




I. Aging-Associated Diseases and Stem Cell Therapy
In 2010, individuals aged 65 years and older constituted approximately 12.9% and 8% of the population in the United States and worldwide, respectively [1]. This number is expected to increase dramatically as millions of individuals from the baby boom generation born between 1945 and 1964, continue to reach this age. Thus, the ability to prevent and treat aging-associated diseases is rapidly becoming a primary focus in various sectors of the biomedical field.
Aging-associated diseases include degenerative conditions affecting tissue and organ function. For example, neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) are conditions marked by the progressive deterioration of structure and function leading to neuronal death. A retinal disorder, age-related macular degeneration, is caused by the gradual degeneration of cells in the macula of the retina and is the leading cause of vision loss in adults over age 55. Conditions such as osteoarthritis and osteoporosis, which are marked by the degeneration of cartilage and bone, respectively, cause the majority of knee, joint, hip, and spine injuries in older individuals.
Aging-associated diseases may also arise from cell dysfunction. Such conditions may include cancer, heart disease, chronic obstructive pulmonary disease (COPD), and diabetes. Cancer is caused by metabolic changes in cells that lead to DNA damage that can fuel the uncontrollable and inappropriate proliferation of cells. The risk of cancer increases significantly with age. Heart disease is typically caused by prolonged exposure of the heart to hypertension, hypercholesterolemia,diabetes, and other cardiovascular risk factors, as well as an age-dependent increase in the prevalence of left ventricular hypertrophy, diastolic dysfunction, and atrial fibrillation [2]. COPD is a group of progressive diseases of the respiratory system that includes emphysema, characterized by the destruction of alveolar cells lining the lung epithelia, and chronic bronchitis, which is caused by abnormal mucus production along the bronchial airways [3]. In the case of ‘adult-onset’ type 2 Diabetes, pancreatic islet β-cell function can be impaired such that insufficient insulin is produced, or cells become resistant to insulin [4].
The prospect of repairing or replacing damaged, dysfunctional or missing cells with new functional cells has shifted the therapeutic paradigm toward restoring tissue function in individuals affected with aging-associated diseases. The primary candidate for the development of these therapies is stem cells, particularly human embryonic stem cells (hESC), which has the capacity to self-renew indefinitely and differentiate into all tissue-specific cell types (Figure 1). In this review, we will describe the derivation, maintenance, and properties of pluripotent hESCs. We will also outline the methods used to induce the generation of specific cell types from hESCs, with primary focus on cell types that are applicable in understanding the pathology, as well as a potential source of cell-based therapies, in aging-associated diseases.
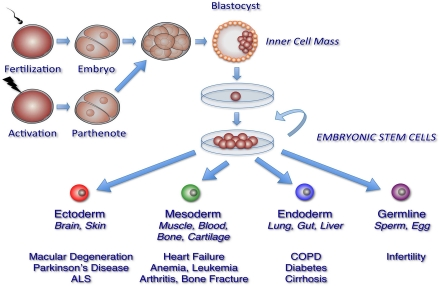
Figure 1. Generation of pluripotent human embryonic stem cell lines. Generation of human embryonic stem cell (hESC) lines involves several steps. Donor embryos are first obtained after in vitro fertilization or by egg activation (parthenogenetic embryos), and allowed to develop in vitro. Pluripotent cells are then isolated either from the inner cell mass of pre-implantation blastocysts or from 4, 8, or 16 -cell stage morulae. Finally, isolated cells are plated in defined hESC medium with or without feeder cell layers to propagate and select for pluripotent cell populations. These processes have resulted in hESC lines able to generate tissues from all three embryonic germ layers and the germline.
II. Human Embryonic Stem Cells: Sources, Maintenance, and Common Properties
Human embryonic stem cells (hESCs) are pluripotent stem cells derived from various stages of embryonic development. hESCs are uniquely capable of proliferating indefinitely and differentiating into all tissue cell types. The unrestricted potential of hESCs has made these cells especially attractive for therapeutic applications. In particular, the regenerative capacity of hESCs could be the key to successful treatment of aging-associated diseases which, as discussed in the previous section, are characteristically marked by progressive dysfunction and/or loss of somatic cells.
Table 1. Methods for differentiating hESCs into specific cell types for treatment of aging-associated diseases
| Clinical Application | Cell Type | Method | Specific Factors and/or Conditions | Ref. | |
|---|---|---|---|---|---|
| Derivation of Endodermal Cells from hESCs | Cirrhosis, Hepatocellular carcinoma, Diabetes-associated liver disease | Hepatocytes | Differentiation of hESC into definitive endoderm, followed by sequential exposure to differentiation factors | FGF, BMP4 hepatocyte growth factor oncostatin M dexamethasone | [35, 36] |
| Diabetes | Pancreatic Islet Progenitors | Activin A, Wnt3A keratinocyte growth factor/FGF7 retinoic acid cyclopamine Noggin | [33] | ||
| Chronic obstructive pulmonary disease | Lung Alveolar Cells | Genetic modification of hESCs followed by spontaneous differentiation | Recombinant keratinocyte growth factor | [38, 39] | |
| Derivation of Mesodermal Cells from hESCs | Prevention and treatment of infection, allograft rejection, allergic and autoimmune diseases, and targeting cancer cells | Dendritic cells | Human embryoid body formation | Serum-free conditions BMP4 | [102] |
| Blood cells | Spin embryoid body formation | Serum-free conditions | [41] | ||
| T and NK cells | Co-culture with stromal cells | Co-culture with stromal M210-B4 cells to enhance expansion of CD34+/CD45+ progenitors | [43] | ||
| Degenerative joint and bone diseases | Chondrocytes | Human embryoid body formation | Micromass of dissociated embryoid bodies BMP2 | [54] | |
| High density culture of dissociated embryoid bodies Ascorbic acid dexamethasone | [57] | ||||
| Directed differentiation on 3D scaffolds | Co-culture with primary chondrocytes poly-D, L-lactide scaffold | [56] | |||
| Heart disease | Cardiomyocytes | Human embryoid body formation | Serum-free conditions bFGF | [46] | |
| Directed differentiation | Activin A BMP4 | [51] | |||
| BMP4 BMP4/bFGF/Activin A VEGF/DKK1 VEGF/DKK1/bFGF | [52] | ||||
| Genetic modification | Cardiac-specific reporters | [49, 53] | |||
| Derivation of Ectodermal Cells from hESCs | Parkinson's disease | Dopaminergic neurons | Co-culture with stromal cells | FGF8 Shh | [61] |
| Formation of neural rosettes | FGF8 Shh | [66] | |||
| Alzheimer's disease, Huntington's disease | Cholinergic neurons | Formation of neurospheres | Shh, FGF8, BMP9 or LHX8/GBX9 overexpression | [68] | |
| ALS | Motor neurons | Formation of neural rosettes | Retinoic acid Shh | [67] | |
| Schwann Cells | Formation of neural rosettes | ciliary neutrotrophic factor neuregulin 1β dbcAMP | [64] | ||
| Oligodendrocytes | Directed differentiation | B27, thyroid hormone retinoic acid, FGF2, EGF, insulin | [65] | ||
| Age-related macular degeneration | Retinal pigment epithelium | Serum-free conditions Activin A, nicotinamide | [73] |
A. Sources and Derivation of hESCs
hESCs are typically derived through the microsurgical removal of the inner cells mass (ICM) of the blastocyst-stage pre-implantation embryo (Figure 1). Cells populating the ICM are pluripotent, in that they are capable of differentiating into the extraembryonic endoderm and the three germ layers that form all tissues of the embryo: ectoderm, mesoderm, and endoderm. Under specific conditions, these cells can proliferate in the undifferentiated state in vitro and retain their pluripotency indefinitely. hESCs have also been derived and established from single blastomeres of the 4- or 8- cell embryo [5-8], 16-cell morula [9, 10], or the ICM of parthenogenetic embryos. A single blastomere is highly totipotent and can generate an entire embryo. Thus, hESCs derived from blastomeres circumvent the ethical controversies surrounding the use of hESCs, since the removal of a single blastomere, in theory, will not impede the ability of the remaining blastomeres to form a normal embryo. Similarly, parthenogenetic embryos, which are generated through artificial fertilization of donor oocytes [11-14], have become highly desirable sources of hESCs because viable embryos are neither created nor destroyed. Furthermore, hESCs derived from parthenotes are especially attractive because these cells are homozygous for major human lymphocyte antigen (HLA) alleles, which could help circumvent immunological rejection that may occur in hESC transplantation therapies (discussed below). Since the initial derivation of pluripotent hESCs from ICM by Thomson and colleagues in 1998 [15], hundreds of hESC lines have been established from various embryonic sources and are now utilized in basic and clinical research worldwide. In the United States, there are over 80 hESC lines that adhere to federal guidelines [16], some of which are now being used in clinical trials.
B. Cellular and Molecular Properties of Pluripotent hESCs
Pluripotent hESCs maintain specific morphological and molecular properties that are shared by the majority of hESC lines. Morphologically, single hESCs have an enlarged nucleus and distinct nucleoli. In culture, proliferating hESCs form compact cell colonies of spherical cells (Figure 2). Under differentiation conditions, these colonies lose their compact morphology and are distinguished by the appearance of flattened cells at the edges as differentiating cells begin to migrate out of the colony. Spontaneous differentiation of hESCs in culture can be controlled with regular supplementation of fresh growth medium [17].
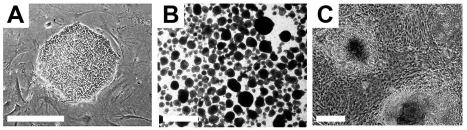
Figure 2. Typical undifferentiated and differentiating hESCs in culture. (A) A compact colony of proliferating pluripotent hESCs can be seen when cultured in defined medium on mouse embryonic fibroblasts. (B) Floating hEBs observed at 3 days after induction of differentiation. (C) Differentiating tissues, including cardiomyocytes, appear within adherent cultures at 48 hours after plating hEBs onto a gelatin-coated culture dish. Bar, 25 μm.
A panel of molecular markers has been identified in 59 independently derived pluripotent hESC lines by the International Stem Cell Initiative, a consortium of stem cell researchers from more than 15 countries [18]. These markers have been routinely used when characterizing pluripotent hESCs. These include genes with known roles in maintaining pluripotency or other developmental processes such as Nanog, POU domain class 5 homeobox 1 protein (POU5F/OCT4), teratocarcinoma-derived growth factor 1 (TDGF1), DNA (cytosine-5-)-methyltransferase 3β (DNMT3β), γ-aminobutyric acid (GABA) A receptor β3 (GABRB3), and growth differentiation factor 3 (GDF4). Additionally, pluripotent hESCs express a number of surface markers such as Stage Specific Embryonic antigens 3 and 4 (SSEA-3, SSEA-4), along with keratin sulfates (TRA-1-60, TRA-1-81, GDTM2, and GCT343) and protein antigens (CD9 and Thy1). Furthermore, hESCs can also be identified based on the expression of alkaline phosphatase, stem cell factor (SCF/c-Kit ligand), and class 1 HLA proteins. Efforts are ongoing to profile the expression of microRNAs in pluripotent hESCs. Although a comprehensive study of miRNA expression is still lacking, several studies have identified a number of microRNA clusters prominently expressed in hESC lines, some of which have established roles in maintaining the pluripotent state of hESCs [19, 20]. These include microRNA (miR)-92b, miR-302 cluster, miR-200c, miR-368, and miR-154* clusters, miR-371, miR372, miR-373*, miR-373, and the miR-515 cluster [21, 22]. Pluripotent hESCs also display distinct epigenetic properties. Generally, the chromatin structure of hESCs is in an open conformation that allows transcription factors to enter and regulate gene expression [23]. In addition, DNA methylation profiles of hESCs are distinguishable from other cell types. Markedly reduced methylation patterns of CpG dinucleotides are specifically present in the promoter regions of pluripotency genes such as OCT4 and Nanog [24]. These unique epigenetic properties of hESCs are necessary to maintain their pluripotent state, and can therefore be used as a hallmark of undifferentiated hESCs.
C. Establishing Pluripotency of hESCs
hESCs are capable of differentiating into cells that constitute the three germ layers. This can be tested using established in vivo and in vitro techniques that are routinely used to determine pluripotency of a hESC line. The most commonly used in vivo method involves the induction of teratoma formation after transplantation of undifferentiated hESCs into immunodeficient mice [25-28]. Teratomas are benign tumors consisting of tissue structures derived from the three embryonic germ layers (Figure 3). Analysis of teratomas formed from engrafted hESCs can be used to determine their differentiation potential. The ability of hESCs to differentiate into various cell types can also be tested in vitro through the formation of embryoid bodies (EBs). EBs are spherical colonies of non-adherent, differentiating hESCs that contain cell populations representative of all three embryonic germ layers. Under suitable conditions, EBs can differentiate into specific cell types. As will be discussed below, EB formation is typically used as an intermediate step when directing hESCs into tissue-specific cell populations.
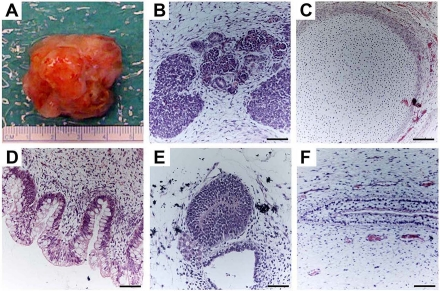
Figure 3. Teratoma formation provides an in vivo assay of hESCs differentiation capacity. Proliferating cultures of hESCs were used to form teratomas by renal capsule grafting using established methods [25-28]. (A) An explanted teratoma is shown. (B-F) Teratomas were sectioned and stained with hematoxylin and eosin to identify embryonic tissues. Representative tissues from all three embryonic germ layers can be seen, including mesoderm (B,C), endoderm (D) and ectoderm (E,F). (B) Nascent renal tubules and glomeruli within bed of primitive renal epithelium. (C) Cartilage surrounded by capsule of condensed mesenchyme. (D) Glandular intestinal structure. (E) Nascent neural tube. (F) Primitive squamous epithelium. Bar, 100 μm.
III. Differentiation of hESCs Into Specific Cell Types
The most promising feature of hESCs is the ability to derive lineage-restricted progenitors that are capable of differentiating into specialized post-mitotic cell types that can be used in cell-based therapies. Furthermore, hESCs provide a virtually inexhaustible source of specific cell populations, due to their ability to divide indefinitely. Current research studies are focused on identifying and refining ways for directing the differentiation of hESCs to enrich for pure, homogenous populations of specific cell types that can be used to either replace damaged cells or coax neighboring cells to function properly. In the following sections, we will provide some examples of how differentiation of hESCs is directed towards tissue-specific cells, particularly those with potential to treat aging-associated diseases.
A. hESC-Derived Endodermal Cells
Endodermal derivatives include cells that populate the lung, liver, pancreas, urinary bladder, pharynx, thyroid, parathyroid, and digestive system. The initial step in generating endodermal cells is the formation of definitive endoderm. D'Amour et al. [29]showed that selective induction of definitive endoderm can be achieved through the addition of high concentrations of Activin A, under low serum conditions, and in a stage-specific manner. Activin A mimics the action of Nodal, a ligand that activates transforming growth factor-β (TGFβ) signaling. The effect of Activin A in inducing definitive endoderm is enhanced when additional factors are present, such as Wnt3a [30], Noggin [31], or when coupled with the suppression of the phosphoinositide 3-kinase pathway [32].
The induction of definitive endoderm from hESCs can lead to the generation of specific progenitor populations such as pancreatic islet β cells, hepatocytes, or alveolar epithelial cells. These are being developed with the intent of treating diseases such as diabetes, liver disease, or lung disease, respectively. Among the most successful examples to date is the generation of pancreatic islet progenitors devised by Kroon et al. [33] through the sequential exposure of hESCs to Activin A and Wnt3A, followed by the addition of keratinocyte growth factor or FGF7 to induce the formation of the primitive gut tube. Subsequently, retinoic acid, cyclopamine, and Noggin are added to inhibit Sonic Hedgehog (Shh) and TGFβ signaling, and thus induce the differentiation of posterior foregut cells, the source of pancreatic cell progenitors. These are cultured further to generate pancreatic endoderm cells. When these cells were engrafted into immunodeficient mice, they displayed the histological and structural characteristics of pancreatic islet β cells and were able to sustain insulin production for at least 100 days. These results have been met with great enthusiasm for their potential in treating diabetes, type 2 diabetes being the most common type affecting more than a quarter of individuals aged 65 years or older in the United States [34].
In a similar manner, hepatocytes are obtained after differentiation of hESCs into definitive endoderm [35, 36]. A highly robust population of functional hepatocytes was generated with the sequential addition of low serum media, collagen I matrix, and hepatic differentiation factors that include FGF, BMP4, hepatocyte growth factor, oncostatin M, and dexamethasone [36]. These cells expressed known markers of mature hepatic cells, exhibited appropriate function, and were able to integrate and differentiate into mature liver cells when injected into mice with liver injury. The ability to differentiate hepatic cells could prove useful in the treatment of a number of liver diseases that are prevalent in aging individuals, such as cirrhosis, hepatocellular carcinoma, and diabetes-associated chronic liver disease [37].
The use of hESCs to treat lung injury has also been an area of active investigation. A significant step towards directed differentiation of lung-specific cells was reported by Wang et al. [38, 39], in which genetically modified hESCs carrying lung-specific reporters under the control of promoters from tissue-specific genes such as surfactant protein C, aquaporin 5 and T1α, resulted in purification of type I and type II alveolar epithelial cells. When engrafted into mice suffering from acute lung injury, these cells exhibited functional properties including the capacity for gas exchange and histological amelioration of lung injury.
B. hESC-Derived Mesodermal Cells
Directing the differentiation of hESCs into mesoderm requires the activation of the TGFβ signaling pathway and can be accomplished through the stepwise and dosage-dependent addition of Activin A, BMP4, and growth factors, such as vascular endothelial growth factor (VEGF) and basic FGF (bFGF) [40]. Mesodermal derivatives have also been successfully obtained by spontaneous differentiation of hESCs through hEB formation without first directing them toward mesoderm. Robust differentiation of hESCs into hematopoietic lineage cells, which give rise to all blood cell types and components of the immune system, has been achieved under serum-free conditions through spin hEB formation [41]. Specific hematopoietic lineage cell types, such as functional dendritic cells, have been successfully differentiated from hESCs through spontaneous hEB formation under serum-free conditions with the addition of BMP4 at specific time points [42]. Hematopoietic progenitor cells that give rise to functional T and natural killer cells capable of targeting human tumor cells both in vitro and in vivo, have also been derived from hESCs co-cultured with stromal cells [43]. Thus, the ability to differentiate hESCs into hematopoietic lineage cells promises to be useful in improving existing therapies that require blood cell transplantation, in fighting cancer, and in immune therapies that require induction of the immune response in an antigen-specific manner [44].
Cardiomyocytes, which represent another therapeutically important derivative of mesoderm, have been successfully generated from hESCs using several methods [45]. Through hEB formation, hESCs can spontaneously differentiate into cardiomyocytes under appropriate culture conditions. These cardiomyocytes exhibit morphological, molecular, and electrophysiological properties similar to adult cardiomyocytes [46], and display quantifiable responses to physiological stimuli reminiscent of atrial, ventricular, and pacemaker/conduction tissue [47-50]. Cardiomyocytes have also been generated by directed differentiation with Activin A and BMP4 on a dense monolayer of hESCs; these cells successfully form functional cardiomyocytes when transplanted in vivo [51]. Another study used additional medium supplements that included VEGF, and the Wnt inhibitor, Dickkopf homolog (DKK1), followed by the addition of bFGF, to promote cardiomyocyte differentiation from hEBs [52]. Success of these studies was measured by the expression of proteins specific for mature cardiac cells such as cardiac troponin T, atrial myosin light chain 2, and the cardiac transcription factors, Tbx5 and Tbx20. Several groups have generated cardiac-specific reporter hESC lines that can be used to test various differentiation strategies [49, 53] and generate specific cardiomyocyte subtypes [49].
hESCs can readily form connective tissue, such as bone or cartilage, as can be appreciated from teratoma formation assays (Figure 3). Thus, hESCs may be a valuable source of cells suitable for connective tissue replacement therapy for diseases such as osteoarthritis and osteoporosis, which are characterized by the breakdown of cartilage in joints and pathological fractures due to low bone density, respectively. Most successful and efficient protocols for directing chondrocyte differentiation from hESCs utilize 3D culture systems created by seeding hESCs at high density leading to formation of a pellet, or by introducing the cells into a synthetic 3D scaffold. Such systems enable cell-cell signaling between the undifferentiated hESCs and mature chondrocytes to stimulate homogeneous and sustained chondrogenic differentiation. For example, single-cell suspension of dissociated hEBs cultured as high-density micromass with BMP2 facilitates chondrocyte formation [54]. hESCs co-cultured with primary chondrocytes or in the presence of osteogenic supplements and polymeric scaffolds yield cartilaginous- or osteogenic-like cells [55, 56]. More recently, feeder-free 3D culture systems have successfully derived multipotent connective tissue progenitors from hESCs yielding tendon-like structures. The engraftment of these in vitro differentiated tendon structures in injured immunosuppressed mice restored ankle joint movements that rely on an intact Achilles tendon [57]. Furthermore, there is evidence to suggest that cell transplantation promotes growth and repair through endogenous cells as well [58].
C. Ectodermal Derivatives of hESCs
The dominant differentiation pathway in hESC cultures leads to the formation of ectoderm, which gives rise to cells of the nervous system and the epidermis. hESC-derived neural progenitor cells are characterized by rosette-like neural structures that form in the presence of FGF2 or EGF through either spontaneous differentiation from an overgrowth of hESCs, or after hEBs are plated onto adherent substrate [59, 60]. These ‘neural rosettes’ have become the signature of hESC-derived neural progenitors capable of differentiating into a broad range of neural cells in response to appropriate developmental signals. Thus, many studies are exploring ways to enhance the formation of neural rosettes to generate enriched populations of specific neural cell types. One example is the use of stromal cell lines [61], which provides ectodermal signaling factors required for neural induction, and promotes the formation of neural rosettes [62, 63].
The withdrawal of FGF2 and EGF, and addition of other factors can lead to the differentiation of neural rosettes into specific neural subtypes. Neural crest stem cells derived from neural rosettes can differentiate into peripheral sympathetic and sensory neurons through the addition of BDNF, GDNF, NGF and dbcAMP, or into Schwann cells in the presence of CNTF, neuregulin 1β and dbcAMP [64]. Neuroglial cells, such as oligodendrocytes, are generated with B27, thyroid hormone, retinoic acid, FGF2, epidermal growth factor, and insulin [65]. Additionally, FGF8 and Shh induce hESC-derived neural progenitors to differentiate into dopaminergic neurons [66], while treatment with Shh and retinoic acid induce motor neuron differentiation [67]. Recently, functional basal forebrain cholinergic neurons have been derived from hESCs through the formation of neurospheres and subsequent exposure to Shh, FGF8, and BMP9, or by overexpression of LHX8 and GBX9 [68].
The ability to differentiate hESCs into neuronal and non-neuronal subtypes has generated much interest due to their potential use in drug testing or cell replacement therapies for a number of neurodegenerative diseases. In particular, the successful derivation of dopaminergic neurons, particularly those of the midbrain subtype, could potentially be used to treat Parkinson's disease, which is marked by the progressive loss and dysfunction of these neurons. In Alzheimer's disease, where the degeneration of basal forebrain cholinergic neurons causes debilitating cognitive dysfunction, hESC-derived cholinergic neurons may also be useful for therapy. However, hESCs may also be helpful without requiring cell replacement. As observed in a clinical trial in which autologous fibroblasts programmed to express human NGF were implanted in the forebrain of individuals with mild Alzheimer's disease, a marked improvement in the rate of cognitive decline was observed [69]. One can imagine exploiting genetically-modified hESC-derived neuronal progenitors that readily engraft and express therapeutic gene products, such as NGF, to prevent the degeneration of cholinergic neurons in the basal forebrain.
Similarly, hESC-derived motor neurons might be used in the treatment of ALS, which is characterized by the progressive loss of motor neurons in the cortex, brain stem, and the spinal cord. Studies of ALS disease models have also suggested that non-neuronal cells, such as oligodendrocytes and Schwann cells, may be involved in the pathogenesis of this disease [70, 71]. Thus, the ability to differentiate hESCs into both neuronal and non-neuronal cells of the central nervous system provides an attractive therapeutic approach.
The efficacy of transplanting hESC-derived oligodendrocytes to treat acute spinal cord injury is now being tested in the first clinical trial with hESCs to be approved by the United States Food and Drug Administration (FDA) [72]. Oligodendrocytes are rapidly lost during acute spinal cord injury leading to demyelination and neuronal loss. In a trial sponsored by Geron Corporation, purified oligodendrocyte progenitor cells derived from hESCs will be injected into the spinal cord of paralyzed patients within two weeks after injury. While this first trial is a safety study, the expectation is that these progenitor cells will terminally differentiate into oligodendrocytes and produce myelin, which insulates neuronal cell membranes and is critical for efficient conduction of neuronal impulse transmission. If successful integration and function of oligodendrocytes is achieved in these studies, it could lead the way toward new treatment approaches for ALS, which manifests in demyelination of degenerating motor neurons.
Retinal pigment epithelium (RPE) cells are another specific cell type derived from neuroectoderm. RPE cells support the neural retina by phagocytosing and renewing the photoreceptor outer segments of rhodopsin. Recent reports have shown that RPE can be induced from hESCs in the presence of nicotinamide and Activin A under serum-free conditions [73]. hESC-derived pigmented cells exhibit the morphological and functional properties of RPE cells after transplantation in an animal model of macular degeneration, a disease caused by dysfunction and loss of RPE. These data have led to the second and third FDA-approved clinical trials using hESCs, these sponsored by Advanced Cell Technology. For these trials, hESC-derived RPEs will be transplanted directly into the degenerating retinae of patients with Stargardt's Macular Dystrophy, a juvenile form of macular degeneration, or Dry Age-Related Macular Degeneration, to rescue visual acuity. The launch of these three clinical trials heralds the translation of hESC research into therapy for degenerative disease, and the fields of stem cell biology and geriatric medicine await the results with great anticipation.
IV. Current Challenges and Potential Solutions for the Therapeutic Use of hESC-Derived Cells
Cellular therapies involving hESCs are in development and have begun to enter clinical trials. The International Stem Cell Banking Initiative (ISCBI) has been created by the International Stem Cell Forum, a group of national and international stem cell research funding bodies, to develop a set of best practices and principles for banking, testing, and distributing hESCs for therapy [74]. In the United States, the FDA also monitors these guidelines and have issued recommendations for reviewers of proposals for stem cell therapeutic trials [75]. It is important to note that these recommendations do not ensure the quality or efficacy of hESC-derived cells used for clinical applications. Rather, these guidelines warrant that the cells used for therapy are reproducible and meet specific criteria to ensure patient safety (Table 2). The major safety concerns for the use of hESCs are discussed in the following sections.
Table 2. Standardization and Quality Control of hESCs for Clinical Use
| Requirement | Methods of Testing |
|---|---|
| Cell line identity | Short Tandem Repeat (STR) testing Human Leukocyte Antigen (HLA) testing |
| Sterility and pathogens | Bacteria/fungi/mycoplasma culture qPCR analysis for murine viral short interspersed elements (SINE) |
| Genetic/chromosomal stability | Single Nucleotide Polymorphism (SNP) analysis G-band karyotype analysis spreads Fluorescent in situ hybridization |
| Epigenetic stability | MicroRNA profiling Methylation analysis X-inactivation |
| Pluripotency | Teratoma formation SSEA-3/4, TRA-1-60, TRA-1-81 detection |
| Quality and differentiation ability | Gene expression profiling qPCR analysis Embryoid body formation |
| Functional assays | Potency Efficacy Lot-to-lot variability |
A. Xenobiotic-Free Conditions
Many of the hESC lines currently in use have been exposed to animal products during isolation and propagation of hESCs in vitro. Under these conditions, hESCs could possess animal viruses and other unknown substances capable of eliciting a detrimental immune response in transplanted hosts. Currently, hESC lines under development for clinical use undergo extensive microbiological testing as strictly recommended by ISCBI. In the United States, the FDA legally requires documentation of the source, the potential genetically modified components, and pathogenic agents in any hESC-derived cell intended for therapeutic use. Thus, avoiding exposure to xenobiotics is an ongoing effort. Recently, replacement media have been developed that would allow maintenance of hESCs in xenobiotic-free conditions. These include xenobiotic-free serum replacements such as Knockout Serum Replacer (Invitrogen) or xenobiotic-free culture media such as HESGRO (Millipore) or TeSR (STEMCELL).
Feeder-free culture systems are now being developed to reduce the risk of contamination with foreign agents when hESCs are cultured on feeder cell layers. Feeder-free and xenobiotic-free defined culture media that consist of a combination of recombinant growth factors known to inhibit differentiation and maintain hESCs in the pluripotent state are now commercially available. However, some reports have associated feeder-free culture conditions with greater chromosomal instability and an increased risk of propagating genetically altered hESCs [76]. For this reason, most hESC labs practice a surveillance program for genomic instability in cultured lines [28, 49].
hESC lines derived using human feeder cells have also been reported. For example, hESC lines have been successfully derived on human fibroblasts generated from neonatal foreskin [77, 78] and adult skin fibroblasts [79]. Some laboratories deriving new lines have moved exclusively to xenobiotic-free conditions [80]. The ability to derive and maintain new hESC lines using human fibroblast feeder cells represents a significant step towards generating clinical-grade hESCs.
B. Genetic Abnormalities In hESC Lines
The best characterized hESC lines to date are among the earliest lines derived. However, they may not be the best lines for therapeutic application as many of these lines were derived using animal products. Chromosomal and genomic instability has been detected in several hESC lines, with acquisition of loss of heterozygosity or copy-number variation in cancer-related genes [81, 82]. Many of these mutations appeared to be induced by prolonged culture, since these changes were not observed in low passage cells. It has been proposed that such karyotypic aberrations occurred with adaptation to the original culture conditions used when the first few lines were being derived and expanded [83]. These observations emphasize the need for complete characterization of hESC lines, particularly the effects of long-term culture, and the design of guidelines for designating therapeutic-grade hESCs.
C. Enrichment, Directed Differentiation, and Purification of hESC-Derived Cells
A primary safety concern when using pluripotent hESCs is their potential to form germ layer tumors. As discussed above, in vivo transplantation of undifferentiated hESCs in mouse models results in teratoma formation. Evidence of tumor formation has also been observed in differentiated hESC derivatives transplanted in vivo [84, 85]. Thus, it is essential that candidate hESC derivatives intended for use in cell transplantation are free of tumorigenic cells. Another concern is the differentiation of hESC-derived cells into unwanted cell types. For example, the engraftment of inappropriate muscle cells into damaged myocardium could alter the electrical activities of recipient tissue, provoking arrhythmias [86]. Thus, developing and further optimizing differentiation and purification protocols are necessary to minimize the generation of unwanted cell types for pre-clinical transplantation experiments and clinical therapy.
As discussed earlier, enrichment of specific cell types can be achieved using molecules introduced at critical time points during culture. However, many of these methods yield only moderate enrichment that is not yet scalable for clinical application. It may be desirable to enrich first for partially differentiated, proliferative hESC intermediates with specific cell fates. These could then be expanded before further differentiation into cells for therapy. For example, the expression of the cell surface antigen, CD133, on proliferating hESCs identifies cells predestined toward a neuroectodermal fate [26]. CD133-positive cells have been selected from cultures of undifferentiated hESCs, and have been observed to differentiate primarily into neuroectodermal cells in vitro and in vivo [26].
In the absence of specific cell surface antigens like CD133 to identify tissue-specific precursors, molecular beacons have been used to select for specific subpopulations of hESCs. King et al. [25] first demonstrated the utility of this system for isolating viable Oct4-expressing pluripotent hESCs in a specific and high-throughput manner. Molecular beacons are single-stranded oligonucleotides that generate fluorescent signals when bound to their target mRNAs, making these cells detectable and selectable by fluoresence-activated cell sorting. More importantly, molecular beacons have a short lifespan within cells and do not alter the function or genomic structure of hESCs. Thus, this method can be used to enrich for desired hESC-derived cell populations or used to select against unwanted cell types, such as undifferentiated hESCs that could form tumors [25].
D. Circumventing Immune Rejection of Transplanted hESC-Derived Cells
Transplanted hESCs encounter immune rejection [87] because both proliferating and differentiated hESCs express class I and II HLA as well as minor histocompatibility antigens at levels sufficient to activate the immune system [87, 88]. Another potential barrier to hESC engraftment can occur through mismatch between hESC donor and recipient ABO blood group antigens [89-91]. While studies to determine the effects of ABO incompatibility on hESC transplantation are still lacking, this has long been a criterion for successful organ transplantation and thus, it is likely that ABO incompatibility between hESC-donor cells and the recipient would also trigger immune rejection.
Ideally, having genetically identical donor and patient cells is the best way to circumvent immune rejection. Thus, there is expressed interest in developing and using somatic cell nuclear transfer to generate patient-specific hESC lines. Using this technique, the DNA obtained from either a patient's skin or muscle cell would be transferred into an unfertilized egg that has had its DNA removed. Subsequently, the egg is artificially fertilized and allowed to develop until it reaches the blastocyst stage to derive hESCs. The resultant hESC line would have an immunologic profile matching the patient and could be used for cell therapy. This technique has been conducted successfully in animals using species-specific ESCs, but derivation of hESCs through somatic cell nuclear transfer has not yet been achieved.
Other strategies to generate hESC lines with the closest match to potential transplant patients include engineering “universal donor hESCs,” a blood antigen O cell in which the expression of HLA is suppressed, or chimeric hematopoietic cells derived from hESCs capable of inhibiting the immune response when co-transplanted with the desired hESC-derived cells [92]. Alternatively, creating banks of hESC lines representing HLA/ABO combinations that match the majority of potential patients has been proposed. Studies have provided estimates on how many hESC lines would be needed in order to support the needs of a specific population. Taylor et al. [93] estimated that approximately 150 hESC lines could provide an HLA match for most of the population in the United Kingdom. Alternatively, approximately 10 parthenote-derived hESC lines that are homozygous for HLA types could be sufficient for a majority of the population. Studies by Nakajima et al. [94] estimated that approximately 170 hESC lines, or 55 hESC lines with homozygous HLA types, would be sufficient for 80% of patients in the Japanese population. These findings demonstrate the feasibility of creating and maintaining a hESC bank with sufficient representation to support a large number of patients. However, in countries such as the United States, many more hESC lines would need to be established to serve its ethnically and genetically diverse population. Given the ethical issues and restrictions on hESC research, and the small number of approved hESC lines currently available, the creation of a hESC bank with a highly diverse collection of cell lines will undoubtedly face enormous challenges.
V. Therapeutic Advantages of hESCs Over Other Stem Cell Sources
While not the focus of this review, other sources of human-derived stem cells are also being explored for use in clinical settings. Among these are adult stem cells and induced pluripotent stem cells (iPSCs). Unlike hESCs, these stem cells can be obtained directly from the individual to be treated. Thus, as a source of cells for therapy, they are able to circumvent the immunocompatibility issues that hamper many non-autologous transplantation therapies. Furthermore, the utilization of these stem cells in both clinical and basic research studies does not face ethical and political issues that otherwise surround the use of embryonic stem cells. However, adult stem cells and iPSCs have significant limitations as well that are potentially overcome by hESCs at this time.
Adult stem cells are derived from non-embryonic tissues and typically reside in their tissue of origin. Similar to hESCs, adult stem cells are capable of self-renewal. However, unlike hESCs, they have restricted potential and are able to differentiate only into cells from the tissue of origin. In some cases, adult stem cells are not able to generate all cell types of the tissue of origin, nor can they sustain growth over time. The latter problem has been encountered specifically in stem cells obtained from aging individuals [95, 96].
iPSCs are generated by reprogramming differentiated somatic cells into a pluripotent state. This can be achieved by inducing the expression of three core reprogramming factors Oct4, Sox2, and Nanog. Several methods have been employed to express these factors and induce pluripotency. Among these are retroviral, lentiviral, and adenoviral transduction, which carry the risk of permanent and harmful genomic integration. Indeed, some established iPSC lines are genetically unstable, exhibiting large-scale genomic rearrangements, copy number variations, and abnormal karyotype even in early passage stages [97, 98]. To minimize the possibility of mutagenesis caused by methods used to introduce the reprogramming factors, integration/plasmid-free strategies have been employed to express these factors and induce pluripotency such as synthetic RNA delivery, RNA virus transduction, or the addition of cell-penetrating purified recombinant proteins [99, 100]. However, these methods are significantly less efficient at generating reprogrammed pluripotent cells in comparison to viral integration. Furthermore, in some iPSC lines reprogrammed using non-integrating viral method, high levels of mutational changes were still observed [97].
In addition to genomic changes, recent studies have also revealed that iPSCs contain epigenetic features that indicate either incomplete or aberrant reprogramming. In particular, iPSC DNA methylation patterns are frequently reminiscent of the somatic cell of origin [101], suggesting that iPSCs are not completely reprogrammed into the naïve pluripotent state seen in hESCs. It is unclear whether the observed genetic instability and epigenetic imprinting accrued during reprograming or was present in the somatic cell of origin. Nevertheless, patient-specific iPSCs may be less suitable for the treatment of aging-associated diseases, since somatic cells from older individuals are more likely to contain genomic mutations and disadvantageous epigenetic programs. Thus, the safety and efficacy of therapeutic iPSCs as currently derived remain to be tested.
VI. Conclusions
As cell replacement therapies are envisioned and realized, their use in the treatment of aging-associated diseases becomes a compelling prospect. hESCs provide much promise as a potential tool in designing such therapies, as well as in drug discovery. It is clear that there are still major scientific challenges as well as ethical and legislative issues that must be addressed. However, it is encouraging to see that clinical trials involving the use of hESCs have begun, and that extensive efforts are underway to efficiently, successfully, and safely differentiate hESCs into specific cell types. These studies will pave the way toward leveraging the therapeutic benefit of hESCs for regenerative medicine, particularly in aging-associated diseases.
Acknowledgments
Our work described in this review has been supported by grants from the National Institutes of Health (HL085377), the California Institute for Regenerative Medicine (RC1-00104), and the Muscular Dystrophy Foundation (186483) to H.S.B., and a fellowship from the National Institutes of Health (HL007544) to O.Y.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. United States Census Bureau. State and County QuickFacts. 2010; http://quickfacts.census.gov/qfd/states/00000.html [Date accessed 12 April 2011]. .
- 2. Ferrari AU, Radaelli A, Centola M. Invited review: Aging and the cardiovascular system. J Appl Physiol. 2003; 95: 2591 -7. [PubMed] .
- 3. Yoshida T and Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007; 87: 1047 -82. [PubMed] .
- 4. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009; 32: S62 -S67. [PubMed] .
- 5. Chung Y, Klimanskaya I, Becker S, Li T, Maserati M, Lu SJ, Zdravkovic T, Ilic D, Genbacev O, Fisher S, Krtolica A, Lanza R. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell. 2008; 2: 113 -7. [PubMed] .
- 6. Geens M, Mateizel I, Sermon K, De Rycke M, Spits C, Cauffman G, Devroey P, Tournaye H, Liebaers I, Van de Velde H. Human embryonic stem cell lines derived from single blastomeres of two 4-cell stage embryos. Hum Reprod. 2009; 24: 2709 -17. [PubMed] .
- 7. Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. Human embryonic stem cell lines derived from single blastomeres. Nature. 2006; 444: 481 -5. [PubMed] .
- 8. Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. Derivation of human embryonic stem cells from single blastomeres. Nat Protoc. 2007; 2: 1963 -72. [PubMed] .
- 9. Strelchenko N, Verlinsky O, Kukharenko V, Verlinsky Y. Morula-derived human embryonic stem cells. Reprod Biomed Online. 2004; 9: 623 -9. [PubMed] .
- 10. Strelchenko N and Verlinsky Y. Embryonic stem cells from morula. Methods Enzymol. 2006; 418: 93 -108. [PubMed] .
- 11. Kim K, Ng K, Rugg-Gunn PJ, Shieh JH, Kirak O, Jaenisch R, Wakayama T, Moore MA, Pedersen RA, Daley GQ. Recombination signatures distinguish embryonic stem cells derived by parthenogenesis and somatic cell nuclear transfer. Cell Stem Cell. 2007; 1: 346 -52. [PubMed] .
- 12. Lin G, OuYang Q, Zhou X, Gu Y, Yuan D, Li W, Liu G, Liu T, Lu G. A highly homozygous and parthenogenetic human embryonic stem cell line derived from a one-pronuclear oocyte following in vitro fertilization procedure. Cell Res. 2007; 17: 999 -1007. [PubMed] .
- 13. Mai Q, Yu Y, Li T, Wang L, Chen MJ, Huang SZ, Zhou C, Zhou Q. Derivation of human embryonic stem cell lines from parthenogenetic blastocysts. Cell Res. 2007; 17: 1008 -19. [PubMed] .
- 14. Revazova ES, Turovets NA, Kochetkova OD, Kindarova LB, Kuzmichev LN, Janus JD, Pryzhkova MV. Patient-specific stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells. 2007; 9: 432 -49. [PubMed] .
- 15. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282: 1145 -7. [PubMed] .
- 16. United States National Institutes of Health. NIH Human Embryonic Stem Cell Registry. 2011; http://grants.nih.gov/stem_cells/registry/current.htm [Date accessed 12 April 2011]. .
- 17. Bodnar MS, Meneses JJ, Rodriguez RT, Firpo MT. Propagation and maintenance of undifferentiated human embryonic stem cells. Stem Cells Dev. 2004; 13: 243 -53. [PubMed] .
- 18. Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, Bello PA, Benvenisty N, Berry LS, Bevan S, Blum B, Brooking J, Chen KG, Choo AB, Churchill GA, Corbel M, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007; 25: 803 -16. [PubMed] .
- 19. Card DA, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, Archer TK. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008; 28: 6426 -38. [PubMed] .
- 20. Sengupta S, Nie J, Wagner RJ, Yang C, Stewart R, Thomson JA. MicroRNA 92b controls the G1/S checkpoint gene p57 in human embryonic stem cells. Stem Cells. 2009; 27: 1524 -8. [PubMed] .
- 21. Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, Ruzzo WL, Ware C, Radich JP, Gentleman R, Ruohola-Baker H, Tewari M. Microrna discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008; 26: 2496 -505. [PubMed] .
- 22. Nishimura M, Naito S, Yokoi T. Tissue-specific mRNA expression profiles of human nuclear receptor subfamilies. Drug Metab Pharmacokinet. 2004; 19: 135 -49. [PubMed] .
- 23. Gan Q, Yoshida T, McDonald OG, Owens GK. Concise review: Epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells. 2007; 25: 2 -9. [PubMed] .
- 24. Lagarkova MA, Volchkov PY, Lyakisheva AV, Philonenko ES, Kiselev SL. Diverse epigenetic profile of novel human embryonic stem cell lines. Cell Cycle. 2006; 5: 416 -20. [PubMed] .
- 25. King FW, Liszewski W, Ritner C, Bernstein HS. High-throughput tracking of pluripotent human embryonic stem cells with dual fluorescence resonance energy transfer molecular beacons. Stem Cells Dev. 2011; 20: 475 -84. [PubMed] .
- 26. King FW, Ritner C, Liszewski W, Kwan HC, Pedersen A, Leavitt AD, Bernstein HS. Subpopulations of human embryonic stem cells with distinct tissue-specific fates can be selected from pluripotent cultures. Stem Cells Dev. 2009; 18: 1441 -50. [PubMed] .
- 27. Ritner C and Bernstein HS. Fate mapping of human embryonic stem cells by teratoma formation. J Vis Exp. 2010; 42: 2036 [PubMed] .
- 28. Gaur M, Ritner C, Sievers R, Pedersen A, Prasad M, Bernstein HS, Yeghiazarians Y. Timed inhibition of p38MAPK directs accelerated differentiation of human embryonic stem cells into cardiomyocytes. Cytotherapy. 2010; 12: 807 -17. [PubMed] .
- 29. D'Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005; 23: 1534 -41. [PubMed] .
- 30. D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006; 24: 1392 -401. [PubMed] .
- 31. Sumi T, Tsuneyoshi N, Nakatsuji N, Suemori H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development. 2008; 135: 2969 -79. [PubMed] .
- 32. McLean AB, D'Amour KA, Jones KL, Krishnamoorthy M, Kulik MJ, Reynolds DM, Sheppard AM, Liu H, Xu Y, Baetge EE, Dalton S. Activin A efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells. 2007; 25: 29 -38. [PubMed] .
- 33. Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, Agulnick AD, D'Amour KA, Carpenter MK, Baetge EE. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008; 26: 443 -52. [PubMed] .
- 34. United Sates Centers for Disease Control and Prevention. National Diabetes Fact Sheet: National estimates and general information on diabetes and prediabetes in the United States. 2011; http://diabetes.niddk.nih.gov/dm/pubs/statistics/index.htm#dd [Date accessed 12 April 2011]. .
- 35. Cai J, Zhao Y, Liu Y, Ye F, Song Z, Qin H, Meng S, Chen Y, Zhou R, Song X, Guo Y, Ding M, Deng H. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007; 45: 1229 -39. [PubMed] .
- 36. Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008; 26: 1117 -27. [PubMed] .
- 37. Hoare M, Das T, Alexander G. Ageing, telomeres, senescence, and liver injury. J Hepatol. 2010; 53: 950 -61. [PubMed] .
- 38. Wang D, Haviland DL, Burns AR, Zsigmond E, Wetsel RA. A pure population of lung alveolar epithelial type II cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2007; 104: 4449 -54. [PubMed] .
- 39. Wang D, Morales JE, Calame DG, Alcorn JL, Wetsel RA. Transplantation of human embryonic stem cell-derived alveolar epithelial type II cells abrogates acute lung injury in mice. Mol Ther. 2010; 18: 625 -34. [PubMed] .
- 40. Evseenko D, Zhu Y, Schenke-Layland K, Kuo J, Latour B, Ge S, Scholes J, Dravid G, Li X, MacLellan WR, Crooks GM. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc Natl Acad Sci U S A. 2010; 107: 13742 -7. [PubMed] .
- 41. Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005; 106: 1601 -3. [PubMed] .
- 42. Kunisaki C, Makino H, Takagawa R, Yamamoto N, Nagano Y, Fujii S, Kosaka T, Ono HA, Otsuka Y, Akiyama H, Ichikawa Y, Shimada H. Surgical outcomes in esophageal cancer patients with tumor recurrence after curative esophagectomy. J Gastrointest Surg. 2008; 12: 802 -10. [PubMed] .
- 43. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, Kaufman DS. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009; 113: 6094 -101. [PubMed] .
- 44. Senju S, Hirata S, Motomura Y, Fukuma D, Matsunaga Y, Fukushima S, Matsuyoshi H, Nishimura Y. Pluripotent stem cells as source of dendritic cells for immune therapy. Int J Hematol. 2010; 91: 392 -400. [PubMed] .
- 45. Wong SS and Bernstein HS. Cardiac regeneration using human embryonic stem cells: Producing cells for future therapy. Regen Med. 2010; 5: 763 -75. [PubMed] .
- 46. Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001; 108: 407 -14. [PubMed] .
- 47. He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: Action potential characterization. Circ Res. 2003; 93: 32 -9. [PubMed] .
- 48. Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, Passier R, Tertoolen L. Differentiation of human embryonic stem cells to cardiomyocytes: Role of coculture with visceral endoderm-like cells. Circulation. 2003; 107: 2733 -40. [PubMed] .
- 49. Ritner C, Wong SS, King FW, Mihardja SS, Liszewski W, Erle DJ, Lee RJ, Bernstein HS. An engineered cardiac reporter cell line identifies human embryonic stem cell-derived myocardial precursors. PLoS One. 2011; 6: e16004 [PubMed] .
- 50. Satin J, Kehat I, Caspi O, Huber I, Arbel G, Itzhaki I, Magyar J, Schroder EA, Perlman I, Gepstein L. Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol. 2004; 559: 479 -96. [PubMed] .
- 51. Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, O'Sullivan C, Collins L, Chen Y, Minami E, Gill EA, Ueno S, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007; 25: 1015 -24. [PubMed] .
- 52. Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008; 453: 524 -8. [PubMed] .
- 53. Kita-Matsuo H, Barcova M, Prigozhina N, Salomonis N, Wei K, Jacot JG, Nelson B, Spiering S, Haverslag R, Kim C, Talantova M, Bajpai R, Calzolari D, Terskikh A, McCulloch AD, Price JH, et al. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS One. 2009; 4: e5046 [PubMed] .
- 54. Toh WS, Yang Z, Liu H, Heng BC, Lee EH, Cao T. Effects of culture conditions and bone morphogenetic protein 2 on extent of chondrogenesis from human embryonic stem cells. Stem Cells. 2007; 25: 950 -60. [PubMed] .
- 55. Bielby RC, Boccaccini AR, Polak JM, Buttery LD. In vitro differentiation and in vivo mineralization of osteogenic cells derived from human embryonic stem cells. Tissue Eng. 2004; 10: 1518 -25. [PubMed] .
- 56. Vats A, Bielby RC, Tolley N, Dickinson SC, Boccaccini AR, Hollander AP, Bishop AE, Polak JM. Chondrogenic differentiation of human embryonic stem cells: The effect of the micro-environment. Tissue Eng. 2006; 12: 1687 -97. [PubMed] .
- 57. Cohen S, Leshansky L, Zussman E, Burman M, Srouji S, Livne E, Abramov N, Itskovitz-Eldor J. Repair of full-thickness tendon injury using connective tissue progenitors efficiently derived from human embryonic stem cells and fetal tissues. Tissue Eng Part A. 2010; 16: 3119 -37. [PubMed] .
- 58. Toh WS, Lee EH, Guo XM, Chan JK, Yeow CH, Choo AB, Cao T. Cartilage repair using hyaluronan hydrogel-encapsulated human embryonic stem cell-derived chondrogenic cells. Biomaterials. 2010; 31: 6968 -80. [PubMed] .
- 59. Reubinoff BE, Itsykson P, Turetsky T, Pera MF, Reinhartz E, Itzik A, Ben-Hur T. Neural progenitors from human embryonic stem cells. Nat Biotechnol. 2001; 19: 1134 -40. [PubMed] .
- 60. Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001; 19: 1129 -33. [PubMed] .
- 61. Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, Studer L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A. 2004; 101: 12543 -8. [PubMed] .
- 62. Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa SI, Sasai Y. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000; 28: 31 -40. [PubMed] .
- 63. Kawasaki H, Suemori H, Mizuseki K, Watanabe K, Urano F, Ichinose H, Haruta M, Takahashi M, Yoshikawa K, Nishikawa S, Nakatsuji N, Sasai Y. Generation of dopaminergic neurons and pigmented epithelia from primate ES cells by stromal cell-derived inducing activity. Proc Natl Acad Sci U S A. 2002; 99: 1580 -5. [PubMed] .
- 64. Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, Tabar V, Studer L. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. 2007; 25: 1468 -75. [PubMed] .
- 65. Nistor GI, Totoiu MO, Haque N, Carpenter MK, Keirstead HS. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia. 2005; 49: 385 -96. [PubMed] .
- 66. Yan Y, Yang D, Zarnowska ED, Du Z, Werbel B, Valliere C, Pearce RA, Thomson JA, Zhang SC. Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells. 2005; 23: 781 -90. [PubMed] .
- 67. Li XJ, Du ZW, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA, Zhang SC. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005; 23: 215 -21. [PubMed] .
- 68. Bissonnette CJ, Lyass L, Bhattacharyya BJ, Belmadani A, Miller RJ, Kessler JA. The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells. 2011; Epub before print 8 Mar 2011. .
- 69. Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005; 11: 551 -5. [PubMed] .
- 70. Forsberg K, Andersen PM, Marklund SL, Brannstrom T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011; Epub before print 3 Feb 2011. .
- 71. Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, Cleveland DW. Schwann cells expressing dismutase active mutant Sod1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci U S A. 2009; 106: 4465 -70. [PubMed] .
- 72. United States National Institutes of Health. A phase 1 safety study of GRNOPC1 in patients with neurologically complete, subacute, spinal cord injury. 2011; http://clinicaltrials.gov/archive/NCT01217008/2011_03_09 [Date accessed 12 April 2011]. .
- 73. Idelson M, Alper R, Obolensky A, Ben-Shushan E, Hemo I, Yachimovich-Cohen N, Khaner H, Smith Y, Wiser O, Gropp M, Cohen MA, Even-Ram S, Berman-Zaken Y, Matzrafi L, Rechavi G, Banin E, et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell. 2009; 5: 396 -408. [PubMed] .
- 74. Crook JM, Hei D, Stacey G. The International Stem Cell Banking Initiative (ISCBI): Raising standards to bank on. In Vitro Cell Dev Biol Anim. 2010; 46: 169 -72. [PubMed] .
- 75. United States Food and Drug Administration. Guidance for FDA reviewers and sponsors: Content and review of chemistry, manufacturing, and control (CMC) information for human somatic cell therapy investigational new drug applications (INDs). 2009; http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Xenotransplantation/ucm074131.htm [Date accessed 12 April 2011]. .
- 76. Catalina P, Montes R, Ligero G, Sanchez L, de la Cueva T, Bueno C, Leone PE, Menendez P. Human ESCs predisposition to karyotypic instability: Is a matter of culture adaptation or differential vulnerability among hesc lines due to inherent properties? Mol Cancer. 2008; 7: 76 [PubMed] .
- 77. Strom S, Holm F, Bergstrom R, Stromberg AM, Hovatta O. Derivation of 30 human embryonic stem cell lines--improving the quality. In Vitro Cell Dev Biol Anim. 2010; 46: 337 -44. [PubMed] .
- 78. Ilic D, Giritharan G, Zdravkovic T, Caceres E, Genbacev O, Fisher SJ, Krtolica A. Derivation of human embryonic stem cell lines from biopsied blastomeres on human feeders with minimal exposure to xenomaterials. Stem Cells Dev. 2009; 18: 1343 -50. [PubMed] .
- 79. Tecirlioglu RT, Nguyen L, Koh K, Trounson AO, Michalska AE. Derivation and maintenance of human embryonic stem cell line on human adult skin fibroblast feeder cells in serum replacement medium. In Vitro Cell Dev Biol Anim. 2010; 46: 231 -5. [PubMed] .
- 80. Genbacev O, Krtolica A, Zdravkovic T, Brunette E, Powell S, Nath A, Caceres E, McMaster M, McDonagh S, Li Y, Mandalam R, Lebkowski J, Fisher SJ. Serum-free derivation of human embryonic stem cell lines on human placental fibroblast feeders. Fertil Steril. 2005; 83: 1517 -29. [PubMed] .
- 81. Lefort N, Feyeux M, Bas C, Feraud O, Bennaceur-Griscelli A, Tachdjian G, Peschanski M, Perrier AL. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat Biotechnol. 2008; 26: 1364 -6. [PubMed] .
- 82. Narva E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, Borghese L, Itskovitz-Eldor J, Rasool O, Dvorak P, Hovatta O, Otonkoski T, Tuuri T, Cui W, Brustle O, Baker D, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010; 28: 371 -7. [PubMed] .
- 83. Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007; 25: 207 -15. [PubMed] .
- 84. Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman SA. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med. 2006; 12: 1259 -68. [PubMed] .
- 85. Wernig M, Benninger F, Schmandt T, Rade M, Tucker KL, Bussow H, Beck H, Brustle O. Functional integration of embryonic stem cell-derived neurons in vivo. J Neurosci. 2004; 24: 5258 -68. [PubMed] .
- 86. Gepstein L, Ding C, Rehemedula D, Wilson EE, Yankelson L, Caspi O, Gepstein A, Huber I, Olgin JE. In vivo assessment of the electrophysiological integration and arrhythmogenic risk of myocardial cell transplantation strategies. Stem Cells. 2010; 28: 2151 -61. [PubMed] .
- 87. Bradley JA, Bolton EM, Pedersen RA. Stem cell medicine encounters the immune system. Nat Rev Immunol. 2002; 2: 859 -71. [PubMed] .
- 88. Drukker M, Katz G, Urbach A, Schuldiner M, Markel G, Itskovitz-Eldor J, Reubinoff B, Mandelboim O, Benvenisty N. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci U S A. 2002; 99: 9864 -9. [PubMed] .
- 89. Chen YT, Dejosez M, Zwaka TP, Behringer RR. H1 and H9 human embryonic stem cell lines are heterozygous for the ABO locus. Stem Cells Dev. 2008; 17: 853 -5. [PubMed] .
- 90. Lee JE, Kang MS, Park MH, Shim SH, Yoon TK, Chung HM, Lee DR. Evaluation of 28 human embryonic stem cell lines for use as unrelated donors in stem cell therapy: Implications of HLA and ABO genotypes. Cell Transplant. 2010; 19: 1383 -95. [PubMed] .
- 91. Molne J, Bjorquist P, Andersson K, Diswall M, Jeppsson A, Strokan V, Rydberg L, Breimer ME. Blood group ABO antigen expression in human embryonic stem cells and in differentiated hepatocyte- and cardiomyocyte-like cells. Transplantation. 2008; 86: 1407 -13. [PubMed] .
- 92. Drukker M. Immunogenicity of human embryonic stem cells: Can we achieve tolerance? Springer Semin Immunopathol. 2004; 26: 201 -13. [PubMed] .
- 93. Taylor CJ, Bolton EM, Pocock S, Sharples LD, Pedersen RA, Bradley JA. Banking on human embryonic stem cells: Estimating the number of donor cell lines needed for HLA matching. Lancet. 2005; 366: 2019 -25. [PubMed] .
- 94. Nakajima F, Tokunaga K, Nakatsuji N. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells. 2007; 25: 983 -5. [PubMed] .
- 95. Phinney DG and Prockop DJ. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair--current views. Stem Cells. 2007; 25: 2896 -902. [PubMed] .
- 96. Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003; 33: 919 -26. [PubMed] .
- 97. Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, Lee JH, Loh YH, Manos PD, Montserrat N, Panopoulos AD, Ruiz S, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011; 471: 63 -7. [PubMed] .
- 98. Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010; 7: 521 -31. [PubMed] .
- 99. Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Scholer HR, Duan L, Ding S. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009; 4: 381 -4. [PubMed] .
- 100. Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009; 4: 472 -6. [PubMed] .
- 101. Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, Downes M, Yu R, Stewart R, Ren B, Thomson JA, Evans RM, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011; 471: 68 -73. [PubMed] .
- 102. Su Z, Frye C, Bae KM, Kelley V, Vieweg J. Differentiation of human embryonic stem cells into immunostimulatory dendritic cells under feeder-free culture conditions. Clin Cancer Res. 2008; 14: 6207 -17. [PubMed] .