



Introduction
Bladder cancer (BCa) is a common malignant tumor of the urinary system. Smoking and occupational exposure to chemical carcinogens are the main risk factors associated with bladder cancer, while other risk factors are less consistently reported [1, 2]. The most predominant pathological subtype is urothelial BCa, which accounts for over 90% of newly diagnosed cases [3]. Around 80% of patients are diagnosed with non-muscle invasive BCa (NMIBC) [4]. Although transurethral resection of bladder tumor (TURBT) has shown great therapeutic effects, NMIBC has a high rate of recurrence and progression after surgical resection [5]. To address this issue, intravesical anti-tumor agents (chemotherapy or immunotherapy) may be given as adjunct treatment after transurethral resection to prevent tumor recurrence and progression in NMIBC [6]. Although immunotherapy has shown promising effects in recent years, it is still at an early stage and has adverse reaction. Therefore, there is an urgent need to identify and establish prognostic markers and therapeutic targets for BCa.
LncRNA, defined as long noncoding RNA, has more than 200 nucleotides that undergoes similar processing as mRNAs [7]. However, lncRNAs lack significant open reading frames. There is growing evidence that lncRNAs take an important part in the gene life cycle as well as cell growth, proliferation, and differentiation [8]. Previous research evidences have shown an intimate association between lncRNAs and the tumorigenicity and metastasis of BCa. For instance, LncRNA RP11-89 has been proven to suppress ferroptosis thereby alleviating the occurrence and progression of BCa, which was expected to serve as a potential therapeutic target for BCa [9]. Another study has reported that LncRNA ELNAT1 may be a hopeful therapeutic target for lymph node (LN) metastatic in BCa, as it could promote lymphangiogenesis and LN metastasis in BCa in a SUMOylation-dependent manner [10].
Necroptosis, a new type of programmed cell death, is regulated by RIPK1-RIPK3-MLKL [11, 12]. This process is involved in pathogen detection and tissue repair. Recent studies suggest that necroptosis participates actively in tumor development, tumor necrosis, tumor metastasis and the tumoral immune response [13]. In breast cancer, RIPK1 could facilitate tumor cell necroptosis to promote metastasis [14]. In colorectal cancer, TRAF6 has been confirmed to stimulate colorectal cell progression through inhibiting the RIPK1/RIPK3/MLKL necroptosis signaling pathway. Therefore, this pathway might represent a new therapeutic target for colorectal cancer [15]. However, the regulatory effects of necroptosis-associated on tumors and its specific mechanisms remain unclear. Thus, identification and exploration of necroptosis-associated lncRNAs (NRlncRNAs) and the identification of prognostic biomarkers among these lncRNAs have enormous potential for clinical application.
In our study, we identified NRlncRNAs with prognostic value to construct a risk model and investigated the correlations between the risk model and the immune microenvironment. We demonstrated that NRlncRNAs with a differential expression in BCa patients could be used as prognostic markers and possible therapeutic targets.
Materials and Methods
Data collection and preprocessing
We acquired the RNA sequencing (RNA-seq) data and corresponding clinical data of BCa patients from The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga/). After removing incomplete data and missed survival information, we obtained sequencing and survival data for 406 BCa patients.
Establishment of prognostic risk model based on NRlncRNAs
Initially, we employed univariate Cox (uni-Cox) regression analysis to discern lncRNAs that were prognostic with a p-value ≤ 0.05. Next, we performed least absolute shrinkage and selection operator (LASSO) regression analysis using a 10-fold cross-validation and a significance threshold of p ≤ 0.05 to pick lncRNAs associated with prognosis. Furthermore, the prognostic-associated lncRNAs selected by the LASSO regression analysis were utilized to establish a risk signature based on multivariate Cox (multi-Cox) proportional hazards regression. The calculation formula for the risk score was as follows:
On the basis of each patient’s median risk score, we categorized all BCa patients into high- and low-risk groups.
Assessment and validation of prognostic risk model
Kaplan-Meier analysis was used to compared the survival situation of two risk groups, followed by the long-rank test. We then performed uni- and multi-Cox regression analyses to ascertain the independence of prognostic factors, including the risk score, age, gender, and pathological grade. Finally, we assessed the accuracy of the model based on the area under the receiver operating characteristic (AUROC) and the concordance index (C-index).
Evaluation of prognostic signature of tumor immune microenvironment
We utilized Gene Set Enrichment Analysis (GSEA) to identify predominantly enriched signaling pathways in the two risk groups. Statistical significance criteria were set at p < 0.05 and FDR < 0.25. We subsequently assessed the variations in immune and stromal cell expressions among patients in the two risk groups and calculated StromalScore, ImmuneScore, and ESTIMATEScore (StromalScore + ImmuneScore) for all patients by the “limma” and “estimate” R packages. To evaluate the infiltration of immunocytes and determine the relative content, we employed Single-Sample GSEA (ssGSEA) and the “GSVA” R package. Lastly, we detected the expression levels of immune checkpoints in high- and low-risk groups and the tumor mutation burden (TMB) scores in two risk groups were ascertained according to somatic mutation data.
Cluster analysis based on prognostic NRlncRNAs
On the basis of NRlncRNA expression levels associated with the risk model, we utilized the “ConsensusClusterPlus” R package to identify potential molecular subgroups responsive to immunotherapy. This analysis ultimately revealed three distinct subforms. Next, we used various packages such as “survival,” “Rzsne,” “limma,” and “reshape2” to estimate and analyze these three distinct subforms.
Assessing chemotherapy and targeted agent response
To gauge the efficacy of the risk signature in predicting responses to chemotherapy and targeted therapy, we utilized the “oncoPredict” package to examine the correlation between the risk score and the half-maximal inhibitory concentration (IC50) of chemotherapy drugs.
Cell lines and cell culture
The two BCa cell lines (T24,5637) and the human immortalized uroepithelial cell line (SV-HUC-1) were all bought from American Type Culture Collection (ATCC) (USA). T24 and 5637 were maintained in RPMI 1640 (Hyclone, China) supplemented with 10% fetal bovine serum media. SV-HUC-1 was cultured in Ham’s F-12K (Hyclone, China) with 10% fetal bovine serum media. An incubator with 5% CO2 was applied to culture all cells at 37°C.
Quantitative real time PCR
We extracted total RNA of two bladder cancer cell lines and human immortalized uroepithelial cell line with TRIzol reagent (Thermo Fisher Scientific, USA). Then we used the reverse transcription kit from Servicebio (China) to obtain cDNAs. After that, the RT-qPCR was conducted on the Quantagene q225 real-time PCR system.
Statistics analysis
In this study, R software (version 4.2.2) was employed for statistical analyses, and the Wilcoxon rank-sum test was used to assess differences between the two subgroups. Statistical significance was defined as p < 0.05 for all two-tailed tests.
Data availability statement
The datasets of TCGA cohort for this study can be found in the (The Cancer Genome Atlas Program) (https://portal.gdc.cancer.gov/, accessed on 10 July 2023).
Results
Identification of NRlncRNAs in BCa patients
Initially, expression data were retrieved from the TCGA-BLCA database, encompassing 19 normal and 406 tumor samples. A total of 1973 necroptosis-related lncRNAs (NRlncRNAs) were identified, meeting the criteria of a correlation coefficient greater than 0.4 and p < 0.001, based on the gene expression matrix. Through a differential expression analysis, we selected 296 NRlncRNAs with |log2 fold change (FC)| > 1 and p < 0.05 (Figure 1A, 1B). The correlation between necroptosis-related genes and NRlncRNAs was visually represented in Figure 1C, 1D.
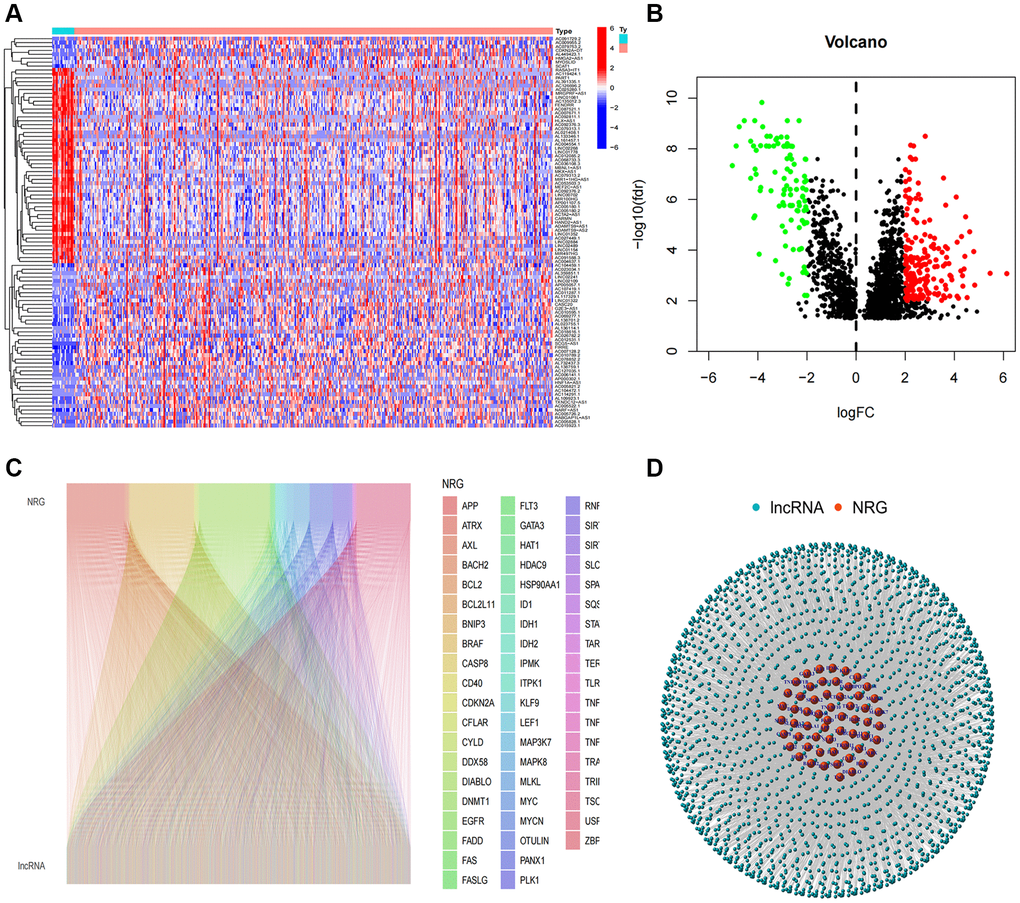
Figure 1. Identification of necroptosis-related lncRNAs. (A) Heatmap of differentially expressed NRlncRNAs based on their expression levels; (B) The volcano diagram of differentially expressed NRlncRNAs; (C) Sankey diagram of necroptosis-related genes and lncRNAs; (D) The network of necroptosis genes and lncRNAs.
Construction of the NRlncRNAs risk model
Following a uni-Cox regression analysis, 10 NRlncRNAs related with overall survival (OS) were pinpointed (Figure 2A). Subsequent LASSO and multi-Cox regression analyses led to the identification of 6 NRlncRNAs for the development of a prognostic model (Figure 2B–2E). The correlation between these 6 lncRNAs and necroptosis genes was displayed in Figure 2F. After using the expression levels and regression coefficients of the 6 lncRNAs, each BCa patient’s risk score was computed using the following formula:
Furthermore, we conducted a survival analysis of the 6 NRlncRNAs and observed their potential as independent prognostic factors for BCa patients (Figure 2G–2L).
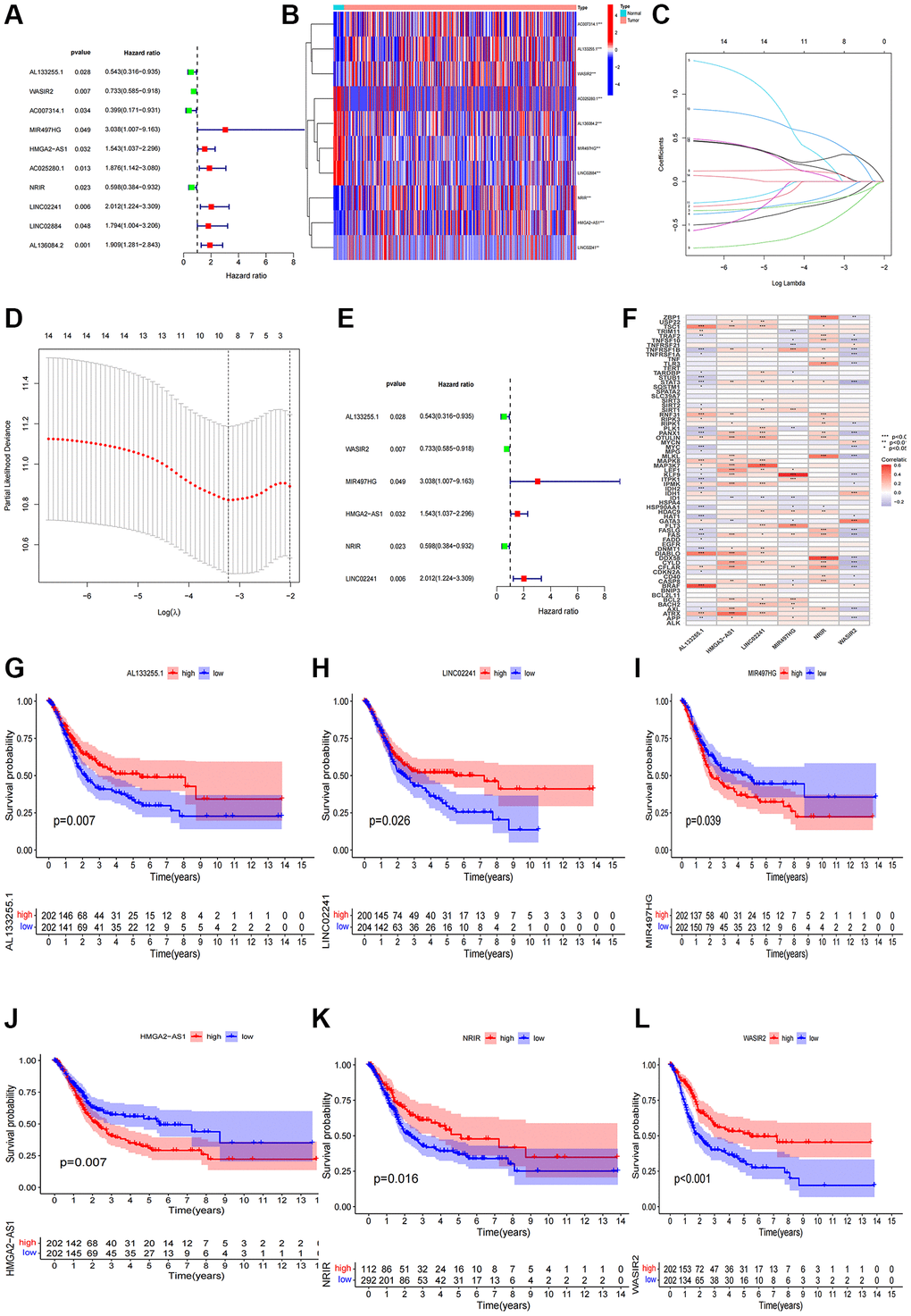
Figure 2. Construction of the NRlncRNAs risk model. (A) The forest plot of prognostic lncRNAs obtained by uni-Cox regression analysis; (B) Heatmap of 15 prognostic lncRNAs expression; (C, D) The final NRlncRNAs of the risk model was confirmed from LASSO regression analysis; (E) The forest plot of six NRlncRNAs of the risk model; (F) Correlations between NRlncRNAs of the risk model and necroptosis-related genes; (G–L) Kaplan–Meier curve of OS analyzed for the six NRlncRNA.
Assessment and validation of the prognostic risk model
After categorizing all BCa patients into low- and high-risk groups based on their median risk scores, we compared their survival status and time. The results revealed a more favorable prognosis for patients in the low-risk group compared to those in the high-risk group. To confirm the applicability of the risk model, we randomly divided the entire cohort (n = 404) into training and testing sets. As expected, consistent with the results in the entire cohort, patients in the high-risk group exhibited worse prognoses compared to those in the low-risk group in both the training and testing sets (Figure 3A–3L). Furthermore, several clinical features, including age (Figure 4A, 4B), gender (Figure 4C, 4D), grade (Figure 4E, 4F), and stage (Figure 4G–4H) confirmed these results. The AUROC values of 1-, 3-, and 5-year survival rates were displayed in Figure 5A. Notably, the AUC value of the risk model surpassed that of individual clinical features (Figure 5B, 5C). Uni- and multi-Cox regression analyses revealed the hazard ratios (HR) of 1.516 and 1.441 for the risk score (Figure 5D, 5E). These findings underscored the significance of the risk score as a robust predictor of prognosis, independent of clinical factors. Moreover, we created a nomogram that integrated the comprehensive landscape of the risk score and independent clinical features to predict 1-, 3-, and 5-year OS (Figure 5F). Calibration charts demonstrated high consistency between actual observation results and the nomogram’s predictions for 1-, 3-, and 5-year OS (Figure 5G). Overall, our results affirm the high accuracy of the risk model in predicting OS for BCa patients.
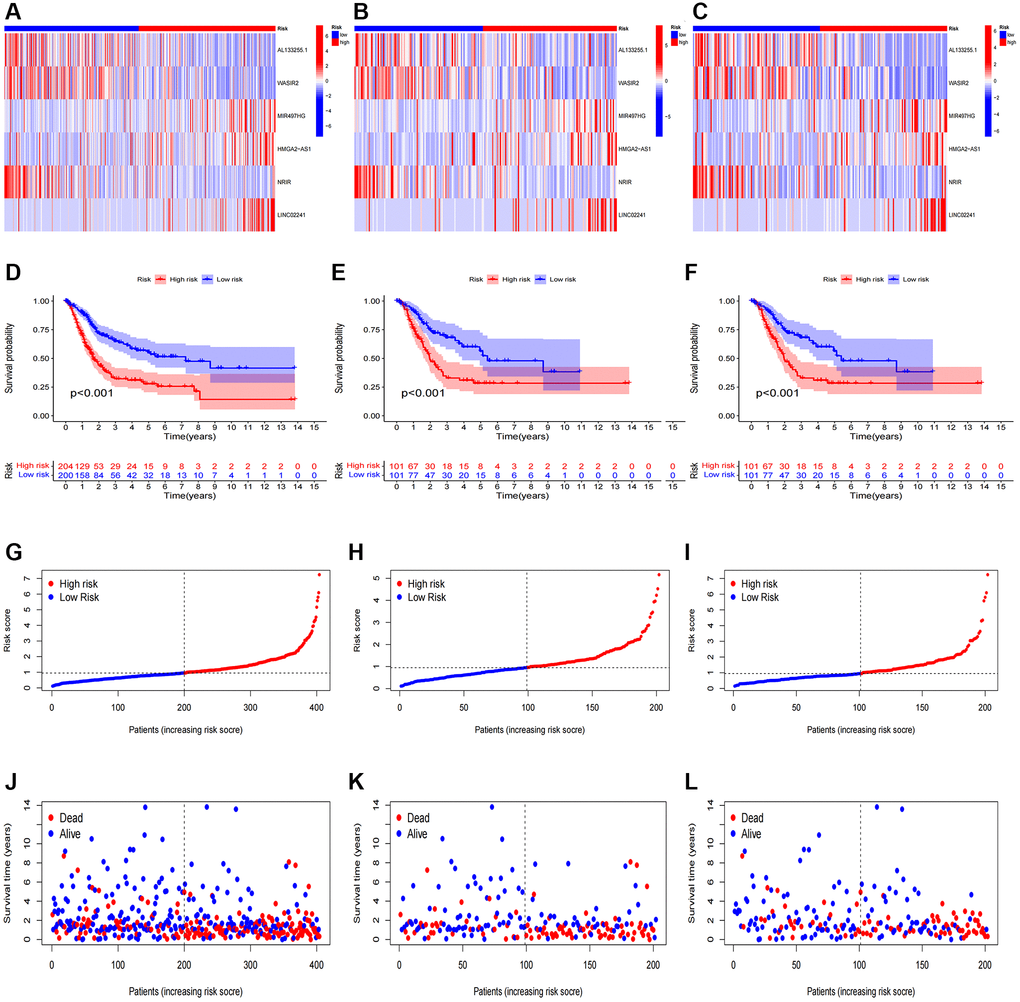
Figure 3. Prognosis evaluation and internal verification of the risk model. Heatmaps of six lncRNA expressions (A–C), risk model (D–F), survival time and survival status (G–I), and Kaplan-Meier survival curves of patients (J–L) in the entire, train, and test cohorts, respectively.
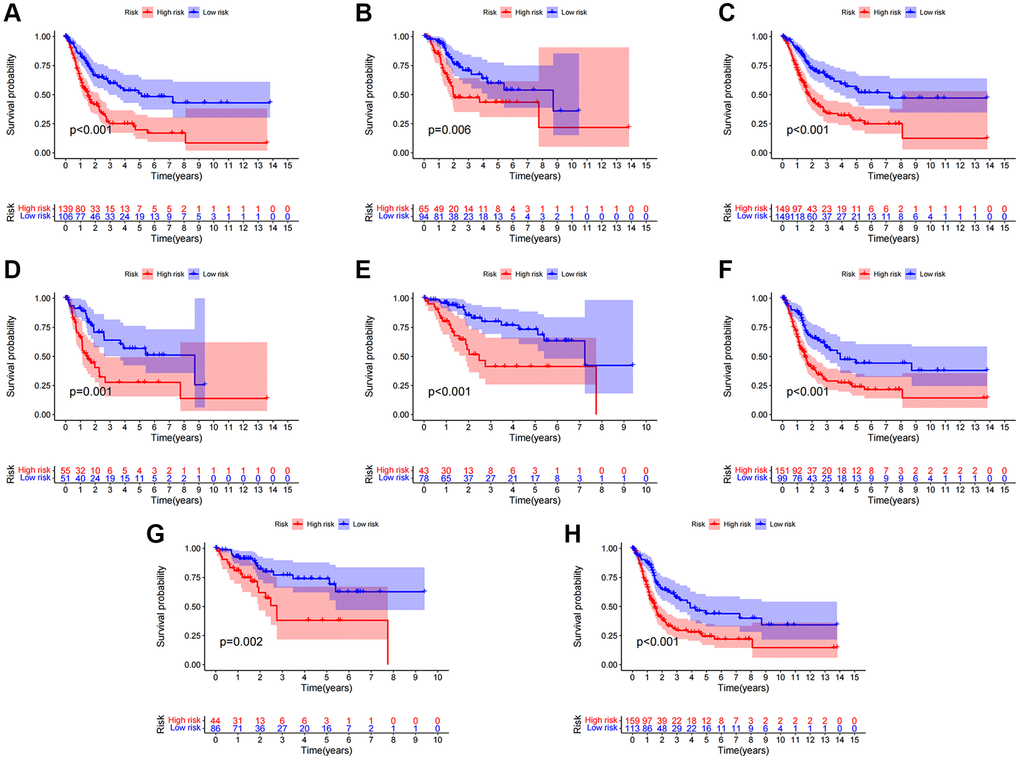
Figure 4. Clinical significance of the risk model. (A, B) Patients with age >65 and age ≤65; (C, D) Female patients and male patients; (E, F) Patients with stages I–II and stages III–IV; (G, H) Patients with stages T1-2 and stages T3-4.
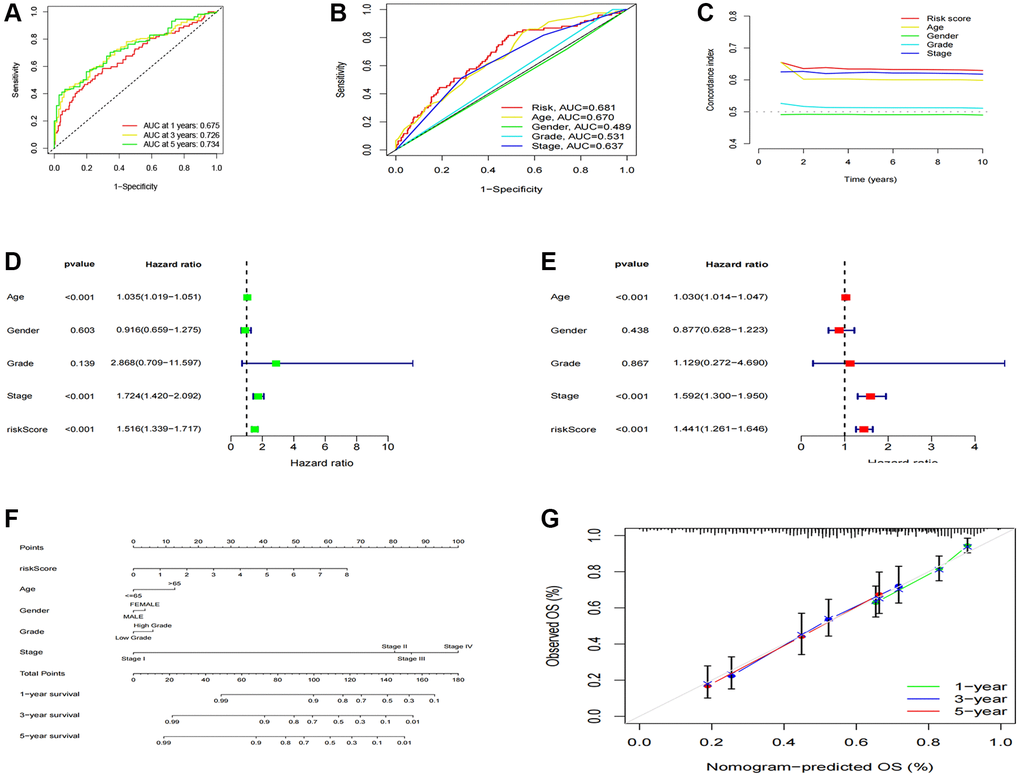
Figure 5. Construction and validation of the nomogram. (A) 1-, 3-, and 5-years ROC curves of the entire cohort; (B) The ROC curves of risk score and clinical features; (C) The C-index curves of the risk model; (D) Uni-Cox analyses of clinical factors and risk score with OS; (E) Multi-Cox analyses of clinical and risk score with OS; (F) 1-, 3-, and 5-year OS of BC patients predicted by the nomogram; (G) The calibration plots for predicting 1-, 3-, and 5-year OS.
Correlations of the risk score with immune factors and the tumor immune microenvironment
We conducted GSEA to explore gene enrichment between two risk groups (Figure 6A). In the high-risk group, we found significant enrichment of ECM receptor interaction, focal adhesion, melanoma, pathways in cancer and regulation of actin cytoskeleton, suggesting a close association with tumor and immune-related pathways. Further investigation into the relationship between risk scores and tumor-infiltrating immunocytes revealed a higher abundance of immunocytes in the high-risk group (Figure 6B). In addition, StromalScore, ImmuneScore, and ESTIMATEScore were all higher in the high-risk group compared to the low-risk group (Figure 6C–6E). To delve deeper into the association between risk scores and immunocytes and functions, we quantified the infiltrating levels of 16 immune cells and 13 immune functions. Our findings demonstrated that most immunocytes exhibited higher scores in the high-risk group, including B cells, CD8+ T cells, DCs, immature dendritic cells, macrophages, mast cells, neutrophils, pDCs, T helper cells, T follicular helper cells, Th1 cells, tumor-infiltrating lymphocyte, and T regulatory cells (Figure 6F). Furthermore, various immune pathways, including APC co-inhibition, APC co-stimulation, CCR, checkpoint, cytolytic activity, human leukocyte antigen, inflammation-promoting, para inflammation, T cell co-inhibition and T cell co-stimulation, scored higher in the high-risk group compared to the low-risk group (Figure 6G). Above conclusions suggested heightened activity of these immune functions in the high-risk group, implying that patients in this group might be more responsive to immunotherapy. We also analyzed the relationship between risk score and immune checkpoints, revealing that certain immune checkpoints, including FAS, STAT3, AXL, KLF9, EGFR, MYC, STUB1, APP were more enriched in the high-risk group while TRIM11, ID1, CD40, RIPK3, GATA3, TNFRSF21 revealed a higher enrichment in the low-risk group (Figure 6H). The elevated expression of these immune checkpoints suggested a potentially enhanced response to immunotherapy for these patients.
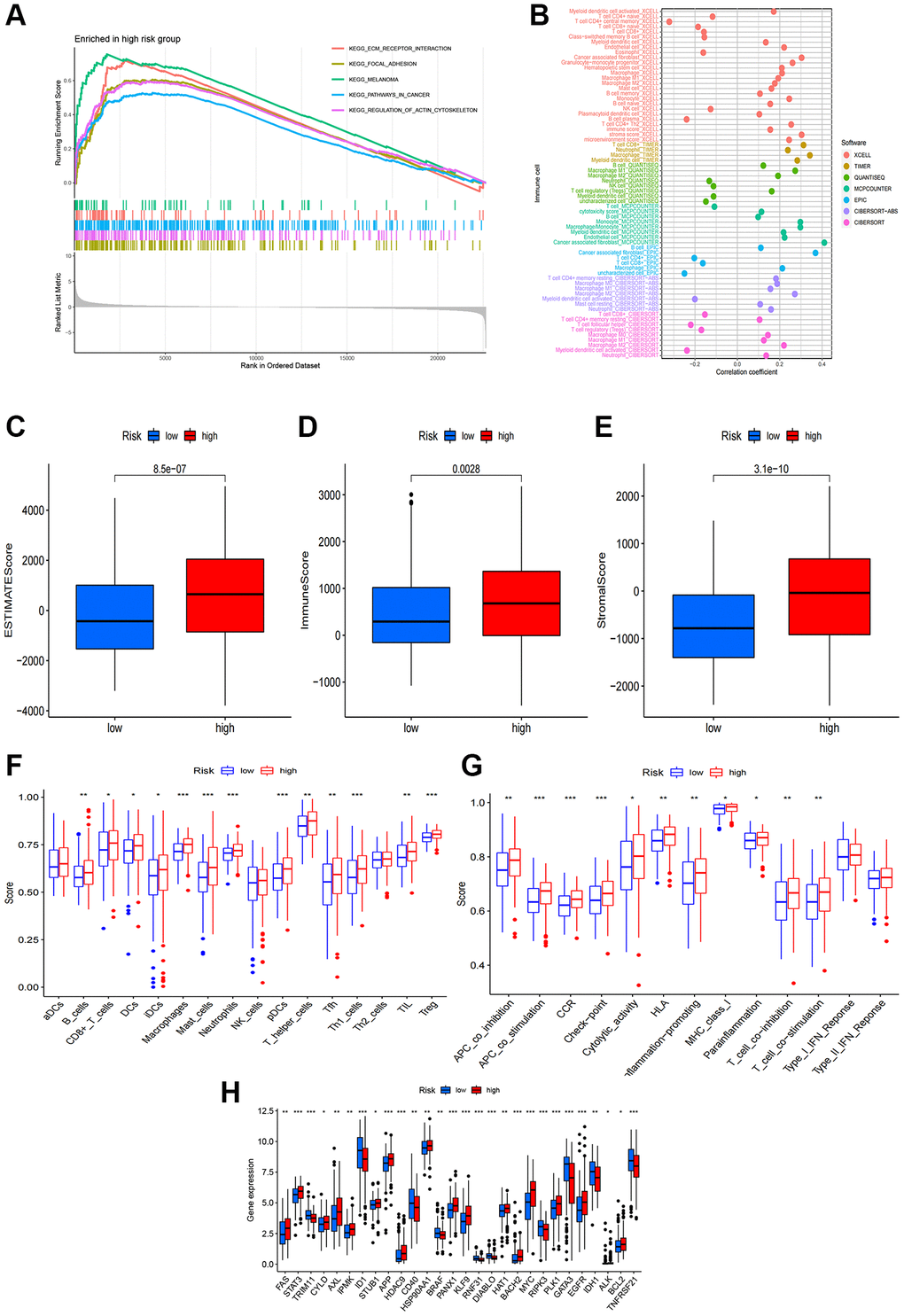
Figure 6. Correlations of the risk score with tumor immune microenvironment. (A) GSEA of the significantly enriched pathways in the high-risk group; (B) The immune cell bubble of risk groups; (C–E) The boxplots of the comparison of ESTIMATEScore, ImmuneScore and StromalScore, respectively, between low-risk and high-risk groups; (F, G) The infiltration level of 16 immune cells and 13 immune functions. (H) The expression of common immune checkpoints in the risk groups. *p < 0.05, **p < 0.01, ***p < 0.001.
Cluster analysis based on risk signature
To assess the immune microenvironment and response of different tumor subtypes, we employed cluster analysis to delineate distinct subtypes. Using the “ConsensusClusterPlus” package (Figure 7A), we categorized the patients into three clusters. Our analysis revealed that most patients in the high-risk group were clustered in cluster 1 and 3, while the majority of patients in cluster 2 belonged to the low-risk group (Figure 7B). Furthermore, utilizing survival analysis, cluster 2 exhibited better overall survival compared to clusters 1 and 3 (Figure 7C). To confirm the distinction between these three clusters, we performed PCA and t-SNE, which yielded clear separation of the clusters (Figure 7D–7G). We then conducted immunological factor and tumor microenvironment analyses, which revealed that cluster 1 and 3 had higher Immunescore, Stromalscore, and ESTIMATEscore than clusters 2 (Figure 7H–7J), consistent with our previous results. In addition, we examined the variations in infiltrating immunocytes across the clusters using severe immune platforms and represented them in a heatmap (Figure 7K). Interestingly, we found that a majority of immune checkpoints had lower expression in cluster 2 (Figure 7L).
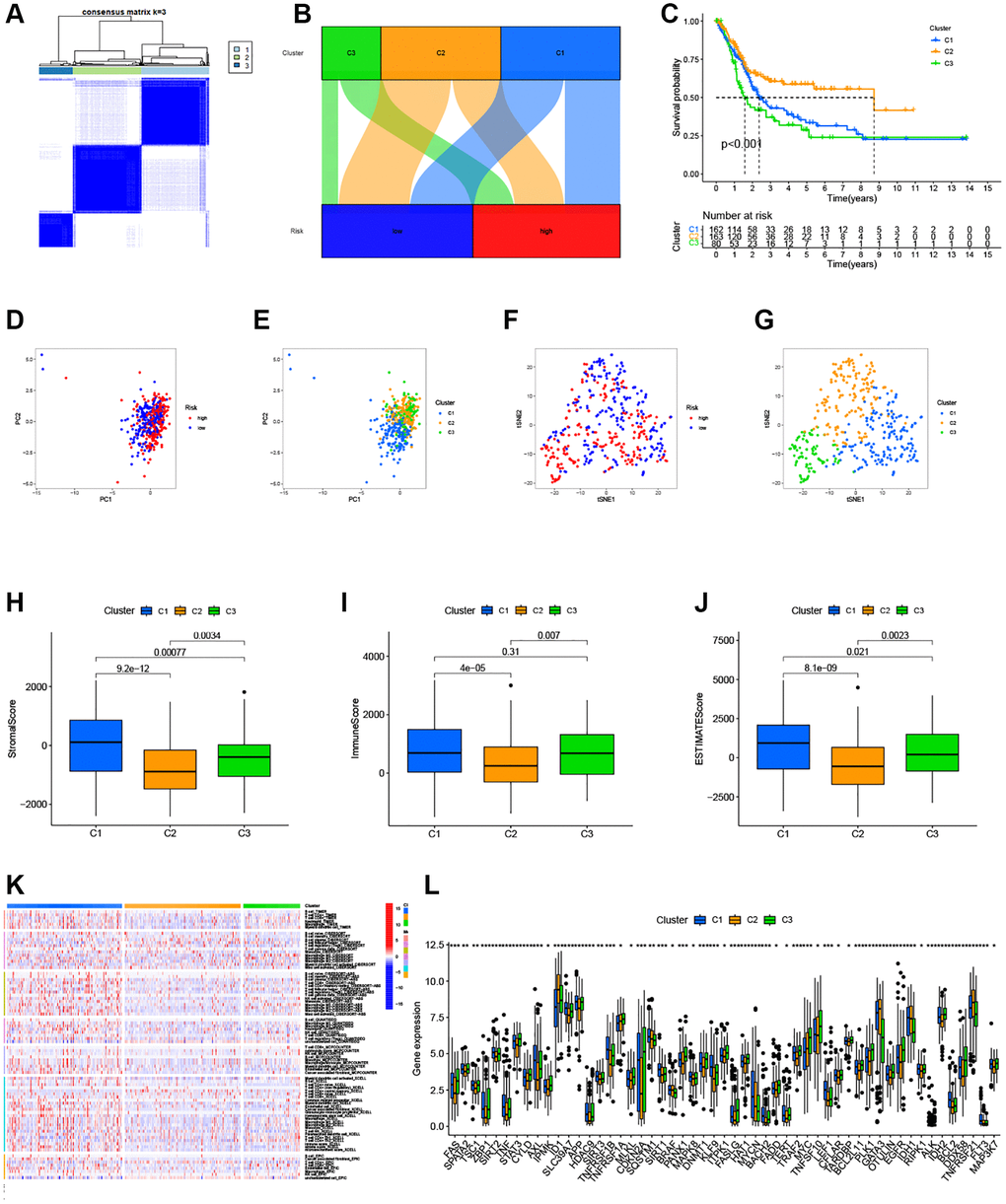
Figure 7. Clusters analysis for the risk model. (A) BC patients were divided into three clusters according to the consensus clustering matrix (k = 3); (B) Sankey diagram of the three clusters; (C) Kaplan–Meier curve of OS analyzed for the three clusters; (D, E) The PCA of risk groups and clusters; (F, G) The t-SNE of risk groups and clusters; (H–J) Immune-related scores in clusters; (K) Heatmap of immune cells in clusters; (L) The expression of immune checkpoints in clusters.
Tumor mutation burden (TMB) analysis
In our study, we found that 184 (93.4%) of 197 samples in the low-risk group exhibited a wider TMB compared to the high-risk group (92.08%), suggesting that immunotherapy might have a greater impact on the low-risk group. Furthermore, prominent genetic changes were noted in genes such as PIK3CA, TP53, TTN, and MUC16, each with a mutation frequency exceeding 15% (Figure 8A, 8B). Based on the survival curves, the low-TMB group showed superior overall survival than the high-TMB group (Figure 8C). Additionally, a robust correlation between TMB and risk score was evident, as demonstrated in Figure 8E, 8F (R = 0.13, p = 0.016). Combining these two parameters, we observed that patients who exhibited the lowest risk score and lowest TMB resulted the best prognosis, while those with the highest risk score and highest TMB resulted the worst prognosis (Figure 8D).
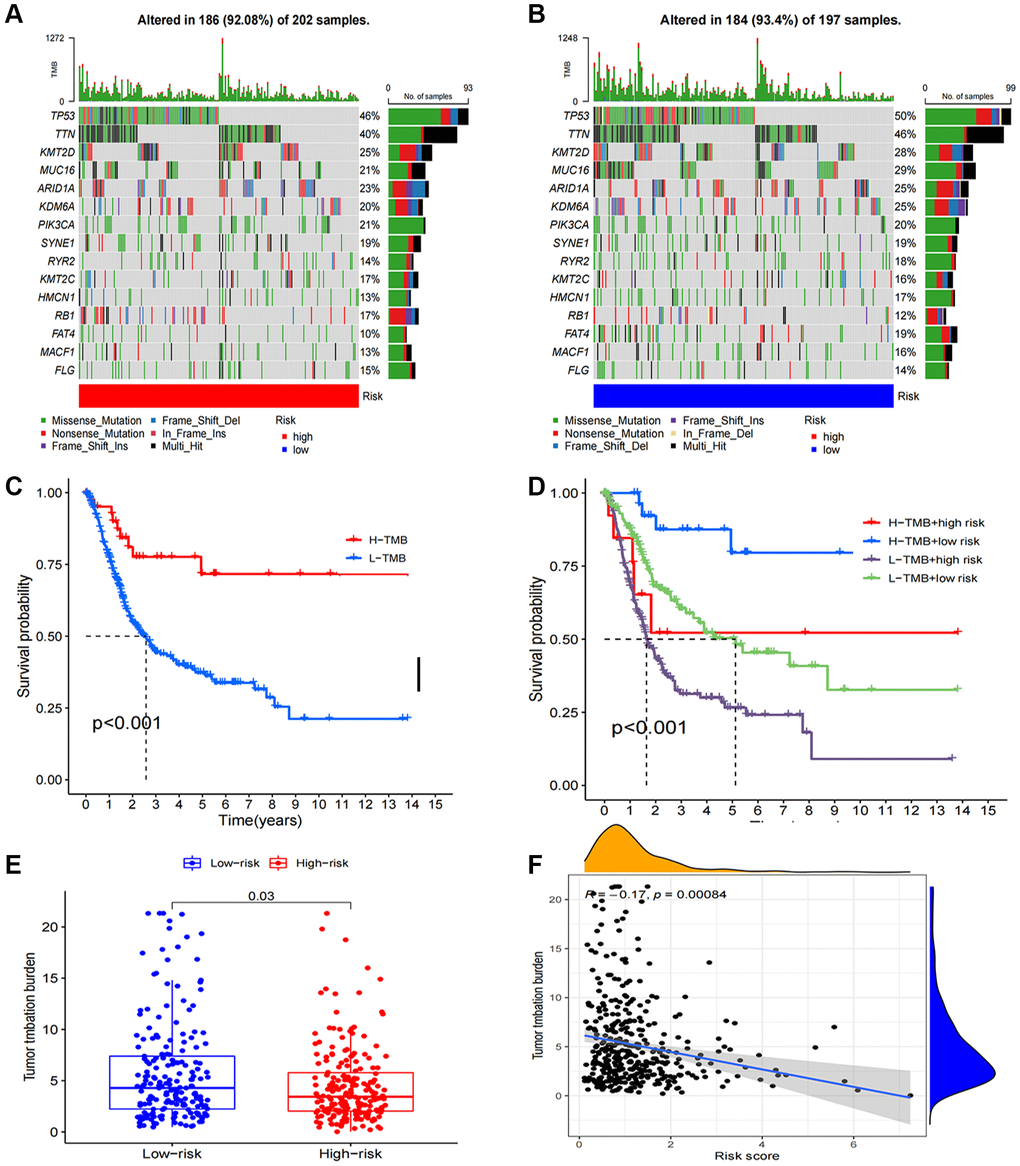
Figure 8. Tumor mutation burden calculation analysis. (A, B) Tumor somatic mutation waterfall graph in high-risk group and low-risk group; (C) Kaplan–Meier curve of OS analyzed for TMB; (D) Kaplan–Meier curve of OS analyzed for TMB and risk groups; (E) The relationship between TMB and risk groups; (F) Correlation analysis of the risk score and TMB.
Potential effects of chemotherapy in BCa
We assessed the sensitivity of chemotherapy drugs in BCa patients by determining their IC50 values. Our findings revealed that the IC50 values for Alisertib, Buparlisib, Dasatinib, and Cisplatin were lower in the high-risk group, implying a correlation between the high-risk status and increased sensitivity to these drugs. Conversely, patients with low-risk status exhibited significantly lower IC50 values for Sorafenib and EPZ5676 (Figure 9A–9F).
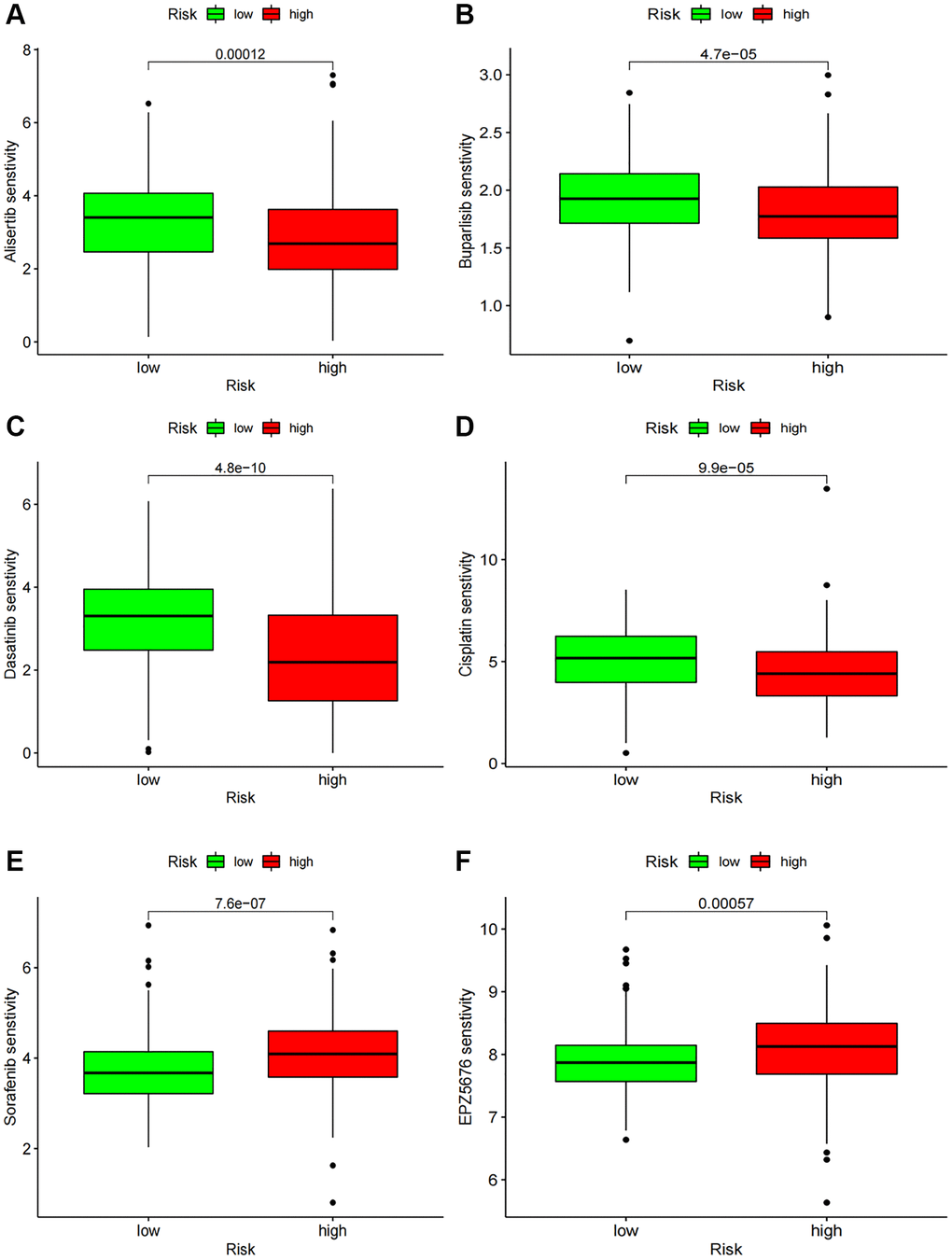
Figure 9. Chemotherapy drug sensitivity. (A–F) IC50 of several chemotherapy drugs in BC. (A) Alisertib; (B) Buparlisib; (C) Dasatinib; (D) Cisplatin; (E) Sorafenib; (F) EPZ5676.
Expression of prognostic NRlncRNAs in bladder cancer
We conducted RT-qPCR to detect the expression levels of NRlncRNAs in the risk model. The outcomes revealed that LINC02241, NRIR, MIR497HG and WASIR2 exhibited higher expression in BCa cell lines compared to normal urinary epithelial cells (Figure 10A–10D).
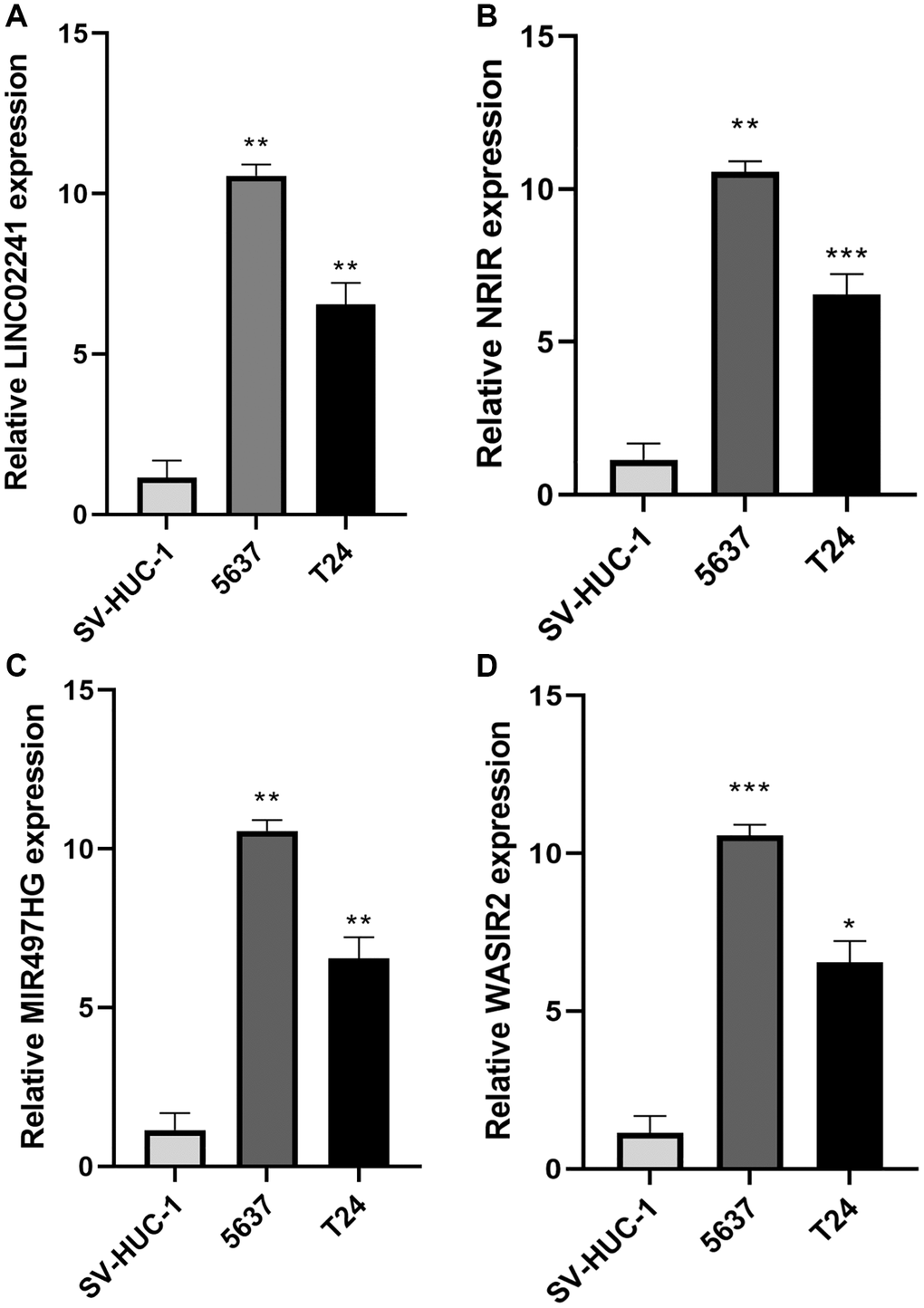
Figure 10. Expression of necroptosis-related genes in bladder cancer. (A–D) Relative expression of LINC02241, NRIR, MIR497HG and WASIR2 in 2 BCa cell lines (5637, T24) and SV-HUC-1 cell line. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
As widely recognized, resistance of cell apoptosis is a major cause of chemotherapy failure during cancer treatment [16]. Necroptosis, an alternative form of regulated necrotic cell death independent of caspases, and acts as an alternative mode for overcoming resistance to programmed cell death via apoptosis [17]. Necroptosis has a dual role in tumors. On the one hand, key mediators in the necroptosis pathway involve the promotion of tumor progression and metastasis [18, 19], while on the other hand, necroptosis can protect against tumor development when apoptosis is compromised [20].
While numerous studies have focused on developing lncRNA signatures for assessing cancer prognosis [21, 22], limited research has explored the role of NRlncRNAs in the tumor microenvironment and prognosis. In our study, we explored the intricate correlation between the tumor microenvironment, immune cell infiltration, immunological checkpoints, and NRlncRNAs. Our findings might provide valuable insights for the clinical diagnosis and treatment of BCa.
First, we collected 296 NRlncRNAs with varying levels of expression. Using LASSO, uni- and multi-Cox regression analyses, we identified 6 NRlncRNAs (ALI33255.1, WASIR2, MIR497HG, HMGA2-AS1, NRIR, and LINC02241) and built a risk model for NRlncRNAs. The LASSO algorithm calculated the risk score for the OS outcome of each patient, to classify all BCa patients into high- and low-risk groups. Notably, patients in the high-risk group exhibited a worse prognosis than their low-risk counterparts. The risk model demonstrated favorable discrimination performance in predicting BCa patient prognosis. Furthermore, our findings were aligned with prior studies emphasizing the importance of lncRNA-related signatures in BCa prognosis prediction [23–25]. Our model exhibited well sensitivity in predicting BCa patient survival, as evidenced by clinicopathological analysis, survival analysis, PCA, and TMB analysis.
Recent studies have emphasized the integral relationship between tumor immune microenvironment and tumor initiation and progression [26–28]. Immunocyte infiltration serves as a regulator of tumor progression, influencing prognosis by reshaping the tumor microenvironment [29]. In our study, we observed an interrelation between the infiltration levels of certain immunocytes and the risk score. Additionally, using the “ConsensusClusterPlus” package, patients were classified into three clusters based on our risk model. Cluster 2 had significantly reduced immunological checkpoint expression compared to clusters 1 and 3, suggesting an association between consensus clustering and immunological microenvironment. Since immunotherapy has proven to be an important treatment for BCa, identifying patients who can derive maximum benefit from immunotherapy remains crucial [30]. TMB, reflecting the number of somatic mutations per megabase of genomic sequence, aids in identifying cancer patients likely to respond to immune checkpoint inhibitors [31]. Our results showed that patients that possessed the lowest TMB experienced the best prognosis and were more likely to benefit from chemotherapy drugs. In summary, these findings might offer a promising method for optimizing immunotherapy for BCa patients.
Our study still had some limitations. First, we were unable to perform external validation analysis, as the 6 identified lncRNAs could not be found in other databases. Second, while our study primarily relied on bioinformatics analysis, further experiments were necessary to establish the mechanism of the NRlncRNAs in BCa.
In conclusion, our study unveils a novel signature crafted from NRlncRNAs, predicting survival in BCa patients, and offering insights into the immune microenvironment and chemotherapy treatment.
Author Contributions
Conceptualization, Zhiyuan Chen and Lei Wang; Data curation, Zhengyu Jiang, Jun Jian; Formal analysis, Jingsong Wang, Zhengyu Jiang and Kai Wang; Funding acquisition, Lei Wang; Investigation, Qingyuan Zheng; Methodology, Jingsong Wang, Zhengyu Jiang and Kai Wang; Project administration, Zhiyuan Chen, Xiuheng Liu, Rui Yang and Lei Wang; Software, Qingyuan Zheng; Supervision, Zhiyuan Chen, Xiuheng Liu, Rui Yang and Lei Wang; Validation, Jingsong Wang, Zhengyu Jiang and Kai Wang; Visualization, Jingsong Wang, Zhiyuan Chen, Xiuheng Liu, Rui Yang and Lei Wang; Writing – original draft, Jingsong Wang and Zhengyu Jiang; Writing – review and editing, Xiuheng Liu, Rui Yang and Lei Wang.
Acknowledgments
We thank our colleagues in the Department of Urology and Pathology, Renmin Hospital of Wuhan University, for their support of this work, as well as all colleagues involved in model development and data collection.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
This research was funded by “National Natural Science Foundation of China, Grant No. 82000639”.
References
- 1. Burger M, Catto JW, Dalbagni G, Grossman HB, Herr H, Karakiewicz P, Kassouf W, Kiemeney LA, La Vecchia C, Shariat S, Lotan Y. Epidemiology and risk factors of urothelial bladder cancer. Eur Urol. 2013; 63:234–41. https://doi.org/10.1016/j.eururo.2012.07.033 [PubMed]
- 2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71:209–49. https://doi.org/10.3322/caac.21660 [PubMed]
- 3. Patel VG, Oh WK, Galsky MD. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J Clin. 2020; 70:404–23. https://doi.org/10.3322/caac.21631 [PubMed]
- 4. Babjuk M, Burger M, Capoun O, Cohen D, Compérat EM, Dominguez Escrig JL, Gontero P, Liedberg F, Masson-Lecomte A, Mostafid AH, Palou J, van Rhijn BWG, Rouprêt M, et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (Ta, T1, and Carcinoma in Situ). Eur Urol. 2022; 81:75–94. https://doi.org/10.1016/j.eururo.2021.08.010 [PubMed]
- 5. Teoh JY, Kamat AM, Black PC, Grivas P, Shariat SF, Babjuk M. Recurrence mechanisms of non-muscle-invasive bladder cancer - a clinical perspective. Nat Rev Urol. 2022; 19:280–94. https://doi.org/10.1038/s41585-022-00578-1 [PubMed]
- 6. Flaig TW, Spiess PE, Abern M, Agarwal N, Bangs R, Boorjian SA, Buyyounouski MK, Chan K, Chang S, Friedlander T, Greenberg RE, Guru KA, Herr HW, et al. NCCN Guidelines® Insights: Bladder Cancer, Version 2.2022. J Natl Compr Canc Netw. 2022; 20:866–78. https://doi.org/10.6004/jnccn.2022.0041 [PubMed]
- 7. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013; 154:26–46. https://doi.org/10.1016/j.cell.2013.06.020 [PubMed]
- 8. Lim LJ, Wong SYS, Huang F, Lim S, Chong SS, Ooi LL, Kon OL, Lee CG. Roles and Regulation of Long Noncoding RNAs in Hepatocellular Carcinoma. Cancer Res. 2019; 79:5131–9. https://doi.org/10.1158/0008-5472.CAN-19-0255 [PubMed]
- 9. Luo W, Wang J, Xu W, Ma C, Wan F, Huang Y, Yao M, Zhang H, Qu Y, Ye D, Zhu Y. LncRNA RP11-89 facilitates tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer. Cell Death Dis. 2021; 12:1043. https://doi.org/10.1038/s41419-021-04296-1 [PubMed]
- 10. Chen C, Zheng H, Luo Y, Kong Y, An M, Li Y, He W, Gao B, Zhao Y, Huang H, Huang J, Lin T. SUMOylation promotes extracellular vesicle-mediated transmission of lncRNA ELNAT1 and lymph node metastasis in bladder cancer. J Clin Invest. 2021; 131:146431. https://doi.org/10.1172/JCI146431 [PubMed]
- 11. Vucur M, Ghallab A, Schneider AT, Adili A, Cheng M, Castoldi M, Singer MT, Büttner V, Keysberg LS, Küsgens L, Kohlhepp M, Görg B, Gallage S, et al. Sublethal necroptosis signaling promotes inflammation and liver cancer. Immunity. 2023; 56:1578–95.e8. https://doi.org/10.1016/j.immuni.2023.05.017 [PubMed]
- 12. Zhe-Wei S, Li-Sha G, Yue-Chun L. The Role of Necroptosis in Cardiovascular Disease. Front Pharmacol. 2018; 9:721. https://doi.org/10.3389/fphar.2018.00721 [PubMed]
- 13. Najafov A, Chen H, Yuan J. Necroptosis and Cancer. Trends Cancer. 2017; 3:294–301. https://doi.org/10.1016/j.trecan.2017.03.002 [PubMed]
- 14. Baik JY, Liu Z, Jiao D, Kwon HJ, Yan J, Kadigamuwa C, Choe M, Lake R, Kruhlak M, Tandon M, Cai Z, Choksi S, Liu ZG. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun. 2021; 12:2666. https://doi.org/10.1038/s41467-021-23004-3 [PubMed]
- 15. Lin P, Lin C, He R, Chen H, Teng Z, Yao H, Liu S, Hoffman RM, Ye J, Zhu G. TRAF6 regulates the abundance of RIPK1 and inhibits the RIPK1/RIPK3/MLKL necroptosis signaling pathway and affects the progression of colorectal cancer. Cell Death Dis. 2023; 14:6. https://doi.org/10.1038/s41419-022-05524-y [PubMed]
- 16. Zou Y, Xie J, Zheng S, Liu W, Tang Y, Tian W, Deng X, Wu L, Zhang Y, Wong CW, Tan D, Liu Q, Xie X. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int J Surg. 2022; 107:106936. https://doi.org/10.1016/j.ijsu.2022.106936 [PubMed]
- 17. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, Jin K, Ni Q, Yu X, Liu C. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019; 18:100. https://doi.org/10.1186/s12943-019-1029-8 [PubMed]
- 18. Park S, Hatanpaa KJ, Xie Y, Mickey BE, Madden CJ, Raisanen JM, Ramnarain DB, Xiao G, Saha D, Boothman DA, Zhao D, Bachoo RM, Pieper RO, Habib AA. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009; 69:2809–16. https://doi.org/10.1158/0008-5472.CAN-08-4079 [PubMed]
- 19. Park S, Zhao D, Hatanpaa KJ, Mickey BE, Saha D, Boothman DA, Story MD, Wong ET, Burma S, Georgescu MM, Rangnekar VM, Chauncey SS, Habib AA. RIP1 activates PI3K-Akt via a dual mechanism involving NF-kappaB-mediated inhibition of the mTOR-S6K-IRS1 negative feedback loop and down-regulation of PTEN. Cancer Res. 2009; 69:4107–11. https://doi.org/10.1158/0008-5472.CAN-09-0474 [PubMed]
- 20. Feng X, Song Q, Yu A, Tang H, Peng Z, Wang X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma. 2015; 62:592–601. https://doi.org/10.4149/neo_2015_071 [PubMed]
- 21. Liu Z, Guo C, Dang Q, Wang L, Liu L, Weng S, Xu H, Lu T, Sun Z, Han X. Integrative analysis from multi-center studies identities a consensus machine learning-derived lncRNA signature for stage II/III colorectal cancer. EBioMedicine. 2022; 75:103750. https://doi.org/10.1016/j.ebiom.2021.103750 [PubMed]
- 22. Liu Z, Liu L, Weng S, Guo C, Dang Q, Xu H, Wang L, Lu T, Zhang Y, Sun Z, Han X. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat Commun. 2022; 13:816. https://doi.org/10.1038/s41467-022-28421-6 [PubMed]
- 23. Li Z, Li Y, Zhong W, Huang P. m6A-Related lncRNA to Develop Prognostic Signature and Predict the Immune Landscape in Bladder Cancer. J Oncol. 2021; 2021:7488188. https://doi.org/10.1155/2021/7488188 [PubMed]
- 24. Wang J, Shen C, Dong D, Zhong X, Wang Y, Yang X. Identification and verification of an immune-related lncRNA signature for predicting the prognosis of patients with bladder cancer. Int Immunopharmacol. 2021; 90:107146. https://doi.org/10.1016/j.intimp.2020.107146 [PubMed]
- 25. Lian P, Wang Q, Zhao Y, Chen C, Sun X, Li H, Deng J, Zhang H, Ji Z, Zhang X, Huang Q. An eight-long non-coding RNA signature as a candidate prognostic biomarker for bladder cancer. Aging (Albany NY). 2019; 11:6930–40. https://doi.org/10.18632/aging.102225 [PubMed]
- 26. Xu C, Fang H, Gu Y, Yu K, Wang J, Lin C, Zhang H, Li H, He H, Liu H, Li R. Impact of intratumoural CD96 expression on clinical outcome and therapeutic benefit in gastric cancer. Cancer Sci. 2022; 113:4070–81. https://doi.org/10.1111/cas.15537 [PubMed]
- 27. He X, Gu Y, Cao Y, Hu B, Fang H, Fei Y, Lv K, Liu X, Wang J, Lin C, Liu H, Zhang H, Li H, et al. Impact of intratumoural CD73 expression on prognosis and therapeutic response in patients with gastric cancer. Eur J Cancer. 2021; 157:114–23. https://doi.org/10.1016/j.ejca.2021.08.006 [PubMed]
- 28. Gao Z, Wang L, Song Z, Ren M, Yang Y, Li J, Shen K, Li Y, Ding Y, Yang Y, Zhou Y, Wei C, Gu J. Intratumoral CD73: An immune checkpoint shaping an inhibitory tumor microenvironment and implicating poor prognosis in Chinese melanoma cohorts. Front Immunol. 2022; 13:954039. https://doi.org/10.3389/fimmu.2022.954039 [PubMed]
- 29. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013; 14:1014–22. https://doi.org/10.1038/ni.2703 [PubMed]
- 30. Konala VM, Adapa S, Aronow WS. Immunotherapy in Bladder Cancer. Am J Ther. 2022; 29:e334–7. https://doi.org/10.1097/MJT.0000000000000934 [PubMed]
- 31. Merino DM, McShane LM, Fabrizio D, Funari V, Chen SJ, White JR, Wenz P, Baden J, Barrett JC, Chaudhary R, Chen L, Chen WS, Cheng JH, et al, and TMB Harmonization Consortium. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020; 8:e000147. https://doi.org/10.1136/jitc-2019-000147 [PubMed]