



Introduction
The free radical theory of aging proposes that free radicals accelerate the accumulation of damaged structures over time leading to impaired cellular and organismal function during aging [1]. Oxidative stress is driven by reactive oxygen species mainly produced in mitochondria. Superoxide anions, being produced at complex I and III of the electron transport chain [2], are primarily detoxified in mitochondria by the manganese dependent form of superoxide dismutase SOD2 (also called MnSOD). It has been shown that Sod2 over-activation can prolong the lifespan of yeast [3, 4]. Vice versa, impairment or deletion of SOD2 expression induced a significant shortening of the lifespan of Drosophila [5, 6] and mice [7, 8].
Mice carrying a heterozygous deletion of Sod2 (Sod2+/-) are viable but show increased oxidative stress, increased nuclear and mitochondrial DNA modifications, impaired mitochondria function, and increased apoptosis rates [9-13]. Sod2+/- mice exhibit slightly increased rates of cancer but no other features of accelerated aging and have a normal lifespan [13]. These studies indicated that a decrease in the oxidative damage defense system by itself does not induce a significant increase in aging pathology in mice. However, the Sod2+/-mouse provided a unique experimental system to analyze whether an impaired anti-oxidant defense can cooperate with other molecular causes of aging and disease. Although Sod2 polymorphisms were not associated with longevity of centenarians [14], the investigation of this question appears to be highly relevant since SNPs in various components of the oxidative stress pathway are associated with phenotypes of human aging [15].
There is growing evidence that an accumulation of telomere dysfunction and DNA damage contributes to human aging [16, 17]. Several lines of evidence indicate that reduced SOD2 levels and increased ROS could influence cellular and organismal aging in the context of telomere shortening and DNA damage accumulation: (i) Experimental data have shown that ROS can induce different lesions in nuclear DNA including oxidized bases, strand breaks and mutations [18-21]. Thus, ROS may contribute to the generation of nuclear DNA damage and the evolution of aging pathology at organismal level.
(ii) Telomere shortening limits the proliferative capacity of human cells to 50-70 cell divisions by induction of senescence or apoptosis [22-25]. Replicative senescent cells show increased ROS levels [26, 27] and increased oxidative DNA damage [28] indicating that senescence can accelerate ROS induced DNA damage. In addition, increased ROS levels and oxidative modifications to DNA have also been implicated as causal factors inducing senescence [19, 27, 29-33]. In agreement with this hypothesis, it was shown that increased ROS accelerate the rate of telomere shortening in cell culture [34] and that oxidative stress severely limits the replicative potential of mouse cells independently of the presence of telomerase [35]. It remains yet to be investigated whether ROS and telomere shortening cooperate to induce an accumulation of DNA damage and senescence in aging tissues.
(iii) SOD2 level could influence the induction of checkpoints in response to DNA damage or telomere dysfunction. It has been shown that decreased SOD2 expression accelerated p53-induced apoptosis [36], whereas up-regulation of SOD2 protected cells from apoptosis by stabilization of mitochondrial membranes [37]. Both mechanisms could be relevant to aging induced by telomere dysfunction, since the impairment of organ maintenance in response to telomere dysfunction is associated with activation of the p53/p21 signaling pathway and increased rates of apoptosis [38-41].
Laboratory mouse strains are of limited use to identify factors that accelerate aging in the context of telomere dysfunction and DNA damage, since laboratory mice in comparison to humans, have very long telomeres [42]. Laboratory mice show some evidence for an accumulation of DNA damage during aging [43] However, biomarker studies revealed that the level of telomere dysfunction and DNA damage in aging laboratory mice is low compared to human aging [16]. Telomerase knockout (mTerc-/-) mice provided an experimental system to study aging induced by telomere dysfunction and DNA damage [16, 44, 45]. Considering that oxidative stress was shown to shorten telomeres [46], limit stem cell function [47], and induce DNA damage and senescence (see above), we hypothesized that Sod2 haploinsufficiency could affect stem cell pools and aging of telomere dysfunctional mice.
Here we analyzed consequences of a heterozygous deletion of Sod2 on aging of telomerase wild-type mice with long telomeres and third generation (G3) mTerc-/- mice with dysfunctional telomeres. The study shows that heterozygous Sod2 deletion does not affect stem cell function, organ maintenance and lifespan of telomere dysfunctional mice. These results indicate that a reduction in SOD2-dependent anti-oxidant defense does not accelerate aging in the context of telomere dysfunction.
Results
Heterozygous deletion of Sod2 reduces SOD2 protein levels and antioxidant capacity
Sod2+/- mice were crossed through 3 generations with telomerase knockout mice (Suppl. Figure 1) to generate the following cohorts: mTerc+,Sod2+/- mice (n=31); mTerc+, Sod2+/+ mice (n=34); G3 mTerc-/-, Sod2+/- mice (n=58); G3 mTerc-/-, Sod2+/+mice (n=38). The mTerc+ groups were composed of both mTerc+/+ and mTerc+/- mice with long telomeres since they do not phenotypically differ from each other.
In mTerc+ mice, heterozygous deletion of Sod2 correlated with significantly decreased SOD2 protein levels in liver, whereas the decrease in brain and bone marrow did not reach statistical significance (Figure 1A-C). SOD2 protein levels were slightly but not significantly decreased in G3 mTerc-/-,Sod2+/+ mice compared to mTerc+, Sod2+/+ mice. A further decrease occurred in G3 mTerc-/-, Sod2+/- mice resulting in a significant decrease in SOD2 protein levels in all investigated organs of these mice compared to mTerc+, Sod2+/+ mice (Figure 1A-C).
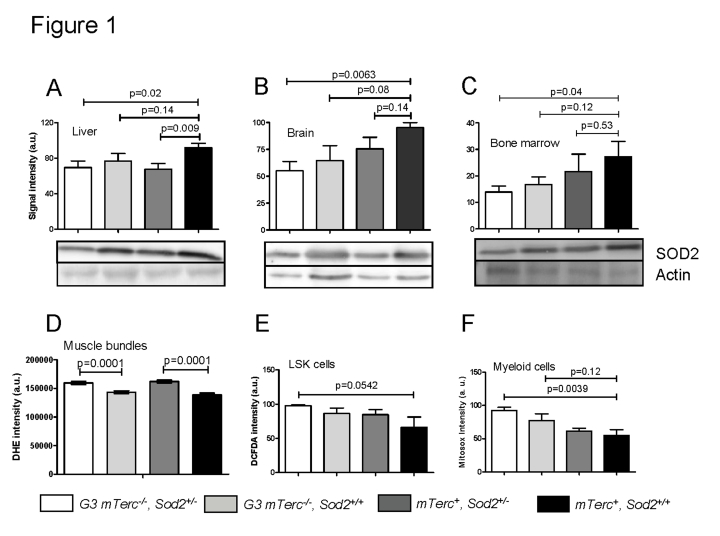
Figure 1.
Western blots
showing SOD2 levels in liver (A), brain (B) and bone marrow (C)
of 12 to 18 months old mice. Lower panels show representative western blots
and upper panels show quantification of normalized SOD2 levels to actin
controls from n=4 mice per group (1 to 2 repeat experiments per sample).
Data is shown in arbitrary units ± SEM. (D) Basal ROS levels in
muscle fibers stained with DHE. Signal quantification of G3
mTerc-/-Sod2+/- (n=235), G3 mTerc-/-(n=211) mTerc+, Sod2+/-(n=270 ) and mTerc+,
Sod2+/+
(n=203) nuclei from 5 mice per genotype. Data is shown as mean fluorescence
intensity ± SEM. (E) Antioxidant capacity of LSK cells. DCFDA loaded
bone marrow cells were incubated with 50 uM of antimycinA and DCFDA
fluorescence was monitored in Lin-Sca+cKit+ populations
by FACS analysis. Data is shown in arbitrary units ± SEM of n=4 mice per
group. (F) Antioxidant capacity of myeloid cells. Mitosox loaded
bone marrow cells were incubated with 20 uM antimycinA and mitosox
intensity monitored in myeloid population by FACS analysis. "Y" axis
denotes arbitrary units for fluorescence intensity of n=5 to 6 mice per
group.
To investigate functional consequences of heterozygous Sod2 deletion, levels of reactive oxygen species (ROS) were analyzed in muscle and hematopoietic cells. For this purpose we used (i) dihydroethidium (DHE), which intercalates in DNA and emits red fluorescent signals in response to oxidation; (ii) mitosox, which localizes to mitochondria and exhibits red fluorescence after superoxide-induced oxidation, and (iii) dichloro-dihydro-fluorescein (DCFDA), which detects a wide range of ROS after removal of its acetate group by oxidation. Heterozygous deletion of Sod2 was associated with increased basal superoxide levels in muscle cells of bothmTerc+and G3 mTerc-/- mice (Figure 1D). Sod2 gene status had no significant effect on basal ROS levels in bone marrow derived stem and progenitorcells (LSK cells: Lineage-negative, Sca1-positive, c-Kit-positive, data not shown). However, stress induced ROS level after treatment with antimycinA (a complex III inhibitor that induces superoxide production) were elevated in bone marrow derived LSK and myeloid cells of G3 mTerc-/-, Sod2+/- mice compared to mTerc+, Sod2+/+ mice (Figure 1E, F). In accordance with the data on SOD2 protein expression, these data on antimycinA induced ROS in hematopoietic and myeloid cells suggested that Sod2 haploinsufficiency cooperated with telomere dysfunction to impair ROS detoxification in G3 mTerc-/-, Sod2+/- mice.
Sod2 heterozygous deletion does not increase mitochondrial dysfunction in aging G3 mTerc-/- mice
Mitochondria are the major source of ROS production in cells and a decrease in anti-oxidant defense can induce mitochondrial damage leading to a decrease in the mitochondrial respiratory capacity [9, 48]. In agreement with these studies, muscles from 8-11 month old mTerc+,Sod2+/- mice had lower state III respiration rates (ADP dependent, normalized to the mitochondrial mass) and lower maximum respiration rates (induced with the mitochondrial uncoupler FCCP) compared to fibers from mTerc+,Sod2+/+ mice (Figure 2A). However, this decrease was ameliorated in muscle fibers of 8-11 month old G3 mTerc-/-, Sod2+/- mice (Figure 2A). Treatment with rotenone, a complex I inhibitor, reduced but did not completely abolish respiration rates of the muscles fibers, indicating ongoing complex I independent respiration. This complex I independent respiration was also significantly reduced in Sod2+/- mice compared to wild type controls (Figure 2A). Again, this Sod2-dependent reduction in respiration rate was rescued in G3 mTerc-/-, Sod2+/- mice.
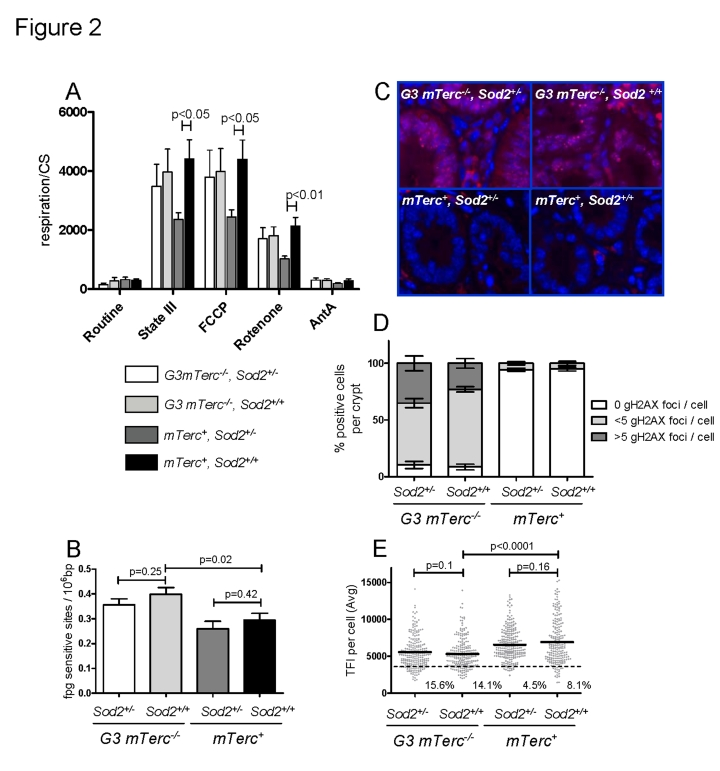
Figure 2. (A)
Mitochondrial respiration of muscle fibers. 10 to 25 mg of permeabilized
bundles were analyzed by high resolution respirometry. Results are
expressed as oxygen consumption per mg of muscle (± SEM) normalized to
citrate synthase activity of n=5 to 6 mice per group. State
III respiration is shown after addition of malate, octanoyl-carnitine, ADP,
glutamate, succinate and cytochrome c. After state III respiration
determination, uncoupled respiration was determined with addition of FCCP
to the respiring fibers. Rotenone and antimycin A were used to inhibit
respiration at complex I and III respectively. (B)
Oxidative modifications (fpg sites) in DNA from bone marrow cells of 12 to
17 month old mice. Data from n=4 to 9 mice per group is shown as number of
lesions per 106 bp ± SEM. (C)
Representative pictures of gH2AX staining in intestinal crypts of
aged mice and bar graphs (D) showing percentage of positive cells
per crypt and number of foci per cell ± SEM of n=4 to 6
mice per group. 200 crypt cells were analyzed per mouse. (E) Telomere length analysis by qFISH in liver
sections of n= 4 to 5 mice per group aged 12 to 18 months old. n=237 G3
mTerc-/-,
Sod2+/-), n=234 (G3 mTerc-/-, Sod2+/+);
n=242 (mTerc+,
Sod2+/-) and n=211 (mTerc+,
Sod2+/+)
nuclei were analyzed for telomere fluorescence intensity (TFI). The black
line indicates the mean TFI value of each genotype and the dotted line the
threshold of critically short telomeres (TFI<3500).
An analysis of basal respiration rates and maximal induced respiration (in response to FCCP treatment) did not reveal a significant influence of Sod2 gene status on the respiratory capacity of total bone marrow cells of 8 to 11 month old mTerc+ and G3 mTerc-/-mice (Suppl. Figure 2A). In this compartment, mitochondria from G3 mTerc-/- mice showed an increased maximal respiratory capacity (FCCP-induced) compared to mTerc+ mice suggesting that telomere dysfunction induced adaptive responses that increase the functional reserve of mitochondria.
Sod2 heterozygous deletion does not increase nuclear DNA damage and telomere shortening in aging G3mTerc-/- mice
To analyze the basal levels of oxidative purine modifications in DNA from total bone marrow cells, we used an alkaline elution assay in combination with formamidopyrimidine-DNA glycoslyase (Fpg) as a probe [49, 50]. The enzyme recognizes 7,8-dihydro-8-oxoguanine (8-oxodG) among other oxidative purine lesions in nuclear DNA [51]. The technique avoids the spontaneous generation of 8-oxodG during DNA isolation and hydrolysis [52]. Our analysis revealed an increase of the basal level of oxidative damage in the nuclear DNA of G3 mTerc-/- mice compared to mTerc+ mice, but heterozygous deletion of Sod2 had no influence (Figure 2B).
Increased ROS levels have been shown to induce DNA double strand breaks and senescence in response to prolonged interferon stimulation [19]. Here, the prevalence of DNA double strand breaks was analyzed by γH2AX staining. γH2AX forms foci at DNA breaks in response to telomere dysfunction and γ-irradiation [53-56]. DNA damage foci were analyzed in intestinal crypts (a proliferative stem cell compartment, which is highly sensitive to telomere dysfunction). In agreement with previous studies, 12-18 month old G3 mTerc-/- mice exhibited significantly higher levels of DNA damage compared to age-matched mTerc+ mice (Figure 2C-D). However, heterozygous deletion of Sod2 did not increase accumulation of DNA damage foci (Figure 2C-D).
Increasing ROS levels can accelerate telomere shortening in cell culture systems [34, 46]. As expected, an analysis of telomere length showed shorter telomeres in liver (Figure 2E) and intestine (Suppl. Figure 2B) of 12 -18 month old G3 mTerc-/- mice compared to age-matched mTerc+ mice. However, Sod2 haplo-insufficiency did not accelerate telomere shortening (Figure 2E and Suppl. Figure 2B). In agreement with the data on oxidative DNA damage, γH2AX-foci, and telomere length, the expression level of serum markers of DNA damage and telomere dysfunction [16] was increased in 12 to 18 month old G3 mTerc-/- mice compared to age-matched mTerc+ mice, but Sod2 haploinsufficiency did not increase serum levels of these biomarkers (Suppl. Figure 2C-E).
Sod2 heterozygosity does not aggravate the impairment of stem cell function, organ maintenance, and the shortening in lifespan of telomere dysfunc-tional mice
Previous studies have shown that telomere dysfunction impairs the maintenance of high turnover organs in aging mTerc-/- mice, specifically affecting the hematopoietic system and the intestinal epithelium [38-40]. In agreement with these studies, 12-15 month old G3 mTerc-/- mice compared to age-matched mTerc+ mice exhibited anemia (Figure 3A), a reduction in bone marrow derived B-lymphopoiesis (Figure 3B, Suppl. Figure 3A-C), a reduction in thymic T-lymphopoiesis (Suppl. Figure 3D, E), and an impaired maintenance and function of hematopoietic stem cells (HSCs) (Figure 3C, D). Heterozygous deletion of Sod2 accentuated the decrease in mature B cells in aging G3 mTerc-/- mice (Figure 3B) but otherwise did not show consistent effects on hematopoietic parameters in mTerc+ and G3 mTerc-/- mice. As shown in previous studies [38, 40, 45], aging telomere dysfunctional mice developed a severe atrophy of intestinal epithelia compared to age-matched mTerc+ mice (Figure 3E, F). Sod2 heterozygosity did not increase the severity of crypt atrophy in aged telomere dysfunctional mice (Figure 3E, F).
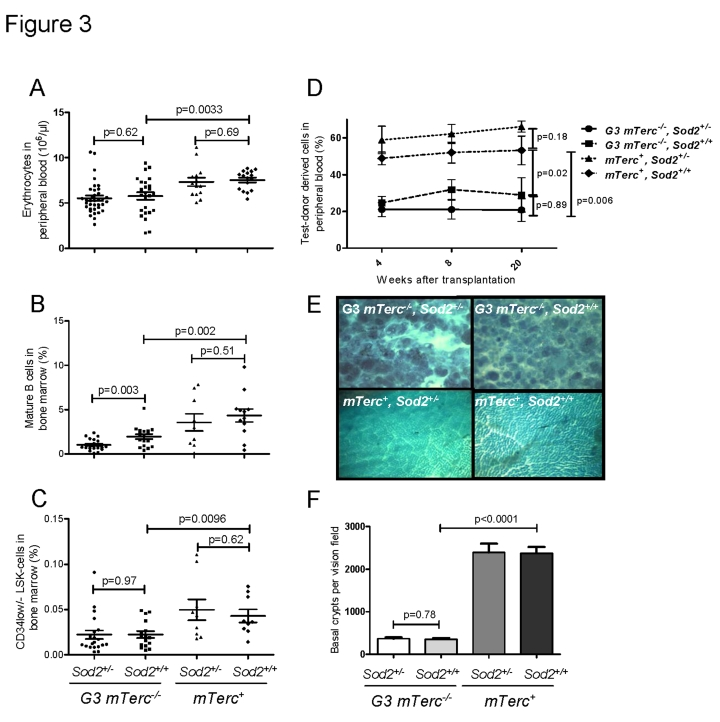
Figure 3. (A)
Number of erythrocytes per ul of peripheral blood ± SEM in 12 to 18
months old mice. (B) Percentage of mature B cells defined as IgD+
IgM+ B220+ CD43- cells in total bone
marrow cells of 12 to 18 months old mice. n=21 (G3 mTerc-/-,
Sod2+/-), n=17 (G3 mTerc-/-, Sod2+/+);
n=9 (mTerc+,
Sod2+/-) and n=12 (mTerc+,
Sod2+/+)
mice per group were analyzed by FACS. (C) Percentage of long term
hematopoietic stem cells defined as Lin- Sca+ cKit+
CD34-/low cells in total bone marrow cells of 12 to 18 months
old mice. n=9 to 20 mice per group were analyzed by FACS. (D)
Competitive transplantation of total bone marrow of Ly5.2 test donor cells
against Ly5.1 competitor cells. 8(10)5 cells of test donor cells
were transplanted along with 4(10)5 competitor cells into 1 to 3
young lethally irradiated recipients per donor. Four different donors were
used per group. White blood cell chimerism was verified at 1, 2 and 5
months after transplantation by FACS analysis. Data is shown as percentage
of donor derived chimerism ± SEM (E) Representative
pictures displaying the large intestine atrophy in telomere dysfunctional
mice wildtype and heterozygous for Sod2. (F) Bar graph
depicting the average number of intestinal crypts per visual field at a
magnification of 40X of whole mounts from n=8 (G3 mTerc-/-,
Sod2+/-), n=7 (G3 mTerc-/-, Sod2+/+);
n=4 (mTerc+, Sod2+/-) and n=4 (mTerc+,
Sod2+/+)
mice per group.
In line with previous results, an impairment in organ maintenance was associated with a shortened lifespan of telomere dysfunctional mice compared to mTerc+ mice (Figure 4 A-C) correlating with an age-dependent decline in body weight (Figure 4 D, E). Heterozygous deletion of Sod2 did not alter weight curves (Figure 4D, E) or survival (Figure 4 A-C) of telomere dysfunctional mice. Specifically, no survival difference was observed between G3 mTerc-/-Sod2+/- mice compared to G3 mTerc-/-, Sod2+/+ mice (median lifespan 72.3 and 69.1 weeks respectively, p=0.75 Figure 4A). Sod2 heterozygosity did also not affect the incidence of spontaneous cancer in aging G3 mTerc-/- mice and mTerc+ mice during the observation period of 20 months (data not shown).
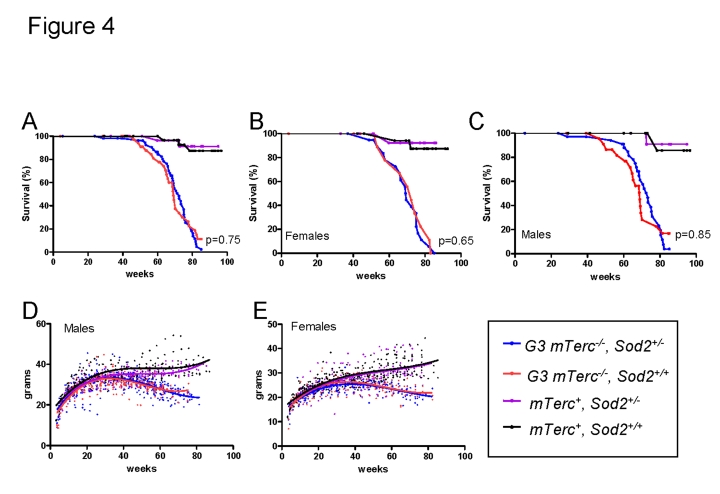
Figure 4. (A)
Kaplan Meyer survival curves for G3 mTerc-/-,
Sod2+/- (n=58); G3 mTerc-/-, Sod2+/+
(n=38); mTerc-/-, Sod2+/-
(n=31)
and mTerc+, Sod2+/+ (n=34). (B)
Survival curves for females G3 mTerc-/-,
Sod2+/- (n=22); G3 mTerc-/-, Sod2+/+
(n=14); mTerc-/-, Sod2+/-
(n=16)
and mTerc+, Sod2+/+ (n=19). (C)
Survival curves for males G3 mTerc-/-,
Sod2+/- (n=36); G3 mTerc-/-, Sod2+/+
(n=24); mTerc-/-, Sod2+/-
(n=15)
and mTerc+, Sod2+/+ (n=15). Dot
blots showing body weight of male (D) and female (E) mice
throughout lifespan in the aging cohorts. Third order polynomial
regression is shown as trendline. All mice were weighed monthly until
death.
Discussion
The current study shows that SOD2 reduction does not affect stem cell function, organ maintenance, and lifespan of telomere dysfunctional mice. These results contrast with studies on mouse models of diseases, where Sod2 hemizygosity exacerbated disease phenotypes as (i) increasing the formation of neurotoxic plaques and tangles in APP and Tg2576 transgenic [57, 58], (ii) reducing the lifespan of G93A transgenic mice - a model for amytrophic lateral sclerosis [59], (iii) increasing diabetic neuropathy in db/db mouse [60], and (iv) increasing endothelial dysfunction in atherosclerosis prone ApoE deficient mice [61]. Together, these findings suggest that in contrast to disease conditions, telomere dysfunction per se does not cooperate with a decrease in anti-oxidant defense to impair organ maintenance and lifespan.
Our experiments show that heterozygous deletion of Sod2 results in diminished SOD2 protein levels and impaired anti-oxidant defense in different organ systems of telomere dysfunctional mice including muscle and hematopoietic cells. However, this deficiency does not lead to an impairment in mitochondrial function in aging telomere dysfunctional mice, whereas it reduces mitochondrial function in aged mTerc+ mice. Previous studies have shown that mitochondrial dysfunction contributes to induction of senescence in fibroblast cultures [26, 62]. The maintenance of mitochondrial function in G3mTerc-/-, Sod2+/- mice suggests that telomere dysfunction might induce adaptive responses that protect mitochondria from oxidative damage possibly involving activation of repair responses. Alternatively, telomere dysfunction may cooperate with mitochondria dysfunction in vivo to induce clearance of dysfunctional cells, thus maintaining a cellular pool with functional mitochondria.
There is an ongoing debate whether mechanisms that increase oxidative stress can contribute to an accumulation of nuclear DNA damage and aging. Studies with cultured human cells suggested that the contribution of mitochondrial-derived ROS to the generation of oxidative modifications in nuclear DNA is small [63]. However, in vivo studies with Sod2 mutant mice have shown that Sod2 haploinsufficiency can increase the level of oxidative modification to nuclear DNA and increase the cancer risk in very old (26 month old mice) [13]. In addition, there is evidence that impairment of DNA repair systems results in elevated ROS-mediated nuclear DNA damage, cellular senescence, and cancer formation [33, 64, 65].
In the current study, the maximal lifespan of telomere dysfunctional (G3 mTerc-/-) mice was limited to 19 months. During this observation period, Sod2 haploinsufficiency did not accelerate the accumulation of oxidative DNA base damage or DNA double strand breaks in both telomere dysfunctional mice and mTerc+ mice. These data indicate that heterozyogous deletion of Sod2 does not cooperate with telomere dysfunction to accelerate the accumulation of nuclear DNA damage. The results from previous studies suggest that telomere independent factors may cooperate with Sod2 haploinsufficiency in long lived wild type mice to increase nuclear damage. It appears to be surprising that nuclear DNA damage was not increased in G3 mTerc-/-, Sod2+/-mice, although the mice showed increased intracellular ROS in muscle and impaired anti-oxidant defense in response to stress induced ROS. Possible explanations include that (i) removal of oxidative DNA lesion in repair proficient mice is sufficiently efficient to counteract effects of moderately increased ROS or (ii) diffusion of mitochondria derived ROS to the nucleus is limited irrespective of Sod2 gene status. In agreement with the data on unchanged rates of nuclear DNA damage, the current study did not detect a cancer promoting effect of Sod2 deficiency in telomere dysfunctional mice and mTerc+ mice during the observation period of 19 month.
Together, the current study provides the first experimental evidence that an impairment of SOD2-dependent anti-oxidant defense does not cooperate with telomere dysfunction to aggravate organismal aging.
Materials and Methods
Mouse crosses and survival. Sod2+/- mice [7] acquired from Jackson laboratories (stock number 002973) was crossed with mTerc-/-mice for 3 generations in order to create the following experimental cohorts G3 mTerc-/-, Sod2+/- (n=58); G3 mTerc-/-, Sod2+/+ (n=38); mTerc+, Sod2+/- (n=31) and mTerc+, Sod2+/+ (n=34). Mice were kept in a pathogen free environment where they had free access to food and water.
Mice were sacrificed by CO2 asphyxiation when presented deteriorated health condition or loss of 30% of body weight. Organs were quickly removed and either frozen down in dry ice or fixed in 4% paraformaldehyde (PFA) for paraffin embedding.
Whole mounts of colon were prepared as previously described [66].
FACS analysis. FACS analysis was performed on freshly isolated bone marrow cells that were stained for 15 min on ice with the appropriate antibody cocktail. Cells were analyzed using an LSRII or FACS Calibur machine.
Bone marrow competitive transplantation. One to three young C57/BL6 mice per donor were retroorbitally transplanted after lethal irradiation (12Gy) with 8x105 donor (Ly5.2) and 4x105 competitor (Ly5.1) bone marrow cells. Four 15-months-old male mice per genotype were used as donors and four 12-month old Ly5.1 female mice were used as competitors in the experiment.
Chimerism was checked at one, 2 and 5 months after transplantation in white blood cells collected from retroorbital bleeding.
γ-H2AX staining in intestine sections. Three um paraffin sections were stained with primary anti γ-H2AX (Millipore 06-636) overnight in PBS, washed three times and incubated for 30 min with secondary anti mouse IgG labeld with Cy7. Slides were kept at 4°C until analysis.
Oxidative modifications to DNA. Quantification of the basal levels of oxidative purine modifications in bone marrow cells isolated from the various mouse strains was carried out by the alkaline elution assay originally described by [49] with modifications reported previously [50, 67]. Fpg-sensitive sites detection was performed in 1x106 bone marrow cells per mouse as previously described [68].
ROS measurement in bone marrow cells. Mitosox staining: Freshly isolated bone marrow cells were loaded in staining media with Mitosox (Molecular probes. Cat. No. M36008) 5uM final concentration and incubated for 30 min at 37°C. The cells were washed once with PBS and antimycinA (Sigma Cat No. 8674) was added for a final concentration of 20uM. The cells were incubated 10 minutes, filtered and immediately analysed by FACS.
DCFDA staining: Freshly isolated bone marrow cells were stained for LSK (except for CD34) loaded with DCFDA 0.5 uM (Molecular probes Cat. No. C6827) and incubated for 5 min at 37°C. The cells were washed once with PBS and antimycinA (Sigma Cat No. 8674) was added for a final concentration of 50uM. The cells were incubated 10 minutes, filtered and immediately analyzed by FACS.
ROS measurement in muscle bundles. Muscle fibers were incubated with DHE at a final concentration of 40 μM in PBS for 30 min at 37°C. After staining, the tissue was washed in PBS and fixed using 2.2 % formaldehyde in 0.1 M Sorensen phosphate buffer (pH 7.1). Confocal images were collected with 40x objective.
Protein analysis. Whole cell extracts were prepared in RIPA buffer with cocktail of protease inhibitors and reducing agents (NaVO3 1mM, DTT 1mM, PMSF 1mM, proteinase cocktail inhibitor ROCHE Cat. No. 11836153001). SOD2 levels were determined using AntiSOD2 antibody (Santa Cruz, SC-30080) and actin levels with antiActin (Santa Cruz SC-1615).
High-resolution respirometry. Mitochondrial respiration was performed in intact bone marrow cells and permeabilized muscle bundles as described [62, 69].
Muscle bundles: Respirometry of saponin-permeabilized muscle fibers was performed with the Oxygraph-2k (OROBOROS instruments) using between 10 and 25 mg of biopsy material. Measurements were performed at 37°C in the range of 200-400 μM oxygen, to avoid oxygen limitation. The experiments were performed in MiRO5 buffer (110 mM sucrose, 60 mM potassium lactobionate, 0.5 mM EGTA, 1 g/l BSA fat free, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, pH 7.1).
Defined respiratory states were obtained by a multiple substrate-inhibitor titration regime: malate 2mM, octanoylcarnitine 1 mM, ADP 5 mM, glutamate 10 mM, succinate 10 mM, cytochrome c 10 μM, FCCP (stepwise, increments of 0.25 μM up to final concentration of maximally 1.25 μM), rotenone 0.5 μM, and antimycin A 2.5 μM. Cytochrome c was added to verify the intactness of the outer mitochondrial membrane after saponin permeabilization. No visible stimulatory effect of cyt. c was observed in our conditions. If necessary, re-oxygenations were performed with pure oxygen.
Bone marrow cells: Approximately 7x106 freshly isolated bone marrow cells were resuspended in 3 ml of Iscoves Modified Dulbecco's Medium (Gibco) and applied for high-resolution respirometry as above. The experimental regime started with routine respiration (defined as endogenous respiration without additional substrates or effectors). After observing steady-state respiratory flux, the ATP synthase inhibitor oligomycin (1 μg/ml) was added, followed by uncoupling of oxidative phosphorylation by stepwise titration of FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) up to optimum concentrations in the range of 2.5-4 μM. Finally, respiration was inhibited by complex I and compex III inhibitors rotenone (0,5 μM) and antimycin A (2,5 μM) respectively.
The mitochondrial respiration data were normalized to the mitochondrial mass marker enzyme Citrate Synthase (CS) activity spectrophotometrically determined [70].
Quantitative Fluorescence In Situ Hybridization (qFISH). qFISH analysis was performed as previously described [71] in 5 uM liver and small intestine sections.
Software and analysis of data. FACS results were analyzed using FlowJo 7.2.2. Statistical analysis of the results was performed using Excel 2003 and GraphpadPrism 5.0 and image analysis with ImageJ 1.39. Chemicapt 5000 ver 15.01 was used for acquisition of images from gels and western blots. Telomere fluorescence intensity was analyzed using the TFL-Telo software from Peter Lansdorp
Supplementary Materials
(A) Mating scheme to generate the double mutant G3 mTerc-/-, Sod2+/-.
(A) Mitochondrial respiration of bone marrow cells. 107 bone marrow cells were analyzed by high resolution respirometry of n=5 to 7 mice per group. Results show normalized respiration of one million cells to citrate synthase activity ± SEM. (B) Telomere length analysis by qFISH in small intestine sections of n= 4 to 5 mice per group aged 12 to 18 months old. n=177 (G3 mTerc-/-, Sod2+/-), n=192 (G3 mTerc-/-, Sod2+/+); n=151 (Sod2+/-) and n=167 (Sod2+/+) nuclei were analyzed for telomere fluorescence intensity (TFI). The black line indicates the mean TFI value of each genotype and the dotted line the threshold of critically short telomeres (TFI<4000). Aging and DNA damage markers EF1-α (C), CRAMP (D) and chitinase (E) were quantified by ELISA in plasma of old age matched G3 mTerc-/-, Sod2+/- (n=16); G3 mTerc-/-, Sod2+/+ (n=14); mTerc-/-, Sod2+/- (n=8) and mTerc+, Sod2+/+ (n=10) and young WT (yWT) mice (n=5). Values are arbitrary units ± SEM.
Bone marrow of 12 to 18 month old mice was evaluated for: (A) Percentage of PreB cells defined as IgD- IgM- B220+ CD43- cells in total bone marrow cells. (B) Percentage of ProB cells defined as CD19+ B220+ LinB- AA4.1+ cells in total bone marrow cells. (C) Percentage of PreproB cells defined as CD19- B220+ LinB- AA4.1+ cells in total bone marrow cells. (D) Representative FACS blot showing the reduction of thymic T-lymphopoiesis and thymic atrophy in aged telomere dysfunctional mice. (E) Bar graphs showing the number of thymocytes ± SEM in n= 5 mice per group aged 12-15 months old.
Acknowledgments
This project was funded by the DFG (Ru745/10-1). BE is supported by the Deutsche Forschungsgemeinschaft (EP11/5). LMG was also supported by the Hannover Biomedical Research School (HBRS).
Conflicts of Interest
The authors have no conflict of interests to declare.
References
- 1. Harman D Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956; 11: 298 -300. [PubMed] .
- 2. Cadenas E Mitochondrial free radical production and cell signaling. Molecular Aspects of Medicine. 2004; 25: 17 -26. [PubMed] .
- 3. Fabrizio P , Liou LL , Moy VN , Diaspro A , Selverstone Valentine J , Gralla EB and Longo VD. SOD2 Functions Downstream of Sch9 to Extend Longevity in Yeast. Genetics. 2003; 163: 35 -46. [PubMed] .
- 4. Fabrizio P , Pletcher SD , Minois N , Vaupel JW and Longo VD. Chronological aging-independent replicative life span regulation by Msn2/Msn4 and Sod2 in Saccharomyces cerevisiae. FEBS Letters. 2004; 557: 136 -142. [PubMed] .
- 5. Duttaroy A , Paul A , Kundu M and Belton A. A Sod2 Null Mutation Confers Severely Reduced Adult Life Span in Drosophila. Genetics. 2003; 165: 2295 -2299. [PubMed] .
- 6. Kirby K , Hu J , Hilliker AJ and Phillips JP. RNA interference-mediated silencing of Sod2 in Drosophila leads to early adult-onset mortality and elevated endogenous oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99: 16162 -16167. [PubMed] .
- 7. Lebovitz RM , Zhang H , Vogel H , Cartwright J Jr , Dionne L , Lu N , Huang S and Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. PNAS. 1996; 93: 9782 -9787. [PubMed] .
- 8. Li Y , Huang TT , Carlson EJ , Melov S , Ursell PC , Olson JL , Noble LJ , Yoshimura MP , Berger C , Chan PH , Wallace DC and Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995; 11: 376 -381. [PubMed] .
- 9. Williams MD , Van Remmen H , Conrad CC , Huang TT , Epstein CJ and Richardson A. Increased Oxidative Damage Is Correlated to Altered Mitochondrial Function in Heterozygous Manganese Superoxide Dismutase Knockout Mice. J Biol Chem. 1998; 273: 28510 -28515. [PubMed] .
- 10. Melov S , Coskun P , Patel M , Tuinstra R , Cottrell B , Jun AS , Zastawny TH , Dizdaroglu M , Goodman SI , Huang TT , Miziorko H , Epstein CJ and Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. PNAS. 1999; 96: 846 -851. [PubMed] .
- 11. Kokoszka JE , Coskun P , Esposito LA and Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. PNAS. 2001; 98: 2278 -2283. [PubMed] .
- 12. Van Remmen H , Williams MD , Guo Z , Estlack L , Yang H , Carlson EJ , Epstein CJ , Huang TT and Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001; 281: H1422 -H1432. [PubMed] .
- 13. Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003; 16: 29 -37. [PubMed] .
- 14. De Benedictis G , Carotebuto L , Carrieri G , De Luca M , Falcone E , Rose G , Calvacanti S , Corsonello F , Feraco E , Baggio G , Bertolini S , Mari D , Mattace R , Yashin AI , Bonafe M and Franceschi C. Gene/longevity association studies at four autosomal loci (REN, THO, PARP, SOD2). Eur J Hum Genet. 1998; 6: 534 -541. [PubMed] .
- 15. Starr JM , Shiels PG , Harris SE , Pattie A , Pearce MS , Relton CL and Deary IJ. Oxidative stress, telomere length and biomarkers of physical aging in a cohort aged 79 years from the 1932 Scottish Mental Survey. Mechanisms of Ageing and Development. 2008; 129: 745 -751. [PubMed] .
- 16. Jiang H , Schiffer E , Song Z , Wang J , Zuerbig P , Thedieck K , Moes S , Bantel H , Saal N , Jantos J , Brecht M , Jeno P , Hall MN , Hager K , Manns MP , Hecker H , Ganser A , Doehner K , Bartke A , Meissner C , Mischak H , Ju Z and Rudolph KL. Proteins induced by telomere dysfunction and DNA damage represent biomarkers of human aging and disease. PNAS. 2008; 105: 11299 -11304. [PubMed] .
- 17. Nalapareddy K , Jiang H , Guachalla LM and Rudolph KL. Determining the influence of telomere dysfunction and DNA damage on stem and progenitor cell aging - what markers can we use. Experimental Gerontology. 2008; 43: 998 -1004. [PubMed] .
- 18. Birnboim HC and Kanabus-Kaminska M. The production of DNA strand breaks in human leukocytes by superoxide anion may involve a metabolic process. Proceedings of the National Academy of Sciences of the United States of America. 1985; 82: 6820 -6824. [PubMed] .
- 19. Moiseeva O , Mallette FA , Mukhopadhyay UK , Moores A and Ferbeyre G. DNA Damage Signaling and p53-dependent Senescence after Prolonged beta-Interferon Stimulation. Mol Biol Cell. 2006; 17: 1583 -1592. [PubMed] .
- 20. Shacter E , Beecham EJ , Covey JM , Kohn KW and Potter M. Activated neutrophils induce prolonged DNA damage in neighboring cells. Carcinogenesis. 1988; 9: 2297 -2304. [PubMed] .
- 21. Wiseman H and Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996; 313: 17 -29. [PubMed] .
- 22. Bodnar AG , Ouellette M , Frolkis M , Holt SE , Chiu CP , Morin GB , Harley CB , Shay JW , Lichtsteiner S and Wright WE. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science. 1998; 279: 349 -352. [PubMed] .
- 23. Harley CB , Futcher AB and Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345: 458 -460. [PubMed] .
- 24. Shay JW , Pereira-Smith OM and Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Experimental Cell Research. 1991; 196: 33 -39. [PubMed] .
- 25. Hayflick L and Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961; 25: 585 -621. .
- 26. Passos J , Saretzki G , Ahmed S , Nelson G , Richter T , Peters H , Wappler I , Birket MJ , Harold G , Schaeuble K , Birch-Machin MA , Kirkwood TBL and von Zglinicki T. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biology. 2007; 5: e110 [PubMed] .
- 27. Unterluggauer H , Hampel B , Zwerschke W and Jansen-Durr P. Senescence-associated cell death of human endothelial cells: the role of oxidative stress. Exp Gerontol. 2003; 38: 1149 -1160. [PubMed] .
- 28. Chen Q , Fischer A , Reagan JD , Yan L and Ames BN. Oxidative DNA Damage and Senescence of Human Diploid Fibroblast Cells. PNAS. 1995; 92: 4337 -4341. [PubMed] .
- 29. Sasaki M , Ikeda H , Sato Y and Nakanuma Y. Proinflammatory cytokine-induced cellular senescence of biliary epithelial cells is mediated via oxidative stress and activation of ATM pathway: A culture study. Free Radic Res. 2008; 42: 625 -632. [PubMed] .
- 30. Chen QM Replicative senescence and oxidant-induced premature senescence. Beyond the control of cell cycle checkpoints. Ann N Y Acad Sci. 2000; 908: 111 -125. [PubMed] .
- 31. Chen Q and Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A. 1994; 91: 4130 -4134. [PubMed] .
- 32. Kim KS , Kang KW , Seu YB , Baek SH and Kim JR. Interferon-[gamma] induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mechanisms of Ageing and Development. 2009; 130: 179 -188. [PubMed] .
- 33. Rai P , Onder TT , Young JJ , McFaline JL , Pang B , Dedon PC and Weinberg RA. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc Natl Acad Sci U S A. 2009; 106: 169 -174. [PubMed] .
- 34. von Zglinicki T , Saretzki G , D÷cke W and Lotze C. Mild Hyperoxia Shortens Telomeres and Inhibits Proliferation of Fibroblasts: A Model for Senescence. Experimental Cell Research. 1995; 220: 186 -193. [PubMed] .
- 35. Parrinello S , Samper E , Krtolica A , Goldstein J , Melov S and Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5: 741 -747. [PubMed] .
- 36. Drane P , Bravard A , Bouvard V and May E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene. 2001; 20: 430 -439. [PubMed] .
- 37. Epperly MW , Sikora CA , DeFilippi SJ , Gretton JA , Zhan Q , Kufe DW and Greenberg JS. Manganese Superoxide Dismutase (SOD2) Inhibits Radiation-Induced Apoptosis by Stabilization of the Mitochondrial Membrane. Radiation Research. 2002; 157: 568 -577. [PubMed] .
- 38. Choudhury AR , Ju Z , Djojosubroto MW , Schienke A , Lechel A , Schaetzlein S , Jiang H , Stepczynska A , Wang C , Buer J , Lee HW , von Zglinicki T , Ganser A , Schirmacher P , Nakauchi H and Rudolph KL. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007; 39: 99 -105. [PubMed] .
- 39. Lee HW , Blasco MA , Gottlieb GJ , Horner JW , Greider CW and DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998; 392: 569 -574. [PubMed] .
- 40. Schaetzlein S , Kodandaramireddy NR , Ju Z , Lechel A , Stepczynska A , Lilli DR , Clark AB , Rudolph C , Kuhnel F , Wei K , Schlegelberger B , Schirmacher P , Kunkel TA , Greenberg RA , Edelmann W and Rudolph KL. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell. 2007; 130: 863 -877. [PubMed] .
- 41. Chin L , Artandi SE , Shen Q , Tam A , Lee SL , Gottlieb GJ , Greider CW and DePinho RA. p53 Deficiency Rescues the Adverse Effects of Telomere Loss and Cooperates with Telomere Dysfunction to Accelerate Carcinogenesis. Cell. 1999; 97: 527 -538. [PubMed] .
- 42. Prowse KR and Greider CW. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92: 4818 -4822. [PubMed] .
- 43. Sedelnikova OA , Horikawa I , Zimonjic DB , Popescu NC , Bonner WM and Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004; 6: 168 -170. [PubMed] .
- 44. Blasco MA , Lee HW , Hande MP , Samper E , Lansdorp PM , DePinho RA and Greider CW. Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell. 1997; 91: 25 -34. [PubMed] .
- 45. Rudolph KL , Chang S , Lee HW , Blasco M , Gottlieb GJ , Greider C and DePinho RA. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell. 1999; 96: 701 -712. [PubMed] .
- 46. von Zglinicki T Oxidative stress shortens telomeres. Trends in Biochemical Sciences. 2002; 27: 339 -344. [PubMed] .
- 47. Ito K , Hirao A , Arai F , Takubo K , Matsuoka S , Miyamoto K , Ohmura M , Naka K , Hosokawa K , Ikeda Y and Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006; 12: 446 -451. [PubMed] .
- 48. Mansouri A , Muller FL , Liu Y , Ng R , Faulkner J , Hamilton M , Richardson A , Huang TT , Epstein CJ and Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mechanisms of Ageing and Development. 2006; 127: 298 -306. [PubMed] .
- 49. Kohn KW , Erickson LC , Ewig RAG and Friedman CA. Fractionation of DNA from mammalian cells by alkaline elution. Biochemistry. 1976; 15: 4629 -4637. [PubMed] .
- 50. Pflaum M , Will O and Epe B. Determination of steady-state levels of oxidative DNA base modifications in mammalian cells by means of repair endonucleases. Carcinogenesis. 1997; 18: 2225 -2231. [PubMed] .
- 51. Bjelland S and Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003; 531: 37 -80. [PubMed] .
- 52. Collins AR , Cadet J , Moller L , Poulsen HE and Vi±a J. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells. Archives of Biochemistry and Biophysics. 2004; 423: 57 -65. [PubMed] .
- 53. Fagagna Fdd , Reaper PM , Clay-Farrace L , Fiegler H , Carr P , von Zglinicki T , Saretzki G , Carter NP and Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003; 426: 194 -198. [PubMed] .
- 54. Rogakou EP , Pilch DR , Orr AH , Ivanova VS and Bonner WM. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J Biol Chem. 1998; 273: 5858 -5868. [PubMed] .
- 55. Satyanarayana A , Greenberg RA , Schaetzlein S , Buer J , Masutomi K , Hahn WC , Zimmermann S , Martens U , Manns MP and Rudolph KL. Mitogen Stimulation Cooperates with Telomere Shortening To Activate DNA Damage Responses and Senescence Signaling. Mol Cell Biol. 2004; 24: 5459 -5474. [PubMed] .
- 56. Takai H , Smogorzewska A and de Lange T. DNA Damage Foci at Dysfunctional Telomeres. Current Biology. 2003; 13: 1549 -1556. [PubMed] .
- 57. Li F , Calingasan NY , Yu FM , Mauck WM , Toidze M , Almeida CG , Takahashi RH , Carlson GA , Beal MF , Lin MT and Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. Journal of Neurochemistry. 2004; 89: 1308 -1312. [PubMed] .
- 58. Melov S , Adlard P , Morten K , Johnson F and Golden T. Mitochondrial Oxidative Stress Causes Hyperphosphorylation of Tau. PLoS ONE. 2007; 2: e536 [PubMed] .
- 59. Florian LM MnSOD deficiency has a differential effect on disease progression in two different ALS mutant mouse models. Muscle & Nerve. 2008; 38: 1173 -1183. [PubMed] .
- 60. Vincent AM , Russell JW , Sullivan KA , Backus C , Hayes JM , McLean LL and Feldman EL. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Experimental Neurology. 2007; 208: 216 -227. [PubMed] .
- 61. Ohashi M , Runge MS , Faraci FM and Heistad DD. MnSOD Deficiency Increases Endothelial Dysfunction in ApoE-Deficient Mice. Arterioscler Thromb Vasc Biol. 2006; 26: 2331 -2336. [PubMed] .
- 62. Hutter E , Renner K , Pfister G , Stöckl P , Jansen-Dürr P and Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004; 380: 919 -928. [PubMed] .
- 63. Hoffmann S , Spitkovsky D , Radicella JP , Epe B and Wiesner RJ. Reactive oxygen species derived from the mitochondrial respiratory chain are not responsible for the basal levels of oxidative base modifications observed in nuclear DNA of mammalian cells. Free Radical Biology and Medicine. 2004; 36: 765 -773. [PubMed] .
- 64. Friedberg EC Nucleotide Excision Repair and Cancer Predisposition : A Journey from Man to Yeast to Mice. Am J Pathol. 2000; 157: 693 -701. [PubMed] .
- 65. Neumann A , Sturgis E and Wei Q. Nucleotide excision repair as a marker for susceptibility to tobacco-related cancers: A review of molecular epidemiological studies. Molecular Carcinogenesis. 2005; 42: 65 -92. [PubMed] .
- 66. Rudolph KL , Millard M , Bosenberg MW and DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001; 28: 155 -159. [PubMed] .
- 67. Epe B and Hegler J. Oxidative DNA damage: endonuclease fingerprinting. Methods Enzymol. 1994; 234: 122 -131. [PubMed] .
- 68. Osterod M , Larsen E , Le Page F , Hengstler JG , Van Der Horst GT , Boiteux S , Klungland A and Epe B. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene. 2002; 21: 8232 -8239. [PubMed] .
- 69. Hutter E , Skovbro M , Lener B , Prats C , Rabol R , Dela F and Jansen-Durr P. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell. 2007; 6: 245 -256. [PubMed] .
- 70. Srere PA Colowick SP and Kaplan NOP. Citrate synthase Methods in Enzymology. London Academic Press 1969; .
- 71. Satyanarayana A , Wiemann SU , Buer J , Lauber J , Dittmar KE , Wustefeld T , Blasco MA , Manns MP and Rudolph KL. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003; 22: 4003 -4013. [PubMed] .