Is thymocyte development functional in the aged?
Abstract
T cells are an integral part of a functional immune system with the majority being produced in the thymus. Of all the changes related to immunosenescence, regression of the thymus is considered one of the most universally recognised alterations. Despite the reduction of thymic size, there is evidence to suggest that T cell output is still present into old age, albeit much diminished; leading to the assumption that thymocyte development is normal. However, current data suggests that recent thymic emigrant from the aged thymus are functionally less responsive, giving rise to the possibility that the generation of naïve T cell may be intrinsically impaired in the elderly. In light of these findings we discuss the evidence that suggest aged T cells may be flawed even before exiting to the periphery and could contribute to the age-associated decline in immune function.
The role of thymocyte development in T cell immunosenescence
One of the most universally recognised
changes of the ageing immune system is the dramatic regression of the thymus;
which in part is responsible for the observed clinical features of
immunosenescence [1,2,3]. The
features of age-related thymic atrophy involve a reduction in tissue mass, loss
of tissue structure and abnormal architecture and a decline in thymocyte numbers
leading to a reduction in naïve T cell output [3,4,5].
Despite the decline in the number of T cells exiting the thymus [6,7], there are
no discernable changes in the number of T cells in the periphery with age [8], which
appears to be tightly regulated by homeostatic mechanisms [9,10]. However,
with increasing age peripheral T cells exhibit altered phenotypes, loss of
diversity and modifications in responses, which have been correlated to
shortened telomere and is related to replicative senescence [11,12,13].
These changes are (in part) a consequence of reduced naïve T cell output;
however new evidence has revealed that recent thymic emigrants (RTE) from the aged thymus exhibit reduced proliferative and
functional activity [7,14,15];
thereby further contributing to T cell senescence. Specifically, aged RTE
undergo phenotypic maturation with delayed kinetics [7] and exhibit
a decreased proliferative capacity and a weak expression of early activation
markers together with a lower production of IL-2 [7].
Furthermore, aged RTE are defective in increasing intracellular calcium
concentration following TCR crosslinking [14] and exhibit
reduced helper and memory activity [15]. Moreover,
these studies also question the notion regarding whether T cell development is
functionally active in the aged thymus; which is often assumed. This is largely
based on the observations that there are no age-related differences in the
proportion of the major subpopulations of thymocytes either in mice [16] or
humans [17] and that T
cell output can still be detected in the aged thymus [6,7]. This
frequently leads to the belief that there is only a quantitative decline but no
age-associated qualitative changes in thymopoiesis. However, with these recent
studies showing intrinsic functional defects in aged RTE, it suggests that
these newly generated T cells are already compromised prior to entry into the
periphery indicating that various stages of differentiation are altered in an
age-dependent manner.
An overview of thymopoiesis
T cell development involves a series of
sequential developmental steps requiring instructions from the specialised
thymic microenvironment to regulate phases of proliferation, gene rearrangement
and selection [18,19]. Each
maturational stage is reflected by changes in gene and protein expression,
which in turn is mirrored by modifications of cell surface markers, enabling
the identification of thymocytes at various phases of development [20]. Briefly,
thymocyte progenitors entering the thymus are identified by the absence of
either co-receptor molecules CD4 and CD8 and are referred to as double negative
(DN) thymocytes [20]. Within
this subset several critical events occur, including commitment to the T cell
lineage and cellular proliferation [21].
Subsequently, thymocytes become double positive (DP) for the expression of CD4
and CD8 with further maturation dependent on proceeding past positive and
negative selection. Positive and negative selection facilitates the generation
of functionally responsive and self-tolerant T cells [22,23], whereby
DP thymocytes then mature into either single positive (SP) CD4+ T
helper cells or SP CD8+ cytotoxic T lymphocytes before being
exported into the periphery [24].
The effect of age on the phenotype and function of
developing thymocytes
Whilst it is widely acknowledged that there
is a decline in the frequency and absolute number and precursor activity of
early thymic progenitors (ETP) in older mice [1,25,26,27], it is often attributed to alterations in haematopoietic stem cells [28,29]. However,
there is increasing evidence to suggest that the defects in ETP are due to cell
intrinsic deficits that arise from exposure to an ageing thymic
microenvironment [25,26,30,31].
For instance, there is an increase in the frequency of ageing ETP undergoing
apoptosis in older mice [25,26] which
is accompanied by a significant reduction in frequency of Ki67+ ETP
in the aged thymus [25]; therefore
these observations may account for the reduction in ETP number with age.
Furthermore, these properties of ETP appear to be governed by signals derived
from the thymus. Intravenously injected lineage negative-enriched bone marrow
from young mice into sublethally irradiated one month old and 18 month old mice
showed absolute number of donor cells was similar in young and older hosts
after three days [31]. However,
seven to ten days after injection, the number of donor cells in older thymi was
severely reduced compared to those identified in younger thymi, suggesting a
decline in their proliferative capacity [31]. In
addition, when fetal thymi were grafted onto the kidney capsule of young and
old mice, the thymic grafts had similar total thymic cellularity despite the
native thymus from older animals still having significantly lower actual and
subset numbers [30], suggesting
the age-associated alterations in ETP is related to intrathymic changes.
Moreover, it would not be unreasonable to assume that the defects that arise in
the aged ETP, could also lead to the acquisition of further aberrations
throughout thymopoiesis.
ETP are contained within the earliest stages of the DN
subset and various reports have proposed several changes within this
subpopulation; however, the results have not been consistent. Some groups have
observed an increase only in the proportion of DN1 thymocytes but not other
significant changes [30], while
others have depicted an increase in DN1 and a subsequent decrease in DN3 subset
[32].
In contrast, different laboratories have described an increase at the DN3 stage
and a decrease in DN4 thymocytes [16], whilst
no significant differences have been reported in percentage of DN thymocytes by
other groups [33]. These
discrepancies could arise from the different strains of mice analysed and the
timepoints examined. Nevertheless, there is data to indicate that the DN
subpopulation is subject to phenotypical and functional alterations with age.
Interestingly a number of groups, including our own (Figure 1), have observed
an increase in the expression of CD3, the signalling transduction complex of
the T-cell receptor (TCR), within the DN compartment [34].
Corresponding to CD3 upregulation, these cells appear to express high levels of
CD44 [34]. Previously
a population of CD44+CD24-CD3+ DN cells has
been described, which accumulates in older mice, and it has been suggested that
these cells belong to a separate lineage [35]; perhaps
representing NK1.1+ thymocytes, which display a similar phenotype [36].
Interestingly, a similar population has been identified in adult murine bone
marrow and have been associated with a role in downregulation of haematopoiesis
[37]. Therefore,
this expanding population may not only represent an alternate lineage but may
have deleterious affects on developing thymocytes.
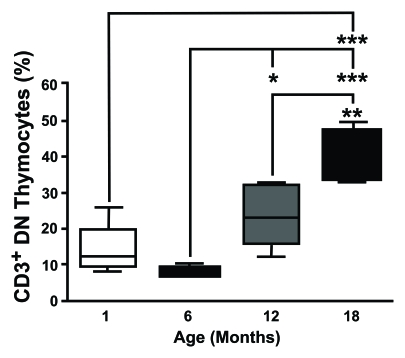
Figure 1. CD3 expression on DN thymocytes shows an age-dependent increase. Thymocytes from different aged
mice were stained with anti-CD3, anti-CD4 and anti-CD8 mAb, analysed by
flow cytometry and CD3 on DN cells was determined gating the appropriate
population. This study revealed that the proportion of CD3+ DN
thymocytes showed an age-dependent increase. (One month n=5; six months
n=5; 12 months n=8; 18 months n=4). *P<0.05; **P<0.01;
***P<0.001.
Despite an increase in the proportion of
DN thymocytes expressing CD3, there is a declining trend in the percentage of
CD3+ thymocytes from both humans [17] and mice [38]. This is
accompanied by a significant decrease in CD3 median fluorescence index (MFI) on
murine thymocytes with age, corresponding to the average number of complexes
per cell (Figure 2). This alteration could have gross implications for the
developing thymocytes. Considering that the CD3 complex is integral for relaying
TCR signals [39], a decrease
in the number of CD3 molecules would affect the ability of T cells to respond
to such TCR-dependent signals and hence impair thymopoiesis [40]. Indeed,
studies by Li and colleagues showed that murine thymocytes stimulated with
ConA, which acts through the TCR, together with interleukin-2 (IL-2) displayed
an age-related decline in proliferation as measured by trititated thymidine
incorporation [16]. A similar
finding was also observed using rat thymocytes [41]. Cell cycle
analysis by propidium iodine conducted in our laboratory provides further
support for a defect in the proliferative response to ConA and IL-2 by
thymocytes from older mice with the results suggesting the deficiency is an
inability to progress from S phase to the G2/M phase of the cell cycle (Figure 3). Although these studies suggest there is an impairment of TCR-expressing
thymocytes to proliferate, it is unclear whether this reflects a shortcoming in
all thymocyte populations or if this is related to the age-associated decrease
in CD3 expression. However, an in vivo method to assess intrathymic
proliferation in humans, employing T cell receptor excision circle (TREC) ratio
analysis, implied that not all thymocyte populations undergo an age-dependent
deficit to proliferate, and only thymocytes in later stages of maturation are
affected [42]. This
appears to correlate with the changes observed in RTE of older mice, which
display a decline in proliferation and activation [7,14].
Therefore, the proliferative impairments observed in RTE from ageing mice could
arise from intrinsic defects imprinted on the developing T cells in the thymus.
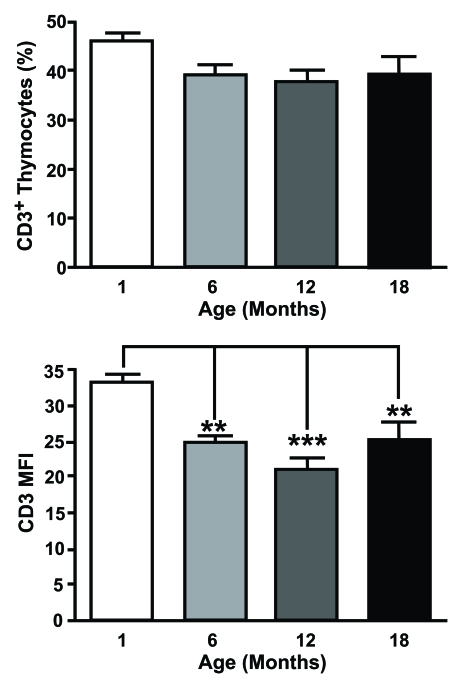
Figure 2. CD3 expression is altered on aged thymocytes. Thymocytes from different aged mice were stained with
anti-CD3 mAb and analysed by flow cytometry. The top histogram shows the percentage of CD3+ cells
positive and the bottom shows mean fluorescent intensity (MFI) of CD3 expression for one month old,
six month old, 12 month old and an 18 month old animals. MFI was obtained by gating on the entire population.
Although there were no age-related changes in the proportion of CD3+ thymocytes, a significant decrease
in the number of CD3 molecules on thymocytes associated with age was observed.
(One month n=5; six months n=5; 12 months n=8; 18 months n=4). **P<0.01; ***P<0.001.
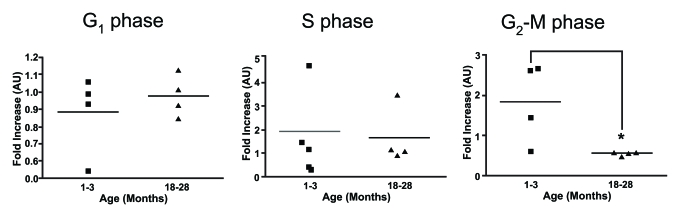
Figure 3. Cell cycle analysis on stimulated thymocytes from young and old mice. The various stages of the cell
cycle in thymocytes from young and old mice following treatment with ConA
and IL-2 after 24 hours was determined by flow cytometry. Data is expressed
as fold increase compared to time zero. It was observed that there was a
significant increase in the proportion of thymocytes from young mice at the
G2-M phase compared to thymocytes from older animals. One month
n=4; 18 months n=4. *P<0.05.
Aged peripheral T cells from either humans or mice
demonstrate an increased resistance to apoptosis [43,44].
Although these cells may represent the most terminally differentiated T cells,
suggesting that increased resistance to apoptosis is the outcome of senescence,
a study investigating in vivo responses to activation induced cell death of T
cells from aged mice implies age-related impairment of apoptosis can occur in
previously unchallenged T cells and is perhaps intrinsically acquired [45]. In
this study, male SCID mice receiving adoptively transferred T cells from old
female HY TCR transgenic mice had a three-fold increase in the percentage of
autoreactive CD8+ HY antigen-reactive T cells in contrast to mice
receiving T cells from young female transgenic mice. Moreover, in our laboratory
we have observed an age-dependent resistant to spontaneous and
dexamethasone-induced apoptosis in murine thymocytes (Figure 4), which has also
been reported in rat thymocytes [41]. Therefore,
the resistance to apoptosis observed in thymocytes from older mice may be
reflected in decreased susceptibility of peripheral T cells to undergo cell
death.
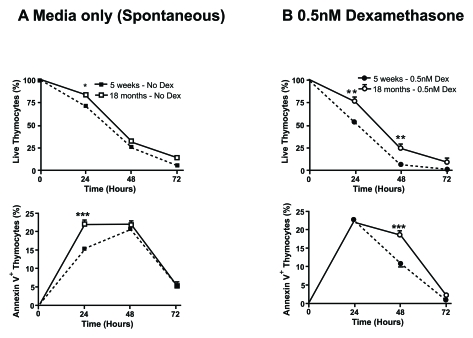
Figure 4. Aged thymocytes have increased resistance to spontaneous and dexamethasone-induced apoptosis. Spontaneous (A) and
dexamethasone (dex)-induced (B) apoptosis at 0.5nM was assessed by
flow cytometry. Graphs show the percentage of viable thymocytes defined as
Annexin V- 7AAD- (top graphs)and
those undergoing early apoptosis as Annexin V+ 7AAD-
(bottom graphs). Closed square/circle with dotted line symbolise
young thymocytes cultured in media or with the addition of 0.5nM dex
respectively. Whereas, open square/circle with solid line signify
thymocytes from 18 month old mice cultured in media or with the addition of
0.5nM dex respectively. The data revealed that there is an
age-associated increased resistance to spontaneous and dex-induced
apoptosis with a higher percentage of viable thymocytes from older mice
compared to younger mice and delayed kinetic of older thymocytes to
initiate apoptosis. Data representative of four experiments. *P<0.05;
**P<0.01.
Collectively these studies corroborate to argue for
the occurrence of age-related deficiencies in T cell development that are
similar to those seen in aged RTE and therefore the abnormalities observed in
these cells are likely to have been acquired during thymopoiesis; primarily due
to a defective microenvironment [15,30,31]. Thus,
thymocytes may be defective before export into the periphery and could
contribute to T cell immunosenescence. However, it is clear that this area
warrants further investigation, including assessing the diversity of thymocyte
receptors with age and evaluating the affect of ageing on selection.
What is the significance of defective thymopoiesis in
the elderly?
Considering these findings, the question then arises,
what are the implications of defective thymopoiesis? Especially, given
the significant decrease in T cell output by the thymus with age [6,7,46] and
that maintenance of the peripheral T cell pool is believed to be predominantly
maintained by homeostatic proliferation [9], how much
can alterations in the properties of newly generated T cells in the elderly
contribute to immunosenescence? The rate of daily export has been determined as
1-2% of the total thymocyte population [47] and is
under control of mechanisms independent of the peripheral T cell pool.
Furthermore, RTE are excluded from the niche-based regulation of peripheral T
cell numbers [48] and are
preferentially selected for survival in the periphery over existing resident T
cells [47]. Therefore,
the thymus is able to influence the T cell pool throughout adult life with
considerable control over the composition of the peripheral T cell pool
repertoire.
Since diversity in the elderly is
dependant on the generation of RTE, defects in their development have as a
profound affect on T cell immunosenescence as those acquired in the periphery.
This has major implication for new and emerging diseases in the elderly, given
the importance of these cells in immune protection. Moreover in light of these
recent findings, methods that are designed to increase thymic output should
also consider targeting the thymic microenvironment. Indeed, where successful
strategies have reversed thymic involution in old mice, they appear to have
done so by targeting the thymic microenvironment [32,49,50].
Impact on the aged thymic microenvironment
The consequence of defective thymopoiesis may also
have more local effects. Thymocytes and the thymic stroma exist in a
bidirectional symbiotic relationship. Several experiments have now provided
evidence that whilst initial patterning of the thymic epithelial compartment is
thymocyte independent, maintenance and continued development requires the
presence of differentiating T cells. Indeed, abrogation of thymopoiesis at
different stages determines the severity and disruption of the thymic architecture
[51,52,53,54]. In mice with defects affecting the later stages of
thymocyte development concerning the DP to SP transition, the thymic medulla,
which is the thymic niche responsible for ensuring tolerance and directing
egression from the thymus, is absent [51,52].
Thymopoiesis blocked at earlier stages of development involving the DN
compartment results in a loss of medulla and cortex, with the latter necessary
to initiate T lineage commitment and provide signals for gene rearrangement and
survival [53,54].
Furthermore, impairment of the bidirectional relationship between thymocytes
and TEC causes alterations in thymic epithelial cell numbers [55].
The absence of either lymphotoxin β rector on thymic epithelial cells, its ligand on
thymocytes or its intracellular signaling molecule nuclear
factor-κB-inducing kinase, results in the disorganization of medullary
thymic epithelial cells [55]. Therefore,
considering the age-related alterations throughout T cell development, it may
induce alterations in the thymic microenvironment. Indeed, we have found a
decline in definitive thymic epithelial cell markers and disruption of the
cortex and medulla [4], concurrent
to alterations in the three dimensional structure [56]. It remains
unclear whether changes in thymopoiesis are cause or effect of the altered
thymic microenvironment, although recent data implies the thymic stroma might
be the initiator [4,30,31].
Nevertheless, thymic involution could be exacerbated by the formation of a
negative feedback loop with deterioration in the stromal compartment
influencing a decline in thymocyte development, which in turn intensifies the
changes in thymic epithelial cells.
Concluding
remarks
The
qualitative contribution of newly generated T cells to the process of
immunosenescence is often overlooked, despite alterations in their quantity
being widely acknowledged. However, considering the evidence, we propose that
in spite of continual T cell output from the thymus throughout life, the thymocytes
from which they are derived are inherently defective and these shortcomings are
acquired during thymopoiesis. Furthermore, we believe that these cells can
significantly contribute to the age-associated changes observed in the
periphery and exacerbate the alterations in thymocyte development through their
interaction with the thymic microenvironment.
Acknowledgments
Many
thanks to Dr. Bradley Cobb for critical reading of the manuscript. We also
thank Drs Andrew Pitsillides, Roberto Souza, Rebecca Collinson, Leanne Saxon,
John Burford, Professors Tim Skerry, Lance Lanyon, and staff at the Royal
Veterinary College BSU for donation of tissue samples and care of the animals.
Additionally, our great appreciation to Shanta Cariese for assistance with the
flow cytometer. D.A is supported by Research into Ageing, A.B.S is supported by
the Thomas Brown Fellowship (University of London).
Conflicts of Interest
The
authors in this manuscript have no conflict of interests to declare.
References
-
1.
Aw
D
, Silva
AB
and Palmer
DB.
Immunosenescence: emerging challenges for an ageing population.
Immunology.
2007;
120:
435
-446.
[PubMed]
.
-
2.
Pawelec
G
, Akbar
A
, Caruso
C
, Solana
R
, Grubeck-Loebenstein
B
and Wikby
A.
Human immunosenescence: is it infectious.
Immunol Rev.
2005;
205:
257
-268.
[PubMed]
.
-
3.
Aspinall
R
and Andrew
D.
Thymic atrophy in the mouse is a soluble problem of the thymic environment.
Vaccine.
2000;
18:
1629
-1637.
[PubMed]
.
-
4.
Aw
D
, Silva
AB
, Maddick
M
, von
Zglinicki T
and Palmer
DB.
Architectural changes in the thymus of aging mice.
Aging Cell.
2008;
7:
158
-167.
[PubMed]
.
-
5.
George
AJ
and Ritter
MA.
Thymic involution with ageing: obsolescence or good housekeeping.
Immunol Today.
1996;
17:
267
-272.
[PubMed]
.
-
6.
Douek
DC
, McFarland
RD
, Keiser
PH
, Gage
EA
, Massey
JM
, Haynes
BF
, Polis
MA
, Haase
AT
, Feinberg
MB
, Sullivan
JL
, Jamieson
BD
, Zack
JA
and Picker
LJ.
Changes in thymic function with age and during the treatment of HIV infection.
Nature.
1998;
396:
690
-695.
[PubMed]
.
-
7.
Hale
JS
, Boursalian
TE
, Turk
GL
and Fink
PJ.
Thymic output in aged mice.
Proc Natl Acad Sci U S A.
2006;
103:
8447
-8452.
[PubMed]
.
-
8.
Hulstaert
F
, Hannet
I
, Deneys
V
, Munhyeshuli
V
, Reichert
T
, De
Bruyere M
and Strauss
K.
Age-related changes in human blood lymphocyte subpopulations. II. Varying kinetics of percentage and absolute count measurements.
Clin Immunol Immunopathol.
1994;
70:
152
-158.
[PubMed]
.
-
9.
Goronzy
JJ
and Weyand
CM.
T cell development and receptor diversity during aging.
Curr Opin Immunol.
2005;
17:
468
-475.
[PubMed]
.
-
10.
Akbar
AN
and Fletcher
JM.
Memory T cell homeostasis and senescence during aging.
Curr Opin Immunol.
2005;
17:
480
-485.
[PubMed]
.
-
11.
Plunkett
FJ
, Franzese
O
, Finney
HM
, Fletcher
JM
, Belaramani
LL
, Salmon
M
, Dokal
I
, Webster
D
, Lawson
AD
and Akbar
AN.
The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation.
J Immunol.
2007;
178:
7710
-7719.
[PubMed]
.
-
12.
Haynes
L
and Swain
SL.
Why aging T cells fail: implications for vaccination.
Immunity.
2006;
24:
663
-666.
[PubMed]
.
-
13.
Nikolich-Zugich
J
T cell aging: naive but not young.
J Exp Med.
2005;
201:
837
-840.
[PubMed]
.
-
14.
Clise-Dwyer
K
, Huston
GE
, Buck
AL
, Duso
DK
and Swain
SL.
Environmental and intrinsic factors lead to antigen unresponsiveness in CD4(+) recent thymic emigrants from aged mice.
J Immunol.
2007;
178:
1321
-1331.
[PubMed]
.
-
15.
Eaton
SM
, Maue
AC
, Swain
SL
and Haynes
L.
Bone marrow precursor cells from aged mice generate CD4 T cells that function well in primary and memory responses.
J Immunol.
2008;
181:
4825
-4831.
[PubMed]
.
-
16.
Li
L
, Hsu
HC
, Grizzle
WE
, Stockard
CR
, Ho
KJ
, Lott
P
, Yang
PA
, Zhang
HG
and Mountz
JD.
Cellular mechanism of thymic involution.
Scand J Immunol.
2003;
57:
410
-422.
[PubMed]
.
-
17.
Bertho
JM
, Demarquay
C
, Moulian
N
, Van
Der Meeren A
, Berrih-Aknin
S
and Gourmelon
P.
Phenotypic and immunohistological analyses of the human adult thymus: evidence for an active thymus during adult life.
Cell Immunol.
1997;
179:
30
-40.
[PubMed]
.
-
18.
Hayday
AC
and Pennington
DJ.
Key factors in the organized chaos of early T cell development.
Nat Immunol.
2007;
8:
137
-144.
[PubMed]
.
-
19.
Anderson
G
and Jenkinson
EJ.
Lymphostromal interactions in thymic development and function.
Nat Rev Immunol.
2001;
1:
31
-40.
[PubMed]
.
-
20.
Ceredig
R
and Rolink
T.
A positive look at double-negative thymocytes.
Nat Rev Immunol.
2002;
2:
888
-897.
[PubMed]
.
-
21.
Fehling
HJ
and von
Boehmer H.
Early alpha beta T cell development in the thymus of normal and genetically altered mice.
Curr Opin Immunol.
1997;
9:
263
-275.
[PubMed]
.
-
22.
von
Boehmer H
, Aifantis
I
, Gounari
F
, Azogui
O
, Haughn
L
, Apostolou
I
, Jaeckel
E
, Grassi
F
and Klein
L.
Thymic selection revisited: how essential is it.
Immunol Rev.
2003;
191:
62
-78.
[PubMed]
.
-
23.
Werlen
G
, Hausmann
B
, Naeher
D
and Palmer
E.
Signaling life and death in the thymus: timing is everything.
Science.
2003;
299:
1859
-1863.
[PubMed]
.
-
24.
Germain
RN
T-cell development and the CD4-CD8 lineage decision.
Nat Rev Immunol.
2002;
2:
309
-322.
[PubMed]
.
-
25.
Min
H
, Montecino-Rodriguez
E
and Dorshkind
K.
Reduction in the developmental potential of intrathymic T cell progenitors with age.
J Immunol.
2004;
173:
245
-250.
[PubMed]
.
-
26.
Heng
TS
, Goldberg
GL
, Gray
DH
, Sutherland
JS
, Chidgey
AP
and Boyd
RL.
Effects of castration on thymocyte development in two different models of thymic involution.
J Immunol.
2005;
175:
2982
-2993.
[PubMed]
.
-
27.
Min
H
, Montecino-Rodriguez
E
and Dorshkind
K.
Effects of aging on early B- and T-cell development.
Immunol Rev.
2005;
205:
7
-17.
[PubMed]
.
-
28.
Tyan
ML
Age-related decrease in mouse T cell progenitors.
J Immunol.
1977;
118:
846
-851.
[PubMed]
.
-
29.
Donnini
A
, Re
F
, Orlando
F
and Provinciali
M.
Intrinsic and microenvironmental defects are involved in the age-related changes of Lin - c-kit+ hematopoietic progenitor cells.
Rejuvenation Res.
2007;
10:
459
-472.
[PubMed]
.
-
30.
Zhu
X
, Gui
J
, Dohkan
J
, Cheng
L
, Barnes
PF
and Su
DM.
Lymphohematopoietic progenitors do not have a synchronized defect with age-related thymic involution.
Aging Cell.
2007;
6:
663
-672.
[PubMed]
.
-
31.
Gui
J
, Zhu
X
, Dohkan
J
, Cheng
L
, Barnes
PF
and Su
DM.
The aged thymus shows normal recruitment of lymphohematopoietic progenitors but has defects in thymic epithelial cells.
Int Immunol.
2007;
19:
1201
-1211.
[PubMed]
.
-
32.
Sutherland
JS
, Goldberg
GL
, Hammett
MV
, Uldrich
AP
, Berzins
SP
, Heng
TS
, Blazar
BR
, Millar
JL
, Malin
MA
, Chidgey
AP
and Boyd
RL.
Activation of thymic regeneration in mice and humans following androgen blockade.
J Immunol.
2005;
175:
2741
-2753.
[PubMed]
.
-
33.
Aspinall
R
and Andrew
D.
Age-associated thymic atrophy is not associated with a deficiency in the CD44(+)CD25(-)CD3(-)CD4(-)CD8(-) thymocyte population.
Cell Immunol.
2001;
212:
150
-157.
[PubMed]
.
-
34.
Thoman
ML
The pattern of T lymphocyte differentiation is altered during thymic involution.
Mech Ageing Dev.
1995;
82:
155
-170.
[PubMed]
.
-
35.
Fowlkes
BJ
and Pardoll
DM.
Molecular and cellular events of T cell development.
Adv Immunol.
1989;
44:
207
-264.
[PubMed]
.
-
36.
Ballas
ZK
and Rasmussen
W.
NK1.1+ thymocytes. Adult murine CD4-, CD8- thymocytes contain an NK1.1+, CD3+, CD5hi, CD44hi, TCR-V beta 8+ subset.
J Immunol.
1990;
145:
1039
-1045.
[PubMed]
.
-
37.
Sykes
M
Unusual T cell populations in adult murine bone marrow. Prevalence of CD3+CD4-CD8- and alpha beta TCR+NK1.1+ cells.
J Immunol.
1990;
145:
3209
-3215.
[PubMed]
.
-
38.
Lau
LL
and Spain
LM.
Altered aging-related thymic involution in T cell receptor transgenic, MHC-deficient, and CD4-deficient mice.
Mech Ageing Dev.
2000;
114:
101
-121.
[PubMed]
.
-
39.
Ohashi
PS
, Pircher
H
, Burki
K
, Zinkernagel
RM
and Hengartner
H.
Distinct sequence of negative or positive selection implied by thymocyte T-cell receptor densities.
Nature.
1990;
346:
861
-863.
[PubMed]
.
-
40.
Yeung
RS
, Penninger
J
and Mak
TW.
T-cell development and function in gene-knockout mice.
Curr Opin Immunol.
1994;
6:
298
-307.
[PubMed]
.
-
41.
Leposavic
G
, Pesic
V
, Kosec
D
, Radojevic
K
, Arsenovic-Ranin
N
, Pilipovic
I
, Perisic
M
and Plecas-Solarovic
B.
Age-associated changes in CD90 expression on thymocytes and in TCR-dependent stages of thymocyte maturation in male rats.
Exp Gerontol.
2006;
41:
574
-589.
[PubMed]
.
-
42.
Dion
ML
, Poulin
JF
, Bordi
R
, Sylvestre
M
, Corsini
R
, Kettaf
N
, Dalloul
A
, Boulassel
MR
, Debre
P
, Routy
JP
, Grossman
Z
, Sekaly
RP
and Cheynier
R.
HIV infection rapidly induces and maintains a substantial suppression of thymocyte proliferation.
Immunity.
2004;
21:
757
-768.
[PubMed]
.
-
43.
Hsu
HC
, Scott
DK
and Mountz
JD.
Impaired apoptosis and immune senescence - cause or effect.
Immunol Rev.
2005;
205:
130
-146.
[PubMed]
.
-
44.
Vallejo
AN
CD28 extinction in human T cells: altered functions and the program of T-cell senescence.
Immunol Rev.
2005;
205:
158
-169.
[PubMed]
.
-
45.
Hsu
HC
, Zhou
T
, Shi
J
, Yang
PA
, Liu
D
, Zhang
HG
, Bluethmann
H
and Mountz
JD.
Aged mice exhibit in vivo defective peripheral clonal deletion of D(b)/H-Y reactive CD8(+) T cells.
Mech Ageing Dev.
2001;
122:
305
-326.
[PubMed]
.
-
46.
Jamieson
BD
, Douek
DC
, Killian
S
, Hultin
LE
, Scripture-Adams
DD
, Giorgi
JV
, Marelli
D
, Koup
RA
and Zack
JA.
Generation of functional thymocytes in the human adult.
Immunity.
1999;
10:
569
-575.
[PubMed]
.
-
47.
Berzins
SP
, Boyd
RL
and Miller
JF.
The role of the thymus and recent thymic migrants in the maintenance of the adult peripheral lymphocyte pool.
J Exp Med.
1998;
187:
1839
-1848.
[PubMed]
.
-
48.
Berzins
SP
, Godfrey
DI
, Miller
JF
and Boyd
RL.
A central role for thymic emigrants in peripheral T cell homeostasis.
Proc Natl Acad Sci U S A.
1999;
96:
9787
-9791.
[PubMed]
.
-
49.
Gray
DH
, Seach
N
, Ueno
T
, Milton
MK
, Liston
A
, Lew
AM
, Goodnow
CC
and Boyd
RL.
Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells.
Blood.
2006;
108:
3777
-3785.
[PubMed]
.
-
50.
Min
D
, Panoskaltsis-Mortari
A
, Kuro
OM
, Hollander
GA
, Blazar
BR
and Weinberg
KI.
Sustained thymopoiesis and improvement in functional immunity induced by exogenous KGF administration in murine models of aging.
Blood.
2007;
109:
2529
-2537.
[PubMed]
.
-
51.
Palmer
DB
, Viney
JL
, Ritter
MA
, Hayday
AC
and Owen
MJ.
Expression of the alpha beta T-cell receptor is necessary for the generation of the thymic medulla.
Dev Immunol.
1993;
3:
175
-179.
[PubMed]
.
-
52.
Surh
CD
, Ernst
B
and Sprent
J.
Growth of epithelial cells in the thymic medulla is under the control of mature T cells.
J Exp Med.
1992;
176:
611
-616.
[PubMed]
.
-
53.
van
Ewijk W
, Hollander
G
, Terhorst
C
and Wang
B.
Stepwise development of thymic microenvironments in vivo is regulated by thymocyte subsets.
Development.
2000;
127:
1583
-1591.
[PubMed]
.
-
54.
Hollander
GA
, Wang
B
, Nichogiannopoulou
A
, Platenburg
PP
, van
Ewijk W
, Burakoff
SJ
, Gutierrez-Ramos
JC
and Terhorst
C.
Developmental control point in induction of thymic cortex regulated by a subpopulation of prothymocytes.
Nature.
1995;
373:
350
-353.
[PubMed]
.
-
55.
Boehm
T
, Scheu
S
, Pfeffer
K
and Bleul
CC.
Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR.
J Exp Med.
2003;
198:
757
-769.
[PubMed]
.
-
56.
Aw
D
, Taylor-Brown
F
, Cooper
K
and Palmer
DB.
Phenotypical and morphological changes in the thymic microenvironment from ageing mice.
Biogerontology.
2009;
10:
311
-322.
[PubMed]
.