Introduction
THCA stands as the most prevalent malignancy within the realm of endocrine disorders, and its global incidence is unequivocally surging [1]. While a majority of THCA patients typically traverse a favorable prognostic trajectory, a subset grapples with formidable challenges such as distant metastasis, recurrent episodes, and suboptimal responses to therapeutic interventions, significantly impinging upon both their quality of life and survival rates [2]. Hence, the imperative quest for novel prognostic markers and therapeutic targets looms large, poised to revolutionize the landscape of THCA management and augment the well-being of affected individuals.
Disulfidptosis, emerging as a distinctive paradigm of cellular demise, operates autonomously from established programmed death pathways encompassing apoptosis, ferroptosis, necroptosis, and copper-induced death [3]. It delineates a rapid cellular demise incited by the perturbation of disulfide equilibrium within cellular confines. Remarkably, amidst glucose-deprived milieus, an anomalous accrual of disulfides, notably cystine, ensues within cells boasting heightened expression of SLC7A11. This accrual precipitates a state of disulfide stress, instigating a surge in disulfide bond densities within the actin-regulated cytoskeleton. Consequently, pronounced cytoskeletal contraction and detachment from the cellular membrane ensue, engendering disruptive distortions in cytoskeletal architecture, culminating inexorably in cellular demise. Ongoing investigations into disulfidptosis predominantly orbit around cancerous cell cohorts exhibiting augmented SLC7A11 expression amidst glucose-deprived microenvironments [4, 5]. Studies have discerned compelling associations linking disulfidptosis with a panoply of malignancies, spanning from bladder cancer [6] to hepatocellular carcinoma [7] and lung adenocarcinoma [8]. Nevertheless, the enigmatic nexus between disulfidptosis, as an incipient mechanism of cellular demise, and THCA remains shrouded in ambiguity, awaiting comprehensive elucidation.
The integration of machine learning methodologies has emerged as a cornerstone in the realm of gene discovery, particularly in discerning genes harboring diagnostic prowess. Harnessing the power of machine learning algorithms has heralded a paradigm shift, markedly amplifying the precision in discerning differentially expressed genes (DEGs) within microarray datasets [9]. In the context of our investigation, we embarked upon the construction of a prognostic framework predicated upon the intricate interplay among the attributes characterizing DRGs. This prognostic model stands poised as a harbinger of elucidating hitherto uncharted biological cascades underpinning the genesis and progression of THCA, thereby unfurling vistas for the identification of novel diagnostic modalities, prognostic markers, and therapeutic targets of paramount clinical significance. The meticulously delineated study protocol is visually encapsulated in Figure 1.
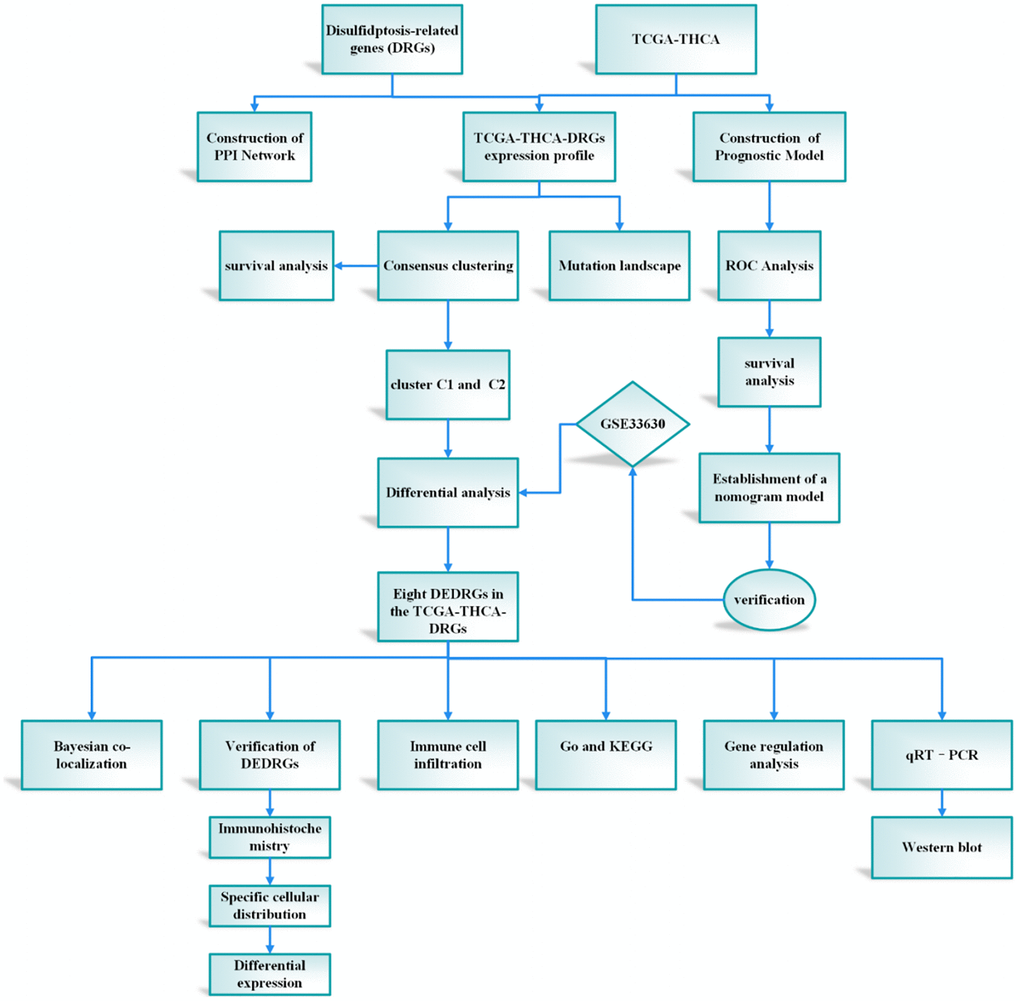
Figure 1. The protocol of our study procedure.
Discussion
THCA, as an endocrine system tumor, has been associated with favorable clinical therapeutic outcomes. However, the potential for cancer cells to evade anti-tumor immune responses raises concerns regarding recurrence or metastasis [23]. Consequently, the search for novel therapeutic targets in THCA remains imperative. Currently, the precise mechanism underlying disulfidptosis in THCA remains elusive. Therefore, through an analysis of the relationship between DRGs and THCA, this study aims to identify new therapeutic targets and provide theoretical insights for the treatment of THCA.
Initially, two distinct subtypes related to DRGs were identified through consensus clustering. Cluster C1, characterized by elevated DRGs levels, exhibited a favorable prognosis, whereas cluster C2, marked by diminished DRGs levels, presented a poorer prognosis. Subsequently, eight DEDRGs (FLNA, IQGAP1, MYH10, ACTN1, ZHX2, INF2, ME1, PDLIM1) were discerned from the TCGA-THCA-DRGs dataset, with seven genes showing up-regulation and one gene displaying down-regulation. Notably, cluster C1 demonstrated heightened expression of DEDRGs, while cluster C2 exhibited diminished expression. Among these genes, FLNA, classified as an actin-binding protein (ABP), plays a pivotal role in cytoskeletal formation and is intricately involved in cellular processes such as adhesion, proliferation, migration, signal transduction, and tumorigenesis [24]. Research has elucidated that the expression of FLNA escalates in invasive breast cancer tissue concomitant with reduced differentiation [25]. Additionally, FLNA exhibits significant mutations in THCA [26]. IQGAP1, an integral player in various biological processes within the human body, plays a pivotal role in modulating cell adhesion, signal transduction, and cell division [27]. Prior investigations have implicated IQGAP1 in the invasion and metastasis of THCA [28]. Moreover, MYH10, a member of the protein-coding gene superfamily, not only participates in normal cellular physiological activities but also bears close association with cancer initiation and progression [29]. Although MYH10’s involvement in bladder cancer and lung cancer has been documented, its relationship with THCA remains unexplored. Furthermore, heightened expression of ACTN1 is intricately linked to tumor cell motility, metastasis, and invasiveness [30]. Silencing ACTN1 expression has been demonstrated to impede the proliferation of hepatocellular carcinoma (HCC) [31]. ZHX2, recognized as a tumor suppressor gene, orchestrates lipid metabolism regulation, suppresses cell proliferation [32], and influences the immune microenvironment [33]. Notably, ZHX2 has been implicated in the occurrence of brain metastasis in THCA [34]. INF2, through its involvement in mitochondrial division, exerts effects on the proliferation and invasion of numerous tumors [35]. Furthermore, INF2 has been shown to inhibit THCA proliferation via the Hippo pathway [36]. ME1 primarily participates in lipid metabolism and tumor progression [37]. Although ME1 serves as a transcriptional target of thyroxine, its association with THCA remains unexplored [38]. PDLIM1, a cytoskeleton protein, interacts with actin stress fibers [39]. Studies have revealed diverse functions of PDLIM1 across various tumor tissues, wherein it can either impede epithelial-mesenchymal transition (EMT) and tumor cell infiltration and metastasis or promote tumor development [40]. Notably, PDLIM1 up-regulation has been observed in THCA [41]. Furthermore, we corroborated the findings related to DEDRGs in the GEPIA and HPA databases, which aligned with the existing literature. Additionally, analysis from the TIMER database revealed a robust correlation between DEDRGs and immune cells. Collectively, these results underscore the close association of DEDRGs with THCA and immune infiltration [23].
In our subsequent analysis, we employed functional enrichment analysis to delve into the potential biological functions and molecular mechanisms orchestrated by DEDRGs. The findings from this endeavor unveiled remarkable enrichments in pivotal pathways, including the MAPK, PPAR signaling pathway, and Proteoglycans in cancer. Among these pathways, the MAPK signaling pathway emerged as a central conduit bridging cell membrane receptors with nuclear events, playing a pivotal role in initiating and propelling various malignancies [42]. Notably, the strikingly elevated BRAF mutation rate, peaking at 76.2% in THCA, underscores its paramount importance in driving the progression of THCA by activating the MAPK signaling pathway [43]. Furthermore, the regulation mediated by PPAR, influencing thyroid peroxidase, thyroglobulin, and thyrotropin receptor gene promoters, facilitates the intricate process of thyroid cell differentiation [44]. These insights shed light on the intricate interplay between signaling pathways and genetic alterations, offering valuable perspectives into the molecular landscape of THCA pathogenesis [24–41].
The tumor microenvironment has garnered recognition as a crucial determinant influencing tumor initiation, progression, treatment response, and prognosis [45]. However, the intricate relationship between genes associated with the tumor microenvironment and THCA remains partially understood. Therefore, we embarked on a comparative analysis of immune cell infiltration between two distinct subtypes. Our findings unveiled that cluster C1, characterized by heightened DRGs levels, exhibited elevated immune cell infiltration, while cluster C2 demonstrated higher immune, estimate, and stromal scores. This led us to hypothesize that cluster C2 signifies an immune rejection type, whereas cluster C1 represents an immune inflammation type. Remarkably, immune checkpoint molecules TIGIT, PD-L1, CTLA4, and CD274 displayed heightened expression in cluster C1. Hence, we conjectured that DEDRGs might modulate disulfidptosis and reshape the immune microenvironment, potentially offering therapeutic targets for THCA.
Following the insightful Bayesian co-localization analysis, PDLIM1 and INF2 emerged as candidates collocated with THCA. However, the genetic correlation between THCA and the remaining five DEDRGs (IQGAP1, MYH10, ACTN1, ZHX2, ME1) did not find substantiation, possibly due to susceptibility to acquired confounding factors. Based on these compelling findings, we selected PDLIM1 and INF2 for experimental validation. Our results revealed that both the mRNA and protein expression levels of INF2 in the control group (FTC-133) were notably lower than those in the normal group, whereas the mRNA and protein expression levels of PDLIM1 in the control group (FTC-133) were markedly higher than those in the normal group (P < 0.05). These experimental validations provide robust confirmation of our study’s hypotheses [42–44].
The establishment of prognostic models represents a pivotal stride in effectively predicting the outcomes of tumor patients [46, 47]. In the realm of molecular characteristics, specialized models like the m1Ascore have been devised to assess individual patients’ m1A modification patterns [48]. Remarkably, our study pioneers a comprehensive analysis delving into potential therapeutic targets, mechanisms, and candidate therapeutic agents for THCA, with a focused exploration into disulfidptosis and the immune microenvironment. Leveraging bioinformatics methodologies, we forecasted that interventions targeting DEDRGs, prognostic genes, and pathways such as MAPK and PPAR could hold transformative potential in THCA treatment by modulating disulfidptosis and the immune microenvironment. In comparison with prior prognostic features, our study’s prognostic model demonstrates a heightened AUC level in the verification set, signifying enhanced predictive accuracy. Moreover, the validation through Bayesian co-localization analysis and in vivo experiments further bolsters the reliability and precision of our findings.
Nevertheless, it is imperative to acknowledge several inherent limitations in our study. Primarily, all our findings stem from publicly available databases, and our experimental validations were exclusively confined to in vitro settings. Thus, the imperative need for further in vivo experiments and additional functional assays to robustly authenticate our results cannot be understated. Secondly, the intricate mechanistic interplay between disulfidoptosis and the tumor microenvironment warrants deeper exploration and elucidation. Sustained research endeavors in these domains will be pivotal in augmenting our comprehension and propelling therapeutic strategies forward for THCA. Thirdly, the construction of a prognostic risk model for THCA based on immune correlation holds significant promise. Such a model could offer comprehensive insights into the survival rate and response to immunotherapy among THCA patients, underscoring the importance of future investigations in this direction [45–48].
AUC: the area under the curve;
BP: Biological process;
CC: Cellular component;
DEGs: differentially expressed genes;
DRGs: disulfidptosis-related genes;
DEDRGs: differentially expressed genes related to disulfidptosis;
GO: Gene Ontology;
GEO: Gene Expression Omnibus;
KEGG: Kyoto Encyclopedia of Genes and Genomes;
MF: Molecular function;
PPI: Protein-protein interaction;
THCA: Thyroid carcinoma.
Jiangyi Yu designed the research. Siyuan Song, Jie Zhou, Li Zhang, Qiling Zhang, Yuqing Sun, Ying Tan, and Xiqiao Zhou analyzed the data and wrote the paper. Siyuan Song performed experiments in this paper. All authors read and approved the submitted version.
We thank all contributors to the TCGA database and developers of the GEO database.
The authors declare no conflicts of interest related to this study.
This study was supported by grants from National Natural Science Foundation of China (Grant No. 82174293 and 82374355), Science and Technology Support Program of Jiangsu Province (ZD202208).