The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1
Abstract
Yeast mother cell-specific aging constitutes a model of replicative aging as it occurs in stem cell populations of higher eukaryotes. Here, we present a new long-lived yeast deletion mutation,afo1 (for aging factor one), that confers a 60% increase in replicative lifespan. AFO1/MRPL25 codes for a protein that is contained in the large subunit of the mitochondrial ribosome. Double mutant experiments indicate that the longevity-increasing action of the afo1 mutation is independent of mitochondrial translation, yet involves the cytoplasmic Tor1p as well as the growth-controlling transcription factor Sfp1p. In their final cell cycle, the long-lived mutant cells do show the phenotypes of yeast apoptosis indicating that the longevity of the mutant is not caused by an inability to undergo programmed cell death. Furthermore, the afo1 mutation displays high resistance against oxidants. Despite the respiratory deficiency the mutant has paradoxical increase in growth rate compared to generic petite mutants. A comparison of the single and double mutant strains for afo1 and fob1 shows that the longevity phenotype of afo1 is independent of the formation of ERCs (ribosomal DNA minicircles). AFO1/MRPL25 function establishes a new connection between mitochondria, metabolism and aging.
Introduction
Yeast (Saccharomyces
cerevisiae) mother cell-specific aging has been shown to be based on the asymmetric
distribution of damaged cellular material including oxidized proteins [1]. The
mother cell progressively accumulates this material and ages depending on the
number of cell division cycles, while the daughter "rejuvenates" and
enjoys a full lifespan. Young daughter cells and old (senescent) mother cells
can be efficiently separated based on their different size, by elutriation centrifugation
[2].
At least
some biochemical and genetic mechanisms of aging are conserved throughout the
evolution of eukaryotes. A prominent hypothesis postulates that the progressive
deterioration of mitochondrial metabolism leads to the production of reactive
oxygen species (ROS) that oxidize vulnerable cellular proteins and lipids,
while damaging the genome. The cell's genetic response to this oxidative stress
may appear as a "genetic program of aging". In this light, some of
the current aging theories could well be interrelated and compatible among each
other (for review, see [3,4]).
The TOR
signaling pathway is highly conserved from yeast to human cells [5]. It
regulates nutrient responses by modulating the nucleo-cytoplasmic shuttling of
transcription factors including Sfp1p, which governs ribosome biosynthesis [6].
Down-regulation of TOR kinase induces entry into stationary phase and
stimulates autophagy, a process that is vital for survival in conditions of starvation
[7]. TOR kinase activity may also be involved in the retrograde response of
cells that adapt their nuclear transcriptome to defects in mitochondrial
respiration [8]. Yeast possesses two closely related proteins, Tor1p and Tor2p,
forming two "TOR complexes" among which only one, TORC1 (containing either
Tor1p or Tor2p and active in growth control), is inhibited by rapamycin. Deletion
of TOR2 is lethal due to its essential function in TORC2 (acting on
determination of cell polarity). Deletion of TOR1 leads to an increase
in mitochondrial respiration and protein density [9,10] and to a 15% increase
in replicative lifespan, thus establishing a link between nutrition, metabolism,
and longevity [11].
In this paper we are presenting a novel
long-lived mutant of yeast that establishes a new connection between
mitochondria, metabolism and aging. The life-prolonging mutation affects a gene
encoding a mitochondrial ribosomal protein, leads to respiratory deficiency,
and relies on TOR1 to confer longevity.
Results
A novel yeast mutant
with reduced replicative aging
We compared the
transcriptome of senescent yeast mother cells (fraction V) with young daughter cells
(fraction II) after separating them by elutriation centrifugation [2].
Senescent cells were found to upregulate 39 genes and to down-regulate 53
transcripts. Deletion mutants [12] corresponding to these 92 genes were tested
for their resistance or hypersensitivity to five different oxidants (hydrogen
peroxide, tert-butyl hydroperoxide (t-BHP), diamide, cumene
hydroperoxide, and menadione). Only two mutants were found to be consistently
resistant against more than one oxidant (and not hypersensitive to any other
oxidant). Among these two mutants only one, deleted for YGR076C/MRPL25 (later
termed AFO1, see below) caused a mother cell-specific lifespan expansion
on the standard media used by us (SC + 2% glucose) (Figure 1). This deletion
mutation conferred resistance to diamide and t-BHP and a somewhat weaker
resistance to hydrogen peroxide, as well as a 50% reduced ROS production (as
compared to the BY4741 ρ0mutant). ROS
production was measured by quantitation of fluorescence signals obtained after dihydroethidium
(DHE) staining. The mutant displayed a 60% increase in the median and a 71% increase
in the maximum lifespan (Figure 1). The mutant only grew on media containing fermentable
carbon sources and hence is respiration deficient. We therefore asked if the
respiratory deficiency caused the increased replicative life span. However, a bona
fide BY4741 ρ0mutant did not
show any extension in replicative life span (as compared to BY4741 WT cells),
meaning that lack of respiration is not sufficient to confer longevity to
mother cells (Figure 1). We also tested if afo1Δ cells displayed
the retrograde response [3,13] by measuring CIT2 transcription and no
effect of the afo1Δ mutation could be discerned
(see Supplementary Figure 1). We conclude that the elongation of lifespan observed here is not
caused by respiratory deficiency and is independent of the retrograde response
as defined by Jazwinski [14] and Butow [15].
AFO1 codes for a mitochondrial ribosomal protein
AFO1 (YGR076C) codes for MrpL25p, identified by proteomic
analysis as a component of the large subunit of the mitochondrial ribosome
[16]. Because of its remarkable longevity phenotype, we re-named the gene AFO1(for aging factor one). A recombinant construct in which Afo1p was fused in
its C-terminus with GFP (Afo1-GFP) was transfected into a heterozygous afo1Δstrain. Tetrad dissection revealed that the Afo1-GFP could replace endogenous Afo1p to enable growth on
a non-fermentable carbon source. Confocal fluorescence microscopy confirmed
that the protein is located in mitochondria irrespective of the cellular age
and the genetic background (supplementary material, Supplementary Figure 2). The deletion
mutant afo1Δ exhibited a ρ0petite phenotype, meaning that it failed to grow
on glycerol media and lacked DAPI-detectable mitochondrial DNA. The mutant also
showed negligible oxygen consumption when growing on glucose (data not shown).
However, in contrast to a bona fide BY4741 ρ0petite mutant, which grew much more slowly than WT
cells on standard media with 2% glucose as carbon source, the afo1Δ
mutant grew as rapidly as WT cells. The growth properties of the mutant and its
metabolic implications will be published in detail elsewhere. The average size
of the afo1Δ mutant cells in exponential phase was equal to that of
WT cells, while cells of the ρ0strain were
about 20% larger. Importantly, disruption of the AFO1 gene in ρ0cells restored rapid growth, hence reversing the
growth defect induced by the absence of mitochondrial DNA.
To obtain definite genetic
proof that the AFO1 deletion caused the resistance against oxidative stress
and the extension of the life span described above, we performed co-segregation
tests in meiotic tetrads after out-crossing the afo1Δ strain in an
isogenic cross. In 10 unselected tetrads, which
all revealed a regular 2:2 segregation, we observed strict co-segregation of
G418 resistance indicating the presence of the gene deletion, respiratory
deficiency, and resistance against hydrogen peroxide stress (Figure 2A). We
also tested the lifespan of all four haploid progeny of one tetrad and found
consistent co-segregation of the deletion allele of afo1 with extended
lifespan (Figure 2B). Furthermore, we also tested if the phenotype of the
mutant might result from changes of the expression of the two neighboring genes
of AFO1. No such effect was apparent (Figure 2C). We conclude that lack
of AFO1 results in long lifespan and the oxidative stress resistance.
![Lifespans of
isogenic strains afo1Δ, wild type
BY4741 and BY4741 ρ0. Lifespans were determined as
described previously [2] by micromanipulating daughter cells and counting
generations of at least 45 yeast mother cells on synthetic complete (SC)
media with 2% glucose as carbon source.](/article/100065/figure/F1/large)
Figure 1. Lifespans of
isogenic strains afo1Δ, wild type
BY4741 and BY4741 ρ0. Lifespans were determined as
described previously [2] by micromanipulating daughter cells and counting
generations of at least 45 yeast mother cells on synthetic complete (SC)
media with 2% glucose as carbon source.

Figure 2A. Segregation of the mutant phenotypes of
afo1Δ in meiotic tetrads after outcrossing and influence of
the genes adjacent to AFO1. 10μl aliquots of the cultured
strains were spotted on SC-glucose and on SC-glucose +
oxidants, as indicated in the figure. Cultures were grown
to OD600 = 3.0 and diluted as indicated. Three out of ten
tetrads tested are shown together with two wild type and
two afo1 deletion strains.
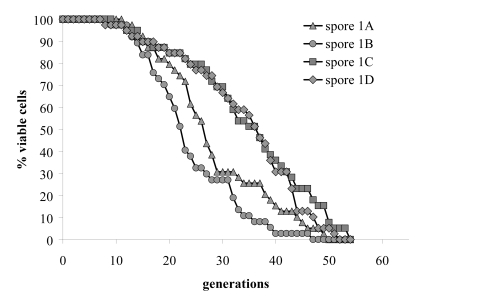
Figure 2B. Replicative lifespans of the four haploid
segregants of one meiotic tetrad were determined.
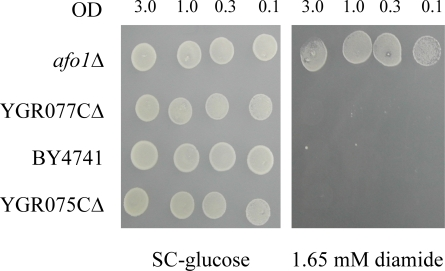
Figure 2C. Deletion strains corresponding to the two
genes adjacent to AFO1 are shown. These deletions have no influence on the resistance to oxidants.
We next addressed the
possibility that the deletion of other mitochondrial ribosomal genes might also
lead to an increase in replicative life span. For this, we investigated the
lifespan, growth properties and oxidative stress resistance of two additional
deletion mutants in the genes MRP17 and PPE1, encoding
mitochondrial ribosomal proteins of yeast. YKL003CΔ (mrp17Δ)
was found to be resistant against diamide, t-BHP and juglone, but was
hypersensitive to hydrogen peroxide and had a normal lifespan. YHR075CΔ (ppe1Δ)
was resistant against diamide, yet had a normal lifespan (Figure 3). Therefore,
the effect of the afo1Δ deletion mutant on lifespan is
gene-specific.
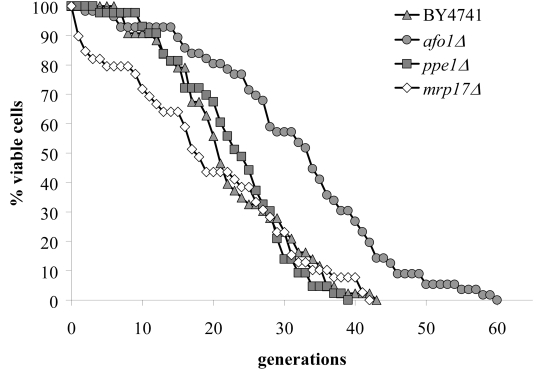
Figure 3. Lifespans of
the strains deleted for ppe1 and mrp17. The single
deletion strains for YKL003C (encoding for Mrp17p) and YHR075C (encoding
for Ppe1p), both of the mitochondrial ribosomal small subunit, were tested
for their lifespan. The strains were constructed in the BY4741 background.
The measured lifespans were not significantly different from wild type (p<0.02).
Longevity mediated by
the afo1 deletion is mediated by the TOR1 pathway
Two independent lines of evidence
revealed that the afo1 deletion confers longevity and oxidative stress
resistance through the TOR1 signaling pathway. First, we chromosomally
integrated a C-terminally GFP-labeled version of the transcription factor,
Sfp1p, at the SFP1 locus under the control of the native promotor in
strains afo1Δ, BY4741 WT and BY4741 ρ0. Sfp1p
is activated by the TOR1 and PKA pathways and is regulated by shuttling between
the nucleus in its active form and the cytoplasm upon deactivation. Sfp1p is a
major regulator of cytoplasmic ribosome synthesis and, consequently, of
cellular growth [6]. As expected, addition of the Tor1p inhibitor rapamycin to WT
cells induced the translocation of Sfp1p from the nucleus to the cytoplasm. In
the bona fide BY4741 ρ0strain, Sfp1p
was found constitutively in the cytoplasm, even in the absence of rapamycin. In
stark contrast, in the afo1Δ mutant, Sfp1p was constitutively
present in the nucleus, and rapamycin failed to induce the nucleo-cytoplasmic
translocation of Sfp1p (Figure 4A). Similar results were obtained with an
alternative Tor1p inhibitor, arsenite [17]. Arsenite
induced the nucleocyto-plasmic translocation of Sfp1p in WT cells,
while Sfp1p stayed in the cytoplasm of ρ0cells and in the nuclei of afo1 mutant cells,
irrespective of the addition of arsenite (Figure 4B). Rapamycin failed to
inhibit the growth of afo1 mutant cells [18]. Altogether, these data
suggested that TOR1 signaling might govern the longevity of afo1 cells.
The relation between TOR1 and AFO1 was further explored by epistasis
experiments using double mutants (Figure 5). The lifespan of the double
deletion strain (afo1Δ, tor1Δ) was similar to the
lifespan of the tor1 deletion strain, i.e. about 15% longer than wild
type (in good agreement with [11]). However, the double mutant afo1Δ,tor1Δ strain aged more rapidly than the single mutant afo1Δ
strain (Figure 5). We conclude that a functional TOR1 gene is needed for
exerting the lifespan-prolonging effect of afo1Δ.
We constructed single and
double knockout afo1Δ, sfp1Δ mutant strains and tested
their mother cell-specific lifespan and oxidative stress resistance. The median
lifespan of sfp1Δ cells was shortened considerably as compared to
WT cells, and the lifespan of the double afo1Δ, sfp1Δ mutant
was longer than that of the sfp1Δ mutant, yet shorter than WT and afo1Δ
(Figure 6A). Hence, the very short lifespan of sfp1Δ mutant cells
is partially rescued by the afo1 mutation. Like the double afo1Δ,sfp1Δ mutant, sfp1Δ cells displayed a major growth
defect. When the sfp1Δ strain was made ρ0with ethidium bromide, cell growth was not further inhibited
(data not shown). Comparison of the strains on plates containing 1.6 mM t-BHP revealed that afo1Δ is
moderately resistant, while single sfp1Δ and double afo1Δ,sfp1Δ mutants exhibited a similar degree of high resistance (Figure 6B). Taken together, these results show that the lifespan-extending effect of afo1Δis most likely independent of the presence of SFP1.
![Influence of rapamycin on subcellular
localization of the transcription factor, Sfp1p. Strains
were grown in liquid SC+2% glucose at 28°C until early
logarithmic phase and rapamycin was added to a final
concentration of 100 nM. This concentration is growth
inhibitory for the wild type strain [6]. Confocal images
were taken at time zero (before addition of rapamycin)
and at 4 h. The chromosomally integrated SFP1-GFP-HIS3
construct [37] was present in the wild type strain BY4741,
was PCR cloned, sequenced and chromosomally integrated at
the SFP1 locus in strains afo1Δ and BY4741 ρ°, respectively.](/article/100065/figure/F4A/large)
Figure 4A. Influence of rapamycin on subcellular
localization of the transcription factor, Sfp1p. Strains
were grown in liquid SC+2% glucose at 28°C until early
logarithmic phase and rapamycin was added to a final
concentration of 100 nM. This concentration is growth
inhibitory for the wild type strain [6]. Confocal images
were taken at time zero (before addition of rapamycin)
and at 4 h. The chromosomally integrated SFP1-GFP-HIS3
construct [37] was present in the wild type strain BY4741,
was PCR cloned, sequenced and chromosomally integrated at
the SFP1 locus in strains afo1Δ and BY4741 ρ°, respectively.
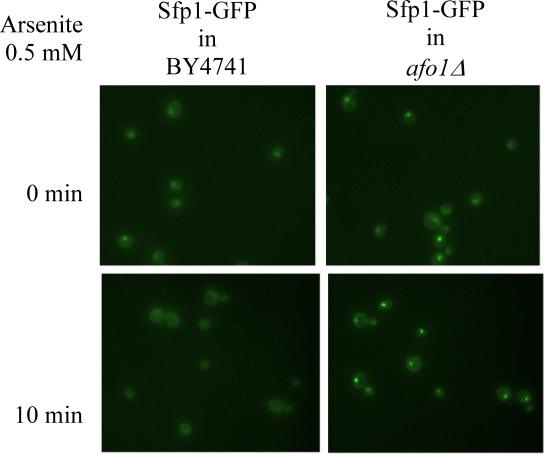
Figure 4B. The same strains as in A were treated
with 0.5 mM arsenite for 10 min.
![Double mutant
experiments of afo1Δ
and
tor1Δ. The TOR1 gene is involved
in nutrient sensing and lifespan determination in yeast [5]. The double
mutant was constructed in an isogenic cross between the two single mutants
in the BY background. Lifespans of the wild type, both single mutants and
the double mutant were determined by micromanipulation. The experiment
shows that an intact TOR1 gene is needed for the lifespan
elongation observed in the afo1Δ strain as the
lifespan of the afo1Δ,
tor1Δ double
mutant strain is not significantly different (ptor1Δ
single
mutant strain.](/article/100065/figure/F5/large)
Figure 5. Double mutant
experiments of afo1Δ
and
tor1Δ. The TOR1 gene is involved
in nutrient sensing and lifespan determination in yeast [5]. The double
mutant was constructed in an isogenic cross between the two single mutants
in the BY background. Lifespans of the wild type, both single mutants and
the double mutant were determined by micromanipulation. The experiment
shows that an intact TOR1 gene is needed for the lifespan
elongation observed in the afo1Δ strain as the
lifespan of the afo1Δ,
tor1Δ double
mutant strain is not significantly different (p<0.02) from the lifespan
of the tor1Δ
single
mutant strain.
Next,
we addressed the question as to whether the longevity phenotype of the afo1 mutation
might originate from suppressing the yeast apoptosis pathway. As shown
previously [2], old mother cells of the wild type display all of the known
markers of yeast apoptosis while these markers are absent from young cells. To
tackle this problem, we isolated young (fraction II) and old cells (fraction V)
from WT and afo1Δ cells by elutriation centrifugation and tested
several markers of apoptosis such as externalization of phosphatidyl serine and
DNA strand breaks (Figure 7). Our data clearly indicated that afo1Δ
cells did not lose the ability to undergo apoptosis. In spite of a 60% longer
median lifespan, senescent mother cells finally succumbed to apoptosis. We
conclude that the components of the programmed cell death pathway that a yeast
cell has at its disposal, do not cause replicative aging, but that vice
versa replicative aging finally leads to cell death via apoptosis.
We also investigated
whether the longevity phenotype of the afo1Δ mutant might be mechanistically
related to the production of extrachromosomal
rDNA minicircles (ERCs) [19,20]. FOB1 encodes
a protein required for the unidirectional replication fork block in rDNA
replication. We analyzed the influence of the fob1 mutation on
longevity, growth, and ERC content of WT and afo1Δ cells. In our
analysis the fob1Δ mutation in the BY4741 strain leads to an
increase of the replicative lifespan by about 5 generations, in good agreement
with previous reports [11,21].
However, we observed a
similar median life span of the fob1Δ, afo1Δ double mutant and
the afo1Δ mutant cells (Figure 8A). As an internal control, both
the fob1Δ single mutant and the fob1Δ, afo1Δ double
mutant exhibited the absence of ERCs even in fraction IV and V old cells, while
a continuous age-dependent increase in ERCs was found, in particular in
fraction IV and V senescent mother cells from WT and afo1Δ cells
(Figure 8B). Thus, the lifespan-extension observed in the afo1Δ strain
occurs in the presence of ERCs and is not further increased when ERCs are
absent, consequently ERCs do not influence longevity in the afo1Δ strain.
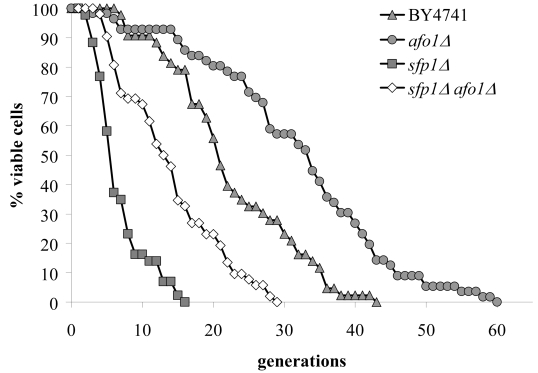
Figure 6A. The double mutant strain, sfp1Δ, afo1Δ
was constructed as described in the Materials and Methods
section, tested for lifespan, and compared with both single
mutant strains and the wild type. The sfp1? strain grows very
slowly although it is respiratory-competent, is highly resistant
to t-BHP and is very short-lived. The short lifespan of sfp1Δ
is partially rescued by afo1Δ.

Figure 6B. The same strains as in A were tested
for resistance against oxidative stress induced by 1.6 mM
and 1.8 mM t-BHP. The strong resistance of the sfp1Δ mutant
strain is not rescued by the afo1 mutation.
Discussion
AFO1, the retrograde response and mitochondrial back-signaling
The retrograde response (as
defined by Jazwinski [14] and Butow [15]) of non-respiring cells is transmitted
through the transcription factor Rtg1/Rtg3p and allows for the transactivation
of genes involved in peroxisome synthesis that compensate for the deficient
amino acid biosynthesis of cells that lack a complete citrate cycle. As an
indicator of the retrograde response, expression of the peroxisomal Cit2p
citrate synthase is usually measured [14,15]. Yeast strains displaying a
strong retrograde response increase their replicative lifespan as ρ0strains over that of the corresponding ρ+strain. The retrograde response is generally
suppressed in 2% glucose but strong on raffinose as sole carbon source [13]. We
have measured CIT2 transcription under the conditions used in this study
and found no increase in the transcript of this gene (Supplementary Figure 1), explaining
why the ρ0strain in the BY4741
series shows the same lifespan as wild type. When raffinose was used as a
carbon source, CIT2 transcription was increased, and as expected, an
increase in the lifespan of the bona fide BY4741 ρ0strain was observed (unpublished observation).
However, during growth on 2% glucose when the retrograde response is absent in
our strain background, we do observe the increase in lifespan described in the
present paper. We therefore conclude that the mechanism leading to this increase
must be different from the retrograde response.
The lifespan elongation
described for the afo1Δ mutant strain depends on a signal
transmitted from mitochondria to the nucleo-cytoplasmic protein synthesis
system and has a strong influence on replicative aging, vegetative growth, and
oxidative stress resistance (see below). We propose to call this regulatory
signaling interaction „mitochondrial back-signaling" to dis-tinguish it from
the retrograde response described by Jazwinski [14] and Butow [15].
Evidence for involvement
of the TOR1 pathway in longevity of the afo1 deletion strain
The nature of the signal created by Afo1p
is unknown, especially since we found this ribosomal protein to be located in
mitochondria in all physiological situations tested, including senescent yeast mother
cells. Nonetheless, two independent lines of evidence support the notion that
increased activity of TOR1 determines the longevity of the afo1 deletion
mutant. First, in the double mutant deleted for both TOR1 and AFO1,
a lifespan is observed that is
only moderately longer than that of the wild type and is identical with the
lifespan of the tor1Δ single mutant (Figure 5). Thus, paradoxically,
the relatively small but significant elongation of the lifespan of ρ+respiring tor1Δ cells depends on
inactivation of Tor1p, while the large increase in lifespan in the
non-respiring afo1Δ cells depends on activity of the Tor1p. Second,
rapamycin fails to abolish the nuclear location of the transcription factor,
Sfp1p, an indicator of Tor1p activity, in afo1Δ cells (Figure 4A). Likewise,
arsenite, another inhibitor of Tor1p [17], fails to abolish the nuclear
location of the transcription factor, Sfp1p, in afo1Δ cells (Figure 4B). Sfp1p is well known to be one of the major metabolic regulators of growth
and ribosome biosynthesis, which is limiting for growth [6]. The data presented
here seem to indicate that Sfp1p activity in the nucleus could be crucial for longevity.
The double mutant experiments shown in Figure 5 indicate that the sfp1Δ
and the afo1Δ mutations exert their influence on longevity
independently of each other. Moreover, we tested TORC1 kinase activity in WT,
ρ0and in afo1Δ
cells (data not shown) and found that the long-lived mutant, like the ρ0strain displayed only very weak TORC1 kinase activity.
These results indicate that the Tor1p activity needed for longevity in the
mutant might be feedback-regulated by Sfp1p and/or
maybe independent of TORC1kinase activity.
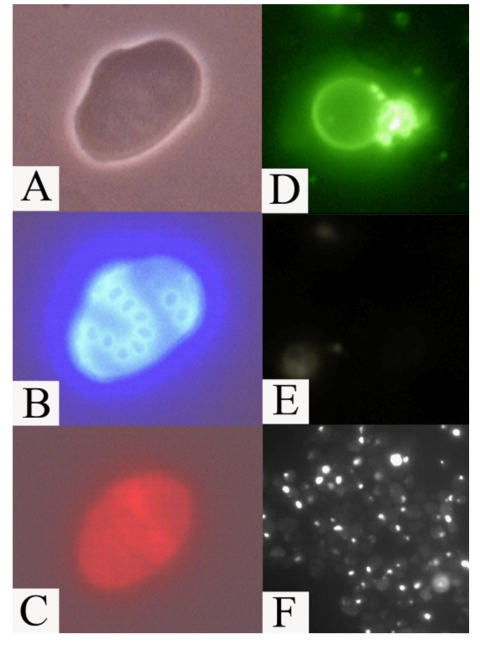
Figure 7. Apoptotic
markers in old mother cells (fraction V) of the mutant afo1Δ strain. (A)
phase contrast; (B) same cell as in A stained with Calcofluor White
M2R; (C) the same cell stained with DHE indicating a high level of
ROS; (D) an old mother cell stained with FITC-annexin V revealing
inversion of the plasma membrane; (E) the same cell as in (D)
shows absence of staining with propidium iodide revealing intact plasma
membrane; (F) TUNEL staining of old afo1Δ cells.
A tentative scheme
describing the genetic interactions of mitochondrial back-signaling that we are
discussing here, is presented in Figure 9.
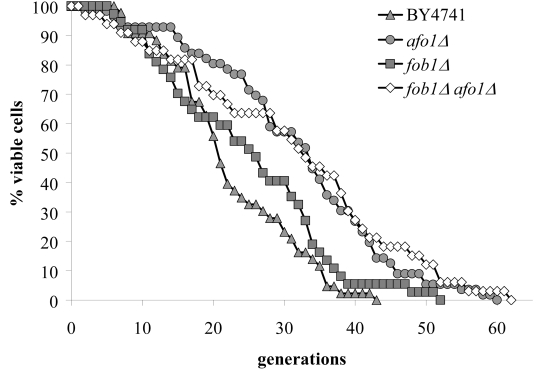
Figure 8A. The two mutations, fob1Δ and afo1Δ were
combined in a haploid strain from a meiotic tetrad obtained
from an isogenic cross. Wild type, the two single mutants
and the double mutant were tested for mother cell-specific
lifespan. The fob1 mutation does not further increase the
lifespan of the afo1 mutant strain (p<0.02).
![Old and young cells of the same strains as
in A were isolated by elutriation centrifugation and
ERCs were analyzed by gel electrophoresis and Southern blotting
with an rDNA-specific probe as described in [19]. Thick arrow:
chromosomal rDNA repeats; Thin arrow: ERCs (minicircles).
Taken together, the results presented in this figure indicate
that longevity in the afo1Δ strain is not influenced by the
fob1-deletion or the presence of ERCs.](/article/100065/figure/F8B/large)
Figure 8B. Old and young cells of the same strains as
in A were isolated by elutriation centrifugation and
ERCs were analyzed by gel electrophoresis and Southern blotting
with an rDNA-specific probe as described in [19]. Thick arrow:
chromosomal rDNA repeats; Thin arrow: ERCs (minicircles).
Taken together, the results presented in this figure indicate
that longevity in the afo1Δ strain is not influenced by the
fob1-deletion or the presence of ERCs.
Has
the function of AFO1 been conserved in evolution?
In this paper we are
presenting evidence for a possibly indirect interaction of the mitochondrial ribosomal
protein, Afo1p, with the TOR1 signaling system of yeast. This
interaction is independent of the primary function of Afo1p in translation. A
reduction of TOR1 signaling in yeast [7], rodent and human cells [22]
suppresses cellular aging in cell culture [22], and increases longevity in mice
[23], worms [24] and fruit flies [25,26]. These effects were shown to be
non-additive with caloric restriction suggesting that the TOR pathway in these
organisms is crucial for transmitting the caloric restriction signal. In
metazoa, cellular life, but not organismic life is possible in the absence
of mitochondrial respiration. It is therefore difficult to draw conclusions as
to the generality of the afo1 mutant-based longevity described in the
present paper.
The protein complement of mitochondrial
ribosomes of both yeast and human cells has been studied [16 and the literature
cited therein, 27-30] and the non-translational or extra-ribosomal functions
(mostly in transcriptional regulation) of ribosomal proteins have been
extensively studied
[31,32]. The published extraribosomal functions mostly concern cytoplasmic,
not mito-chondrial ribosomes. Possible extraribosomal functions have to date
been found for three of the yeast mitochondrial ribosomal proteins only.
Mrps17p and Mrpl37p may play a special role in yeast sporulation [33]. The
mitochondrial ribosomal protein of the small subunit, yDAP-3 [34] is well
conserved between yeast and human cells and besides its translational role has
a distinct function in apoptosis. Its role in the aging process has not been
studied yet.
![Schematic
diagram of genetic interactions involving AFO1 based on the
results presented in this paper. Dashed arrows: genetic interactions for
which a molecular mechanism has not been determined. Both Sfp1p and Rtg1,3p
shuttle to the cytoplasm when Tor1p is inhibited by rapamycin. They are
indicated in bold in the nucleus, where they are active. An activating
influence of the TOR1
kinase
complex on the transcription factor Rtg1/Rtg3 has been
postulated by Dann [5]. Feedback inhibition of Tor1p by nuclear Sfp1p is
indicated. The RAS/cAMP and SCH9 components are omitted for
clarity. Their interaction with the TOR pathway is complex. M,
mitochondrion; N, nucleus; P, peroxisome.](/article/100065/figure/F9/large)
Figure 9. Schematic
diagram of genetic interactions involving AFO1 based on the
results presented in this paper. Dashed arrows: genetic interactions for
which a molecular mechanism has not been determined. Both Sfp1p and Rtg1,3p
shuttle to the cytoplasm when Tor1p is inhibited by rapamycin. They are
indicated in bold in the nucleus, where they are active. An activating
influence of the TOR1
kinase
complex on the transcription factor Rtg1/Rtg3 has been
postulated by Dann [5]. Feedback inhibition of Tor1p by nuclear Sfp1p is
indicated. The RAS/cAMP and SCH9 components are omitted for
clarity. Their interaction with the TOR pathway is complex. M,
mitochondrion; N, nucleus; P, peroxisome.
Afo1p is
a protein of S. cerevisae for which an obvious homolog is known in Neurospora
crassa [16], but which has no easily apparent counterparts in other
eukaryotes (or in E. coli) as judged by sequence similarity alone. It is
therefore impossible presently to draw conclusions about possible functions of
homologs of this protein in aging of higher eukaryotes. However, this may
change when the three-dimensional structure of mitochondrial ribosomes will be
determined and structural and functional homologs of Afo1p in higher eukaryotes
may be found.
Conclusion
In conclusion, we have
shown that deletion of a gene coding for a mitochondrial ribosomal protein of
yeast (AFO1, systematic name: YGR076C) leads not only to respiratory
deficiency (as expected), but also to oxidative stress resistance, very low
internal production of ROS and a substantial (60%) increase in the mother
cell-specific lifespan of the strain. This was unexpected because a bona
fide ρ0strain derived
from the same parental yeast displayed no increase in lifespan. The lifespan
effect of the mutant depends on the presence of a functional TOR1 gene.
The relatively large effect on lifespan which afo1Δ confers is,
however, independent of the presence or absence of ERCs in the aging mother
cells. These experimental results show once again that replicative aging is multifactorial
and that the limiting factor for the determination of the replicative lifespan
may be very different for different strains and for different growth
conditions. The physiological characterization of the long-lived mutant shows a
relationship of the yeast replicative aging process to two cellular processes
that have also been found to determine aging in higher organisms: i) nutritional
signaling through the highly conserved TOR pathway, and ii) generation of and
defense against internally generated oxidative stress molecules (ROS).
Materials and Methods
Media.
The following media were used in this study: complex
medium (YPD) containing 1% yeast extract, 2% (w/v) peptone and 2%
(w/v) D-glucose; synthetic complete glucose medium (SC-glucose) containing 2%
(w/v) D-glucose, 0.17% yeast nitrogen base without amino acids and ammonium sulphate,
0.5% ammonium sulphate and 10 mL complete dropout; synthetic complete raffinose
(SC-raffinose), synthetic complete glycerol medium (SC-glycerol) or synthetic
complete lactate medium (SC-lactate), containing the same ingredients as
SC-glucose, except that 2% (w/v) D-glucose is replaced by 2% (w/v) raffinose,
2% (v/v) glycerol or 3% (w/v) lactate as a carbon source; synthetic minimal
medium (SD) containing 2% (w/v) D-glucose, 0.17% yeast nitrogen base without
amino acids and ammonium sulphate and 0.5% ammonium sulphate. Complete dropout
contains: 0.2% Arg, 0.1%, His, 0.6% Ile, 0.6% Leu, 0.4% Lys, 0.1% Met, 0.6%
Phe, 0.5% Thr, 0.4% Trp, 0.1% Ade, 0.4% Ura, 0.5% Tyr. Agar plates were made by
adding 2% (w/v) agar to the media.
Strains.
S. cerevisiae strains BY4741 and BY4742
(EUROSCARF) were used. For experiments with deletion strains we used the
EUROSCARF deletion mutant collection
(http://www.rz.unifrankfurt.de/FB/fb16/mikro/euroscarf/index.html). Other strains were obtained from the
"Yeast-GFP clone collection" (Invitrogen Cooporation, Carlsbad, California, USA) or the "TetO7promoter collection"
(Open Biosystems, Huntsville, AL, USA). Double mutants were constructed by
isogenic crossing of two single mutants of opposing mating type in the BY4741
background followed by sporulation of the obtained zygote and dissection of
meiotic tetrads. A bona fide ρ0petites strain was made from the BY4741 wild type
-strain as described in [35]. Briefly the strain was grown from a small
inoculum to saturation in synthetic minimal medium (SD) containing the
auxotrophic requirements plus 25μg/mL ethidium bromide. A second culture
was inoculated from the first in the same medium and grown to saturation. This
culture was streaked out for single colonies on YPD plates and checked for
petite character by growth on YPG (complex medium containing 2% (v/v) glycerol
as sole carbon source). To transfer the SFP1-GFP-HIS3 chromosomal
integrated GFP construct into the afo1Δ and bona fide BY4741
ρ0strain, the SFP1-GFP-HIS3 construct was PCR cloned and chromosomally integrated at the SFP1 locus
of the afo1Δ and BY4741 ρ0strains, respectively.
Elutriation.
Cells were separated according to their diameter
using the Beckman elutriation system and rotor JE-6B with a standard
elutriation chamber. Before the separation, the cells were grown in 100 mL of YPD
medium at 28°C on a rotary shaker for 24 h. Then, the cells were harvested at
3000 rpm and resuspended in 1X PBS buffer (8 g of NaCl, 0.2 g of KCl, 1.44 g of
Na2HPO4, 0.24 g of KH2PO4, pH 7.4, in a total volume of 1 L) at 4°C. The
elutriation chamber was loaded with 4.2 mL of cell suspension corresponding to
about 109 cells. To separate cell fractions with different diameters, the chamber
was loaded at a flow rate of 10 mL/min and a rotor speed of 3200 rpm. Cells
with a diameter <5 μm were elutriated (fraction I). To collect fraction
II (diameter 5-7 μm), the flow rate was set to 15 mL/min and a rotor speed
to 2700 rpm. Fraction III (diameter 7-8.5 μm) was elutriated at 2400 rpm.,
fraction IV (diameter 8.5-10 μm) at 2000 rpm. and, finally, fraction V (diameter
10-15 μm) at 1350 rpm. The quality of separation of particular fractions
was verified microscopically. Note that in the separation of the slightly
smaller afo1Δ mutant cells no significant amount of fraction V
cells could be isolated. Therefore, fraction IV was used for ERC determination.
Replicative lifespan.
The replicative lifespan measure-ments were performed
as described previously [2]. All lifespans were determined on defined
SC-glucose media for a cohort of at least 45 cells. Standard deviations of the
median lifespans were calculated according to Kaplan-Meier statistics [36].
Median lifespan is the best-suited single parameter to describe a lifespan
distribution. To determine whether two given lifespan distributions are
significantly different at the 98% confidence level, Breslow, Tarone-Ware and
log-rank statistics were used. All statistical calculations were performed
using the software package SPSS 15.0 (SPSS Inc., Chicago, IL, USA).
Sensitivity to oxidants.
Plate tests for sensitivity to oxidants were
performed by spotting cell cultures onto SC-glucose plates containing various
concentrations of H2O2(2-4 mM) and
t-BHP (0.8-2 mM). Cells were grown to stationary phase in liquid SC-glucose,
serially diluted to OD600values of 3.0;
1.0; 0.3; 0.1 and 10 μL aliquots were spotted onto the appropriate plates.
Sensitivity was determined by comparison of growth with that of the wild-type
strain after incubation at 28°C for three days. RNA preparation and Northern
analysis RNA was prepared from
log-phase cells in SC-glucose and SC-raffinose with the RNeasy Midi Kit (Qiagen,
Vienna, Austria). Heat-denatured RNA samples (10 μg) were separated by
electrophoresis (5 h, 5 V/cm) in a 1.3% (w/v) agarose gel containing 0.6 M
formaldehyde, transferred to a nylon membrane, and immobilized by irradiation
with UV light (UV Stratalinker 1800, Stratagene, La Jolla, CA). Membranes were
pre-hybridized for 2 h at 60°C in 10 mL Church Gilbert solution (0.5 M Na2HPO4, 1 mM
EDTA, 7% (w/v) SDS, 1% BSA) and 100 μl (10 mg/mL stock solution) single-stranded
denatured salmon sperm DNA, and then probed under the same conditions for 16 h
with CIT2 and ACT1 probes which were labelled with 32P-dCTP
by random oligonucleotide priming. After hybridisation, the filters were washed
two times for 15 min with 2xSSC/0.1% SDS at room temperature, followed by two
15-min washes with 0.2xSSC/0.1% SDS at 56°C. Blots were wrapped in Saran Wrap
and exposed for 15 min to an imaging cassette (Fujifilm BAS cassette 2325).
Images were scanned in a Phosphoimager (Fujifilm BAS 1800II) using the
BASreader 2.26 software.
Sfp1-GFP localization experiment.
Strains of interest were grown overnight from a small
inoculum to saturation in SC-glucose medium. These cultures were taken to
inoculate 25 mL of SC-glucose medium in such a way that cultures were in the
early exponential growth phase on the next morning. A sample was taken for confocal
microscopy (cells without rapamycin treatment). The rest of the culture was
treated for 4 hours with 100 nM rapamycin (LC Laboratories, Woburn, MA, USA) and further inoculated at 28°C. Before cells were used for confocal microscopy,
cells were harvested by centrifugation and resuspended in fresh SC-glucose
medium. For the arsenite inhibition experiments, the cells were grown in
synthetic medium to an OD600of 1. As2O3was added to the cells to a final concentration of 0.5
mM. Live fluorescence pictures were taken after 10 minutes incubation at 30°C. Markers
of apoptosis were determined as described in [2]. ERCs were determined as
described in [19].
Supplementary Materials
Northern blots (see also experimental procedures) of
CIT2 showing the absence of the retrograde response [15] in
the afo1Δ strain grown on 2% glucose.
Subcellular localization of AFO1-GFP.
Exponentially growing cells of strain YUG37 [38] transformed
with the Afo1p-GFP construct in pMR2 under a tetracyclin-
regulatable promoter were induced with doxycyclin, stained
with Mitotracker deep red and analyzed with a Leica confocal
microscope. (A) Cells stained with Mitotracker deep red; (B)
the same cells as in (A) stained with Afo1p-GFP; (C) the same
cells in phase contrast; (D) overlay of (A) and (B). (E-H) The
same technique as in (A) to (D) was applied to a senescent
mother cell (fraction V) of the same strain. (I) Strain JC 482
[39] transformed with plasmid pUG35 containing Afo1p-GFP under
control of the MET25 promoter and grown to mid-log phase on
SC-lactate was observed by confocal microscopy to reveal the
mitochondrial localization of the protein.
Acknowledgments
We are grateful to the
Austrian Science Fund FWF (Vienna, Austria) for grants S9302-B05 (to M.B.) and
to the EC (Brussels, Europe) for project MIMAGE (contract no. 512020; to M.B.).
CS was supported by the Herzfelder Foundation, the Austrian Science Fund FWF
(B12-P19966), and grant I031-B by the University of Vienna.
Conflicts of Interest
The authors in this
manuscript have no conflict of interests to declare.
References
-
1.
Aguilaniu
H
, Gustafsson
L
, Rigoulet
M
and Nystrom
T.
Asymmetric inheritance of oxidativelydamaged proteins during cytokinesis.
Science.
2003;
299:
1751
-1753.
[PubMed]
.
-
2.
Laun
P
, Pichova
A
, Madeo
F
, Fuchs
J
, Ellinger
A
, Kohlwein
S
, Dawes
I
, Frohlich
KU
and Breitenbach
M.
Aged mother cells of Saccharomyces cerevisiae show markers of oxidativestress and apoptosis.
Mol Microbiol.
2001;
39:
1166
-1173.
[PubMed]
.
-
3.
Jazwinski
SM
The retrograde response links metabolism with stress responses, chromatindependentgene activation, and genome stability in yeast aging.
Gene.
2005;
354:
22
-27.
[PubMed]
.
-
4.
Laun
P
, Heeren
G
, Rinnerthaler
M
, Rid
R
, Kossler
S
, Koller
L
and Breitenbach
M.
Senescenceand apoptosis in yeast mother cell-specific aging and in higher cells: a short review.
Biochim Biophys Acta.
2008;
1783:
1328
-1334.
[PubMed]
.
-
5.
Dann
SG
and Thomas
G.
The amino acid sensitive TOR pathway from yeast to mammals.
FEBS Lett.
2006;
580:
2821
-2829.
[PubMed]
.
-
6.
Marion
RM
, Regev
A
, Segal
E
, Barash
Y
, Koller
D
, Friedman
N
and O'Shea
EK.
Sfp1 is astress- and nutrient-sensitive regulator of ribosomal protein gene expression.
Proc Natl Acad Sci U S A.
2004;
101:
14315
-14322.
[PubMed]
.
-
7.
Bonawitz
N D
, Chatenay-Lapointe
M
, Pan
Y
and Shadel
GS.
Reduced TOR signaling extendschronological life span via increased respiration and upregulation of mitochondrial geneexpression.
Cell Metab.
2007;
5:
265
-277.
[PubMed]
.
-
8.
Dilova
I
, Aronova
S
, Chen
JC
and Powers
T.
Tor signaling and nutrient-based signals converge on Mks1p phosphorylation to regulate expression of Rtg1.Rtg3p-dependent target genes.
JBiol Chem.
2004;
279:
46527
-46535.
[PubMed]
.
-
9.
Finkel
T
Breathing lessons: Tor tackles the mitochondria.
Aging.
2009;
1:
9
-11.
.
-
10.
Pan
Y
and Shadel
GS.
Extension of chronological life span by reduced TOR signaling requiresdown-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density.
Aging.
2009;
1:
131
-145.
.
-
11.
Kaeberlein
M
, Powers
RW 3rd
, Steffen
K K
, Westman
EA
, Hu
D
, Dang
N
, Kerr
EO
, Kirkland
KT
, Fields
S
and Kennedy
BK.
Regulation of yeast replicative life span by TOR andSch9 in response to nutrients.
Science.
2005;
310:
1193
-1196.
[PubMed]
.
-
12.
Winzeler
EA
, Shoemaker
DD
, Astromoff
A
, Liang
H
, Anderson
K
, Andre
B
, Bangham
R
, Benito
R
, Boeke
JD
, Bussey
H
, Chu
AM
, Connelly
C
and Davis
K.
Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis.
Science.
1999;
285:
901
-906.
[PubMed]
.
-
13.
Kirchman
P A
, Kim
S
, Lai
CY
and Jazwinski
SM.
Interorganelle signaling is a determinant oflongevity in Saccharomyces cerevisiae.
Genetics.
1999;
152:
179
-190.
[PubMed]
.
-
14.
Jazwinski
SM
Rtg2 protein: at the nexus of yeast longevity and aging.
FEMS Yeast Res.
2005;
5:
1253
-1259.
[PubMed]
.
-
15.
Butow
RA
and Avadhani
NG.
Mitochondrial signaling: the retrograde response.
Mol Cell.
2004;
14:
1
-15.
[PubMed]
.
-
16.
Smits
P
, Smeitink
JA
, van den Heuvel
LP
, Huynen
MA
and Ettema
TJ.
Reconstructing theevolution of the mitochondrial ribosomal proteome.
Nucleic Acids Res.
2007;
35:
4686
-4703.
[PubMed]
.
-
17.
Hosiner
D
, Lempiainen
H
, Reiter
W
, Urban
J
, Loewith
R
, Ammerer
G
, Schweyen
R
, Shore
D
and Schuller
C.
Arsenic toxicity to Saccharomyces cerevisiae is a consequence of inhibitionof the TORC1 kinase combined with a chronic stress response.
Mol Biol Cell.
2009;
20:
1048
-1057.
[PubMed]
.
-
18.
Xie
MW
, Jin
F
, Hwang
H
, Hwang
S
, Anand
V
, Duncan
M C
and Huang
J.
Insights into TORfunction and rapamycin response: chemical genomic profiling by using a high-density cellarray method.
Proc Natl Acad Sci U S A.
2005;
102:
7215
-7220.
[PubMed]
.
-
19.
Borghouts
C
, Benguria
A
, Wawryn
J
and Jazwinski
SM.
Rtg2 protein links metabolism andgenome stability in yeast longevity.
Genetics.
2004;
166:
765
-777.
[PubMed]
.
-
20.
Sinclair
DA
and Guarente
L.
Extrachromosomal rDNA circles--a cause of aging in yeast.
Cell.
1997;
91:
1033
-1042.
[PubMed]
.
-
21.
Defossez
PA
, Prusty
R
, Kaeberlein
M
, Lin
SJ
, Ferrigno
P
, Silver
PA
, Keil
RL
and Guarente
L.
Elimination of replication block protein Fob1 extends the life span of yeast mother cells.
Mol Cell.
1999;
3:
447
-455.
[PubMed]
.
-
22.
Demidenko
ZN
, Zubova
SG
, Bukreeva
EI
, Pospelov
VA
, Pospelova
TV
and Blagosklonny
MV.
Rapamycin decelerates cellular senescence.
Cell Cycle.
2009;
8:
1888
-1895.
[PubMed]
.
-
23.
Harrison
DE
, Strong
R
, Sharp
ZD
, Nelson
JF
, Astle
CM
, Flurkey
K
, Nadon
NL
, Wilkinson
JE
, Frenkel
K
, Carter
CS
, Pahor
M
, Javors
MA
and Fernandez
E.
Rapamycinfed late in life extends lifespan in genetically heterogeneous mice.
Nature.
2009;
460:
331
-332.
[PubMed]
.
-
24.
Vellai
T
, Takacs-Vellai
K
, Zhang
Y
, Kovacs
AL
, Orosz
L
and Muller
F.
Genetics: influence of TOR kinase on lifespan in C. elegans.
Nature.
2003;
426:
620
[PubMed]
.
-
25.
Kapahi
P
and Zid
B.
TOR pathway: linking nutrient sensing to life span.
Sci Aging Knowledge Environ.
2004;
2004:
PE34
[PubMed]
.
-
26.
Kapahi
P
, Zid
B M
, Harper
T
, Koslover
D
, Sapin
V
and Benzer
S.
Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway.
Curr Biol.
2004;
14:
885
-890.
[PubMed]
.
-
27.
Gan
X
, Kitakawa
M
, Yoshino
K
, Oshiro
N
, Yonezawa
K
and Isono
K.
Tag-mediated isolationof yeast mitochondrial ribosome and mass spectrometric identification of its newcomponents.
Eur J Biochem.
2002;
269:
5203
-5214.
[PubMed]
.
-
28.
Graack
HR
and Wittmann-Liebold
B.
Mitochondrial ribosomal proteins (MRPs) of yeast.
Biochem J.
1998;
329 ( Pt 3):
433
-448.
[PubMed]
.
-
29.
Grohmann
L
, Graack
HR
, Kruft
V
, Choli
T
, Goldschmidt-Reisin
S
and Kitakawa
M.
ExtendedN-terminal sequencing of proteins of the large ribosomal subunit from yeast mitochondria.
FEBS Lett.
1991;
284:
51
-56.
[PubMed]
.
-
30.
Saveanu
C
, Fromont-Racine
M
, Harington
A
, Ricard
F
, Namane
A
and Jacquier
A.
Identification of 12 new yeast mitochondrial ribosomal proteins including 6 that have noprokaryotic homologues.
J Biol Chem.
2001;
276:
15861
-15867.
[PubMed]
.
-
31.
Naora
H
Involvement of ribosomal proteins in regulating cell growth and apoptosis:translational modulation or recruitment for extraribosomal activity.
Immunol Cell Biol.
1999;
77:
197
-205.
[PubMed]
.
-
32.
Wool
IG
Extraribosomal functions of ribosomal proteins.
Trends Biochem Sci.
1996;
21:
164
-165.
[PubMed]
.
-
33.
Hanlon
SE
, Xu
Z
, Norris
DN
and Vershon
AK.
Analysis of the meiotic role of themitochondrial ribosomal proteins Mrps17 and Mrpl37 in Saccharomyces cerevisiae.
Yeast.
2004;
21:
1241
-1252.
[PubMed]
.
-
34.
Berger
T
, Brigl
M
, Herrmann
JM
, Vielhauer
V
, Luckow
B
, Schlondorff
D
and Kretzler
M.
Theapoptosis mediator mDAP-3 is a novel member of a conserved family of mitochondrialproteins.
J Cell Sci.
2000;
113:
3603
-3612.
[PubMed]
.
-
35.
Fox
TD
, Folley
LS
, Mulero
JJ
, McMullin
TW
, Thorsness
PE
, Hedin
LO
and Costanzo
MC.
Analysis and manipulation of yeast mitochondrial genes.
Methods Enzymol.
1991;
194:
149
-165.
[PubMed]
.
-
36.
Kaplan
EL
and Meier
P.
Nonparametric estimation from incomplete observations.
Am Stat Assoc J.
1958;
53:
:457
-481.
.
-
37.
Huh
WK
, Falvo
JV
, Gerke
LC
, Carroll
AS
, Howson
RW
, Weissman
JS
and O'Shea
EK.
Global analysis of protein localization in budding yeast.
Nature.
2003;
425:
686
-691.
[PubMed]
.
-
38.
Bogengruber
E
, Briza
P
, Doppler
E
, Wimmer
H
, Koller
L
, Fasiolo
F
, Senger
B
, Hegemann
JH
and Breitenbach
M.
Functional analysis in yeast of the Brix protein superfamily involved inthe biogenesis of ribosomes.
FEMS Yeast Res.
2003;
3:
35
-43.
[PubMed]
.
-
39.
Pichova
A
, Vondrakova
D
and Breitenbach
M.
Mutants in the Saccharomyces cerevisiae RAS2gene influence life span, cytoskeleton, and regulation of mitosis.
Can J Microbiol.
1997;
43:
774
-781.
[PubMed]
.