Association for Laboratory Animal Science of Anhui Medical University (No. LLSC20190710) and carried out in accordance with the National Institutes of Health Guide for the Care.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant number: 81671316), 2022 Key Research and Development Plan of Anhui Province (2022e07020029), and Postgraduate Innovation Research and Practice Program of Anhui Medical University (grant number: YJS20230089).
References
- 1. Mason GM, Lokhandwala S, Riggins T, Spencer RMC. Sleep and human cognitive development. Sleep Med Rev. 2021; 57:101472. https://doi.org/10.1016/j.smrv.2021.101472 [PubMed]
- 2. Gupta R, Rawat VS. Sleep and sleep disorders in pregnancy. Handb Clin Neurol. 2020; 172:169–86. https://doi.org/10.1016/B978-0-444-64240-0.00010-6 [PubMed]
- 3. Wilson DL, Barnes M, Ellett L, Permezel M, Jackson M, Crowe SF. Decreased sleep efficiency, increased wake after sleep onset and increased cortical arousals in late pregnancy. Aust N Z J Obstet Gynaecol. 2011; 51:38–46. https://doi.org/10.1111/j.1479-828X.2010.01252.x [PubMed]
- 4. Pires GN, Benedetto L, Cortese R, Gozal D, Gulia KK, Kumar VM, Tufik S, Andersen ML. Effects of sleep modulation during pregnancy in the mother and offspring: Evidences from preclinical research. J Sleep Res. 2021; 30:e13135. https://doi.org/10.1111/jsr.13135 [PubMed]
- 5. Aswathy BS, Kumar VM, Gulia KK. The effects of rapid eye movement sleep deprivation during late pregnancy on newborns’ sleep. J Sleep Res. 2018; 27:197–205. https://doi.org/10.1111/jsr.12564 [PubMed]
- 6. Yu Y, Huang Z, Dai C, Du Y, Han H, Wang YT, Dong Z. Facilitated AMPAR endocytosis causally contributes to the maternal sleep deprivation-induced impairments of synaptic plasticity and cognition in the offspring rats. Neuropharmacology. 2018; 133:155–62. https://doi.org/10.1016/j.neuropharm.2018.01.030 [PubMed]
- 7. Zhao Q, Peng C, Wu X, Chen Y, Wang C, You Z. Maternal sleep deprivation inhibits hippocampal neurogenesis associated with inflammatory response in young offspring rats. Neurobiol Dis. 2014; 68:57–65. https://doi.org/10.1016/j.nbd.2014.04.008 [PubMed]
- 8. Lv J, Xu S, Meng C, Wang Y, Ji L, Li X, Wang X, Li Q. Ferroptosis participated in hippocampal neuroinflammation damage of in offspring rats after maternal sleep deprivation. J Neuroimmunol. 2023; 375:578021. https://doi.org/10.1016/j.jneuroim.2023.578021 [PubMed]
- 9. Zhang YM, Wei RM, Li XY, Feng YZ, Zhang KX, Ge YJ, Kong XY, Liu XC, Chen GH. Long-term environmental enrichment overcomes depression, learning, and memory impairment in elderly CD-1 mice with maternal sleep deprivation exposure. Front Aging Neurosci. 2023; 15:1177250. https://doi.org/10.3389/fnagi.2023.1177250 [PubMed]
- 10. Carvajal-Flores FN, Díaz A, Flores-Gómez GD, de la Cruz F, Flores G. Phenylbutyrate ameliorates prefrontal cortex, hippocampus, and nucleus accumbens neural atrophy as well as synaptophysin and GFAP stress in aging mice. Synapse. 2020; 74:e22177. https://doi.org/10.1002/syn.22177 [PubMed]
- 11. Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002; 23:795–807. https://doi.org/10.1016/s0197-4580(02)00019-2 [PubMed]
- 12. Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013; 62:90–101. https://doi.org/10.1016/j.freeradbiomed.2012.11.014 [PubMed]
- 13. Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019; 710:132933. https://doi.org/10.1016/j.neulet.2017.06.052 [PubMed]
- 14. Ozben T, Ozben S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin Biochem. 2019; 72:87–9. https://doi.org/10.1016/j.clinbiochem.2019.04.001 [PubMed]
- 15. Wang Y, Lin Y, Wang L, Zhan H, Luo X, Zeng Y, Wu W, Zhang X, Wang F. TREM2 ameliorates neuroinflammatory response and cognitive impairment via PI3K/AKT/FoxO3a signaling pathway in Alzheimer’s disease mice. Aging (Albany NY). 2020; 12:20862–79. https://doi.org/10.18632/aging.104104 [PubMed]
- 16. Iorio R, Celenza G, Petricca S. Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation. Antioxidants (Basel). 2022; 11:1199. https://doi.org/10.3390/antiox11061199 [PubMed]
- 17. Wójtowicz S, Strosznajder AK, Jeżyna M, Strosznajder JB. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem Res. 2020; 45:972–88. https://doi.org/10.1007/s11064-020-02993-5 [PubMed]
- 18. Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang XJ, Wang H, Du Y, Guan J, Wang X, Fu J. NAD+ improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J Neuroinflammation. 2021; 18:207. https://doi.org/10.1186/s12974-021-02250-8 [PubMed]
- 19. Han Y, Yang H, Li L, Du X, Sun C. Schisanhenol improves learning and memory in scopolamine-treated mice by reducing acetylcholinesterase activity and attenuating oxidative damage through SIRT1-PGC-1α-Tau signaling pathway. Int J Neurosci. 2019; 129:110–8. https://doi.org/10.1080/00207454.2018.1503183 [PubMed]
- 20. Fu X, Qin T, Yu J, Jiao J, Ma Z, Fu Q, Deng X, Ma S. Formononetin Ameliorates Cognitive Disorder via PGC-1α Pathway in Neuroinflammation Conditions in High-Fat Diet-Induced Mice. CNS Neurol Disord Drug Targets. 2019; 18:566–77. https://doi.org/10.2174/1871527318666190807160137 [PubMed]
- 21. Gubert C, Hannan AJ. Environmental enrichment as an experience-dependent modulator of social plasticity and cognition. Brain Res. 2019; 1717:1–14. https://doi.org/10.1016/j.brainres.2019.03.033 [PubMed]
- 22. Stein LR, O’Dell KA, Funatsu M, Zorumski CF, Izumi Y. Short-term environmental enrichment enhances synaptic plasticity in hippocampal slices from aged rats. Neuroscience. 2016; 329:294–305. https://doi.org/10.1016/j.neuroscience.2016.05.020 [PubMed]
- 23. Zhuang ZQ, Zhang ZZ, Zhang YM, Ge HH, Sun SY, Zhang P, Chen GH. A Long-Term Enriched Environment Ameliorates the Accelerated Age-Related Memory Impairment Induced by Gestational Administration of Lipopolysaccharide: Role of Plastic Mitochondrial Quality Control. Front Cell Neurosci. 2021; 14:559182. https://doi.org/10.3389/fncel.2020.559182 [PubMed]
- 24. Zentall TR. Effect of Environmental Enrichment on the Brain and on Learning and Cognition by Animals. Animals (Basel). 2021; 11:973. https://doi.org/10.3390/ani11040973 [PubMed]
- 25. Zhang X, Yuan M, Yang S, Chen X, Wu J, Wen M, Yan K, Bi X. Enriched environment improves post-stroke cognitive impairment and inhibits neuroinflammation and oxidative stress by activating Nrf2-ARE pathway. Int J Neurosci. 2021; 131:641–9. https://doi.org/10.1080/00207454.2020.1797722 [PubMed]
- 26. Shen J, Li Y, Qu C, Xu L, Sun H, Zhang J. The enriched environment ameliorates chronic unpredictable mild stress-induced depressive-like behaviors and cognitive impairment by activating the SIRT1/miR-134 signaling pathway in hippocampus. J Affect Disord. 2019; 248:81–90. https://doi.org/10.1016/j.jad.2019.01.031 [PubMed]
- 27. Radhakrishnan A, Aswathy BS, Kumar VM, Gulia KK. Sleep deprivation during late pregnancy produces hyperactivity and increased risk-taking behavior in offspring. Brain Res. 2015; 1596:88–98. https://doi.org/10.1016/j.brainres.2014.11.021 [PubMed]
- 28. Han Y, Wang J, Zhao Q, Xie X, Song R, Xiao Y, Kang X, Zhang L, Zhang Y, Peng C, You Z. Pioglitazone alleviates maternal sleep deprivation-induced cognitive deficits in male rat offspring by enhancing microglia-mediated neurogenesis. Brain Behav Immun. 2020; 87:568–78. https://doi.org/10.1016/j.bbi.2020.02.002 [PubMed]
- 29. Wang B, Zeng H, Liu J, Sun M. Effects of Prenatal Hypoxia on Nervous System Development and Related Diseases. Front Neurosci. 2021; 15:755554. https://doi.org/10.3389/fnins.2021.755554 [PubMed]
- 30. Kajantie E. Early-life events. Effects on aging. Hormones (Athens). 2008; 7:101–13. https://doi.org/10.1007/BF03401501 [PubMed]
- 31. Zhang ZZ, Zeng LP, Chen J, Wu YF, Wang YT, Xia L, Yang QG, Wang F, Chen GH. Long-Term Environmental Enrichment Relieves Dysfunctional Cognition and Synaptic Protein Levels Induced by Prenatal Inflammation in Older CD-1 Mice. Neural Plast. 2022; 2022:1483101. https://doi.org/10.1155/2022/1483101 [PubMed]
- 32. Eckert MJ, Abraham WC. Effects of environmental enrichment exposure on synaptic transmission and plasticity in the hippocampus. Curr Top Behav Neurosci. 2013; 15:165–87. https://doi.org/10.1007/7854_2012_215 [PubMed]
- 33. Atrooz F, Salim S. Sleep deprivation, oxidative stress and inflammation. Adv Protein Chem Struct Biol. 2020; 119:309–36. https://doi.org/10.1016/bs.apcsb.2019.03.001 [PubMed]
- 34. Merelli A, Repetto M, Lazarowski A, Auzmendi J. Hypoxia, Oxidative Stress, and Inflammation: Three Faces of Neurodegenerative Diseases. J Alzheimers Dis. 2021; 82:S109–26. https://doi.org/10.3233/JAD-201074 [PubMed]
- 35. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014; 20:1126–67. https://doi.org/10.1089/ars.2012.5149 [PubMed]
- 36. Thangaraj A, Periyasamy P, Guo ML, Chivero ET, Callen S, Buch S. Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy. 2020; 16:289–312. https://doi.org/10.1080/15548627.2019.1607686 [PubMed]
- 37. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014; 94:909–50. https://doi.org/10.1152/physrev.00026.2013 [PubMed]
- 38. Hossain S, Bhowmick S, Jahan S, Rozario L, Sarkar M, Islam S, Basunia MA, Rahman A, Choudhury BK, Shahjalal H. Maternal lead exposure decreases the levels of brain development and cognition-related proteins with concomitant upsurges of oxidative stress, inflammatory response and apoptosis in the offspring rats. Neurotoxicology. 2016; 56:150–8. https://doi.org/10.1016/j.neuro.2016.07.013 [PubMed]
- 39. Tang YF, Duan YJ, Ge RD, Lu X, Gao BY, Guo JW, Jiang S. Environmental Enrichment Protects Against Cognition Deficits Caused by Sepsis-Associated Encephalopathy. J Integr Neurosci. 2022; 22:5. https://doi.org/10.31083/j.jin2201005 [PubMed]
- 40. Ruggiero RN, Rossignoli MT, Marques DB, de Sousa BM, Romcy-Pereira RN, Lopes-Aguiar C, Leite JP. Neuromodulation of Hippocampal-Prefrontal Cortical Synaptic Plasticity and Functional Connectivity: Implications for Neuropsychiatric Disorders. Front Cell Neurosci. 2021; 15:732360. https://doi.org/10.3389/fncel.2021.732360 [PubMed]
- 41. Franchini L, Stanic J, Ponzoni L, Mellone M, Carrano N, Musardo S, Zianni E, Olivero G, Marcello E, Pittaluga A, Sala M, Bellone C, Racca C, et al. Linking NMDA Receptor Synaptic Retention to Synaptic Plasticity and Cognition. iScience. 2019; 19:927–39. https://doi.org/10.1016/j.isci.2019.08.036 [PubMed]
- 42. Lalert L, Ji-Au W, Srikam S, Chotipinit T, Sanguanrungsirikul S, Srikiatkhachorn A, Maneesri-le Grand S. Alterations in Synaptic Plasticity and Oxidative Stress Following Long-Term Paracetamol Treatment in Rat Brain. Neurotox Res. 2020; 37:455–68. https://doi.org/10.1007/s12640-019-00090-2 [PubMed]
- 43. Schmitt U, Tanimoto N, Seeliger M, Schaeffel F, Leube RE. Detection of behavioral alterations and learning deficits in mice lacking synaptophysin. Neuroscience. 2009; 162:234–43. https://doi.org/10.1016/j.neuroscience.2009.04.046 [PubMed]
- 44. Coley AA, Gao WJ. PSD-95 deficiency disrupts PFC-associated function and behavior during neurodevelopment. Sci Rep. 2019; 9:9486. https://doi.org/10.1038/s41598-019-45971-w [PubMed]
- 45. Whitfield DR, Vallortigara J, Alghamdi A, Howlett D, Hortobágyi T, Johnson M, Attems J, Newhouse S, Ballard C, Thomas AJ, O’Brien JT, Aarsland D, Francis PT. Assessment of ZnT3 and PSD95 protein levels in Lewy body dementias and Alzheimer’s disease: association with cognitive impairment. Neurobiol Aging. 2014; 35:2836–44. https://doi.org/10.1016/j.neurobiolaging.2014.06.015 [PubMed]
- 46. Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci USA. 2007; 104:4176–81. https://doi.org/10.1073/pnas.0609307104 [PubMed]
- 47. Wang A, Zou X, Wu J, Ma Q, Yuan N, Ding F, Li X, Chen J. Early-Life Stress Alters Synaptic Plasticity and mTOR Signaling: Correlation With Anxiety-Like and Cognition-Related Behavior. Front Genet. 2020; 11:590068. https://doi.org/10.3389/fgene.2020.590068 [PubMed]
- 48. Hong S, Huang H, Yang M, Wu H, Wang L. Enriched Environment Decreases Cognitive Impairment in Elderly Rats With Prenatal Mobile Phone Exposure. Front Aging Neurosci. 2020; 12:162. https://doi.org/10.3389/fnagi.2020.00162 [PubMed]
- 49. Caldeira GL, Ferreira IL, Rego AC. Impaired transcription in Alzheimer’s disease: key role in mitochondrial dysfunction and oxidative stress. J Alzheimers Dis. 2013; 34:115–31. https://doi.org/10.3233/JAD-121444 [PubMed]
- 50. Halling JF, Pilegaard H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl Physiol Nutr Metab. 2020; 45:927–36. https://doi.org/10.1139/apnm-2020-0005 [PubMed]
- 51. Yang X, Xu S, Qian Y, Xiao Q. Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun. 2017; 64:162–72. https://doi.org/10.1016/j.bbi.2017.03.003 [PubMed]
- 52. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006; 127:397–408. https://doi.org/10.1016/j.cell.2006.09.024 [PubMed]
- 53. Liu X, Tang M, He TY, Zhao S, Li HZ, Li Z, Guo YX, Wang XL. Resveratrol Improves Paclitaxel-Induced Cognitive Impairment in Mice by Activating SIRT1/PGC-1α Pathway to Regulate Neuronal State and Microglia Cell Polarization. Drug Des Devel Ther. 2023; 17:1125–38. https://doi.org/10.2147/DDDT.S400936 [PubMed]
- 54. Yin Z, Gao D, Du K, Han C, Liu Y, Wang Y, Gao X. Rhein Ameliorates Cognitive Impairment in an APP/PS1 Transgenic Mouse Model of Alzheimer's Disease by Relieving Oxidative Stress through Activating the SIRT1/PGC-1α Pathway. Oxid Med Cell Longev. 2022; 2022:2524832. https://doi.org/10.1155/2022/2524832 [PubMed]
- 55. Yu K, Kuang S, Wang C, Wang Y, Liu G, Xie H, Jiang C, Wu J, Wang N, Wu Y. Changes in Mitochondria-Associated Protein Expression and Mitochondrial Function in Response to 2 Weeks of Enriched Environment Training After Cerebral Ischaemia-Reperfusion Injury. J Mol Neurosci. 2020; 70:413–21. https://doi.org/10.1007/s12031-019-01428-3 [PubMed]
- 56. Mahati K, Bhagya V, Christofer T, Sneha A, Shankaranarayana Rao BS. Enriched environment ameliorates depression-induced cognitive deficits and restores abnormal hippocampal synaptic plasticity. Neurobiol Learn Mem. 2016; 134:379–91. https://doi.org/10.1016/j.nlm.2016.08.017 [PubMed]