



Introduction
The Hayflick limit of primary mouse embryo fibroblasts (MEF) is variable since the growth conditions can either reduce or extend it [1-3]. We have reported that primary MEF expanded every three days (6T3 protocol) underwent four population cell doubling and thereafter cell proliferation was fully abolished and SA-β-gal+ cells induced [4]. However it is known that MEF senescence can be induced prematurely, before the end of the physiological lifespan, by cellular stresses such as the over expression [5] or down regulation [6] of single oncogenes and DNA damaging drugs [7-9]. Recently, it has been reported that miRNAs are involved in senescence of MEF as well as of human diploid fibroblasts [10,11].
An unsolved question is which cells are able to escape the in vitro lifespan limit. So far, the molecular characterization of immortalized MEF cell lines has shown that functional silencing of either the INK4a/ARF locus [12] or p53 [13] appears necessary to bypass senescence. A recurrent concept is that mutational events occurring in culture have a key role. If mutations confer a proliferative advantage, mutated cells can bypass the in vitro lifespan limit and rapidly replace the existing population [14,15]. According to this view, the time spent in culture should affect immortalization. We modified the 6T3 expansion protocol of primary MEF [4] by lengthening the interval between passages (15 days versus 3 days) and reiterating it until MEF became immortal. Here we report that: i) the life span was extended up to 8 population cell doubling and immortalization was consequentially delayed; ii) p21 down regulation marks the switch from primary to immortalized MEF; iii) deregulation of genes and miRNAs which control cell proliferation pathways correlate with the immortalization process; iv) down regulated miRNAs can behave as tumor suppressors.
Results
1. The pro senescence axis p53/p21 is disrupted in immortalized MEF
To investigate the relative importance of the time in culture versus the number of population cell doubling in the MEF immortalization process we modified the split time. MEF p0 (passage 0) were thawed, and expanded to obtain MEF p1. Thereafter MEF p1 were collected, diluted at the appropriate concentration (6x105) and grown for 15 days with a medium change every three days (6T15 protocol), before the next trypsinization and expansion. The 6T15 protocol was reiterated 10 times for a total of 150 days in culture. During the first three passages a reduced number of population cell doubling was observed; afterwards a population of proliferating MEF emerged, which rapidly increased (p5) and replaced the preexisting cell population (p10) (Fig. 1A). At the molecular level we found that both p19ARF and p16 were progressively up regulated during passages (with respect to the spontaneous level of MEF p0) (Fig. 1B). Vice versa the expression of p21 was biphasic: while it was up regulated till p3 it decreased thereafter with a clear switch toward under expression at p6 (Fig. 1B,C). As p21 is under the direct control of p53, a disruption of the p53/p21 axis can be hypothesized at the basis of immortalization.
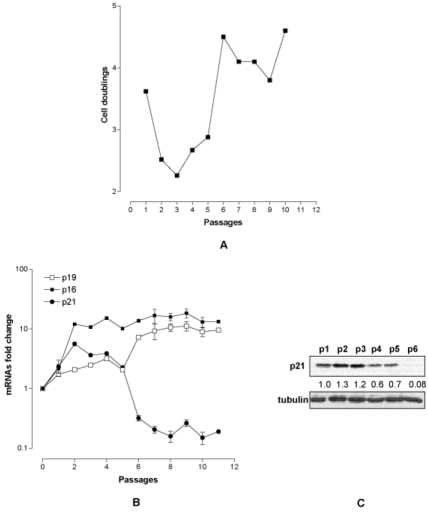
Figure 1. Characterization of MEF under 6T15 propagation regimen. (A) Proliferation curve of MEF expressed as cell doublings per passage. (B) Quantification of p19ARF, p16 and p21 transcripts per passage (each point represent one passage) normalized to that of MEF at passage 0. (C) p21 protein level at the various passages. The value reported under each lane represents the average of two independent experiments.
2. Genome stability and miRNA signatures are modified in immortalized MEF
The down regulation of p21 strongly suggests a loss of p53 activity. In accordance with the hypothesis, FACS analysis showed that while the DNA content distribution curve per cell of MEF p1 and MEF p5 were very similar and typical of diploid cells (Fig. 2A) a massive shift toward cells with higher DNA contents was observed in MEF p6 (Fig. 2B). Hyperdiploid cells, which emerged from diploid primary MEF, are indicative that genomic stability, controlled by p53 [16-18], is lost. It is of note that a stable hyperdiploid cell population is selected within four passages (p6-p10) (Fig. 2C). To strengthen the hypothesis of a functional loss of p53, in parallel we determined the expression profiles of p53 and of miR-34a directly controlled by p53. We found that p53 mRNA (Fig. 2D) and protein levels did not significantly change up to p6 (Fig. 2E); conversely miR-34a appears to increase till p4, starts to decrease at p5 to become markedly down regulated at p6 (Fig. 2D) reinforcing the idea that p53 becomes non-functional.
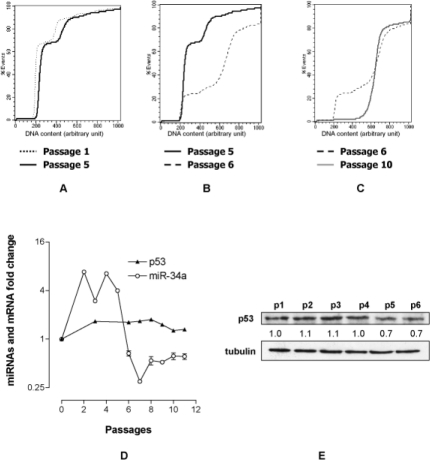
Figure 2. MEF immortalization is characterized by a p53-dependent events. DNA content profile per cell at p1/p5 (A), p5/p6 (B) and p6/p10 (C). (D) Fold change of miR-34a and p53 transcripts at various passages normalized to that of MEF at passage 0. (E) p53 protein level at the various passages. The value reported under each lane represents the average of two independent experiments.
3. The senescence related miRNAs are down regulated in immortalized MEF
As the p53/p21 axis of primary MEF was disrupted in immortalized MEF, we investigated whether the time course of the signatures of miRNAs related to either premature (miR-20a [19]) or replicative (miR-21, miR-28 [20], miR-290 [4]) primary MEF senescence were also modified. The analysis showed that while miR-20a and miR-290 were down regulated till p6 (Fig 3A) miR-21 and miR-28 were up regulated. It is worth noting that the up regulation of miR-21 and miR-28 is in agreement with findings in MEF replicative senescence, while miR-290 down regulation is the opposite of previous observations because we have shown that miR-290 steadily increased when either spontaneous or nocodazole-induced MEF G1 blocked tetraploid cells were present [4]. For that, we examined the cell cycle of p1-p5 MEF. We found that under 6T15 protocol no significant accumulation of G2/M cells was observed (Fig. 3B) suggesting that the failed accumulation of G2/M cells was responsible for the lack of miR-290 up regulation. The miRNA signatures changed markedly after p6: while miR-20a and miR-290 remain down regulated, although to a lesser extent, miR-21 and miR-28 switched from up to down regulation (Fig. 3A). These data suggest that the deregulation of these two senescence related miRNAs, besides miR-34a, is involved in MEF immortalization.
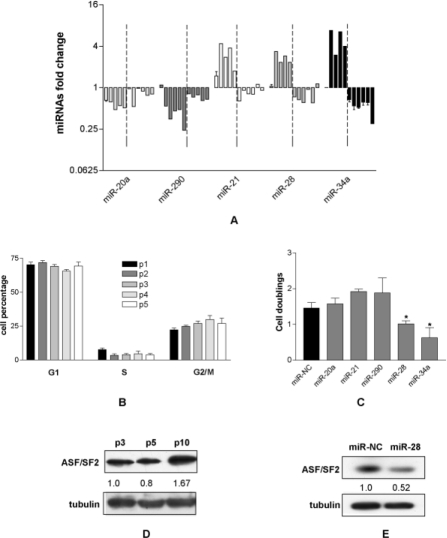
Figure 3. The re-expression of miRNAs down regulated in immortalized MEF reduces cell proliferation. (A) Quantification of miR-20a, miR-21, miR-28, miR-290 and miR-34a per passage normalized to that of MEF at passage 0. Dashed lines indicate the transition from passage 5 to 6. (B) Cell cycle phase distribution (%) of MEF from p1 to p5. (C) Population cell doubling of immortalized MEF after transfection of miR-NC, miR-20a, miR-21, miR-28, miR-290 and miR-34a. Each bar represents the mean ± SD of three biological replicates (*=p<0.05). (D) Expression of ASF/SF2 protein in primary (p3 and p5) and immortalized (p10) MEF. The value reported under each lane represents the average of two independent experiments. (E) Expression of ASF/SF2 after the transfection of immortalized MEF with either miR-NC or miR-28. The value reported under each lane represents the average of two independent experiments.
4. The transfection of the down regulated miRNAs reduces the proliferation of immortalized MEF
To investigate if the deregulated miRNAs play an active role in the immortalization, immortalized MEF (p10) were transfected with miR-21, miR-28 and miR-34. In addition we tested miR-20a and miR-290 whose expression was not affected by the immortalization. The proliferation data showed that miR-20a and miR-290 did not affect cell proliferation as expected, the down regulated miR-28 and miR-34 significantly reduced the proliferation of immortalized MEF with similar efficiency, while miR-21 did not inhibit cell proliferation (Fig. 3C). These data indicate that the re-expression of miR-34a and miR-28 (and consequently of their controlled pathways) was sufficient to reduce the proliferative rate of immortalized MEF and suggest that miR-28, like the tumor suppressor miR-34a, could have a tumor suppressor activity.
5. MiR-28 behaves as tumor suppressor miRNA
It is well known that miR-34a has a widespread tumor suppressor activity [21,22]. We show that it behaved as such in immortalized MEF as its re-expression inhibits cell proliferation, probably by partially restoring the p53 pathway. Interestingly miR-28, as far as we know, is not directly linked to the p53 controlled pathways. We have previously reported that in primary MEF miR-28 targets the proto-oncogenic splicing factor ASF/SF2 and that in turn miR-28 over-expression induces both apoptosis and senescence of primary MEF via ASF/SF2 down regulation [20]. This prompted us to verify the expression of ASF/SF2 in primary and immortalized MEF. As expected the expression of ASF/SF2 was higher in immortalized than in primary MEF (Fig. 3D) suggesting a possible post transcriptional control of miR-28 on ASF/SF2 expression. To confirm the hypothesis, we transfected immortalized MEF (p10) with miR-28 and found 50% reduction of ASF/SF2 expression (Fig. 3E), suggesting that miR-28 control cell proliferation by targeting ASF/SF2. These results strengthen the idea that miR-28 has a tumor suppressor-like activity and might be suitable to be tested in tumor cell lines defective in miR-28 content.
Discussion
The in vitro life span defines the number of population cell doubling which primary cells can undergo in culture. We have reported that MEF under the 6T3 expansion regimen were able to undergo four population cell doubling before achieving senescence. In this work we reported that by modifying the propagation regimen and growth conditions (6T15 protocol) MEF slowed down without reaching a complete cell proliferation block and restarted proliferation after about eight population cell doubling (passage 3). These results suggest that activation of p53 with the consequent induction of p21 in this case may favor proliferation arrest rather than senescence as previously described [23]. Interestingly the in vitro life span of MEF was extended both in terms of population cell doubling and in terms of days in culture, in keeping with the hypothesis that p53 activation, by inducing quiescence rather than senescence may increase the cell life span [24]. The immortalization process is delayed accordingly, suggesting that it does not strictly depend on the number of cell doubling or the time that the cells spend in culture (45 culture days in this case).
The spontaneous immortalization of primary MEF has been attributed to either the inactivation of p53 or the loss of the INK4a/ARF locus (p19ARF, p16) [25], two loci strictly related to cell proliferation. In this work we found that both loci were transcribed and translated during passages whereas p21 shifted from up to down regulation after p5. As p21 expression is mostly p53 dependent we argued that despite p53 continued to be expressed, it may not be functional any more. In support of this conclusion are the down regulation of the p53 dependent miR-34a and the loss of genomic stability revealed by the massive appearance of hyperdiploid cells [16-18]
A recurrent concept is that immortalization is due to mutations induced by DNA damage occurring during the population expansion [14]. The loss of p53 activity found in immortalized MEF could be due to either point mutation or loss of one allele which appears to be sufficient to alter cell growth [15]. In any case the sharp increase of cell proliferation suggests that the immortalization process is caused by the concomitant alteration of multiple pathways. We found that the expression of miR-20a, miR-21, miR-28 and miR-290, all involved in MEF senescence, were deregulated in coincidence with p21 down regulation and increase of cell proliferation. As a high number of genes are potentially targeted by one miRNA [26], we argued that the deregulation of these miRNAs could be in part responsible for the absence of cell proliferation control characteristic of immortal cells. In particular we demonstrate that by replacing miR-28 and miR-34a, under expressed in immortalized MEF, cell proliferation was reduced suggesting that both miRNAs are implicated in immortalization. Evidence in support of an anti proliferative role of miR-28 is already available. We have shown in primary MEF that miR-28 targets the proto-oncogene ASF/SF2, a splicing factor involved in the alternative splicing of many transcripts [27,28] and its over expression induces apoptosis and senescence by down regulating ASF/SF2 [20]. Our results demonstrate that also in immortalized MEF miR-28 targets ASF/SF2 suggesting that its tumor suppressor activity is due to ASF/SF2 inhibition, although other targets cannot be excluded.
The fact that the replacement of miR-28 in immortalized MEF reduced cell proliferation to the same extent as miR-34a further strengthens the hypothesis that miR-28 represents a novel TS miRNA. MiR-34a is a well known TS miRNA able to inhibit cell proliferation of a wide range of tumor cells [21,29-31] as well as of human fibroblasts which extend their replicative capacity when treated with miR-34a antisense [32]. Conversely miR-290 and miR-20a involved in culture and stress induced senescence of primary MEF were not able to inhibit proliferation of immortal MEF in keeping with the idea that miRNAs behave differently in different cellular context [33]. In conclusion the delayed immortalization obtained with this protocol implies that point mutations; due to DNA damage accumulated during population expansion [14], are not the only molecular events at the basis of immortalization. The most notable feature which marks the immortalization process is the drastic p21 down regulation possibly due to p53 functional inactivation. Associated to the sudden down regulation of p21 was the induction of hyperploidy, suggesting that genomic instability and/or epigenetic changes are also responsible for the immortalization process. Interestingly, the switch of p21 expression was accompanied by the change of the signature of miRNAs related to MEF senescence, including the p53-dependent miR-34 and miR-28 [20]. In particular, the down regulated miR-28, behaved as a TS miRNA, when transfected in immortalized MEF, analogously to the well known TS miR-34a, indicating that the comparison of the miRNA signature of primary versus immortalized cells could allow the identification of novel miRNAs with potential tumor suppressor-like activity.
Materials and Methods
Reagents
miRNeasy mini kit, QuantiTect Reverse Trascription Kit, miScript Reverse Transcription Kit, miScript SYBR Green PCR Kit (QIAGEN, Milano, Italy); Dulbecco's Modified Eagle Medium-High Glucose (D-MEM-HG), foetal bovine serum (FBS) (Invitrogen, CA, USA); LightCycler 480 Probes Master, Universal ProbeLibrary LNA Probes; LightCycler 480 SYBR Green I Master (Roche Diagnostic, Mannheim, Germany); X-Gal (5-bromo-4-chloro-3-indolylb-D-galactoside); propidium iodide, anti-α-tubulin (Sigma-Aldrich Corporation, Missouri, USA); anti-p21 (Santa Cruz Biotechnology, Inc.); ECL, Hybond-C extra membranes (Amersham); anti-p53 (Cell Signaling Technology). The anti-ASF/SF2 was a gift of Dr. Adrian Krainer. MiR-20a, miR-21, miR-28, miR-34, miR-290 and miR-NC (negative control) (GenePharma Shanghai, China)
Cells and culture conditions
MEF were isolated from 13.5d mouse embryos, expanded and then replated every three days (6T3 protocol). For 6T15 protocol MEF p1 were collected, diluted at the appropriate concentration (6x105) and grown for 15 days with a medium change every three days, before the next trypsinization and expansion. MEF were grown in Dulbecco's Modified Eagle Medium-high glucose (DMEM-HG)-10% FBS at 37°C in a humidified atmosphere containing 6% CO2.
Cell proliferation
Cell proliferation was measured as number of population cell doubling per passage (CD= ln(Nf/Ni)/ln2) where Nf is the final number of collected cells (day 3) and Ni the initial number of seeded cells (day 0).
MiRNAs transfection
Immortalized MEF (p10) were seeded at cell density of 1.0x105 per 30 mm diameter dish. After 24 hours cells were transfected with either miRNAs under test or a double-stranded oligonucleotide, named miR-NC. Briefly, 15 μl Optimem and 25 μl transfection buffer plus 80 nM miRNA were mixed with a solution of Gene Silencer (5 μl) plus Optimem (25 μl). After 15 minutes incubation, Optimem was added up to 800 μl. After 6 hours the medium was replaced with complete DMEM.
Quantification of miRNAs and genes with Q-Real-time PCR
Total RNA was extracted from 1x106 cells using the miRNeasy mini kit (Qiagen) following the manufacturer's recommendations. To quantify p19ARF, p16 and p21 transcripts, 1μg of total RNA was reverse transcribed using QuantiTect Reverse Trascription Kit (Qiagen). Real-time PCR (qRT-PCR) was carried out with LightCycler 480 (Roche) using LightCycler 480 SYBER Green I Master (Roche). Mature miR-20a, miR-21, miR-28, miR-34a and miR-290 were quantified using the miScript System: 1μg of total RNA was retrotranscribed with miScript Reverse Transcription Kit (Qiagen) and qRT-PCR was carried out using miScript SYBR Green PCR Kit (Qiagen). All reactions were performed in triplicate. Relative quantification of gene expression was calculated with the fit point method. Transcript values were normalized with those obtained from the amplification of the internal controls (GAPDH for transcripts and U6 for miRNAs). The following oligonucleotides were used: p19ARF, forward (F) (5'-CATGGGTCGCAGGTTCTTG-3') and reverse (R) (5'-GCTCGCTGTCCTGG GTCTC-3'); p16, F (5'-CGACGGGCATAGCTTCAG-3') and R (5'-GCTCTGCTCTTGGGATTGG-3'); p21, F (5'-TCCACAGCGATATCCAGACA-3') and R (5'-GGACATCACCAGGATTGGAC-3'); p53, F (5'-ATGCCCATGCTACAGAGGAG-3') and R (5'-AGACTGGCCCTTCTTGGTCT-3'); GAPDH, F (5'-GCCTTCCGTGTTCCTACCC-3'), R (5'-TGCCTGCTTCACCACCTTC-3'); miR-20a, F (5'-TAAAGTGCTTATAGTGCAGGTAG-3'); miR-21, F (5'-TAGCTTATCAGACTGATGTTGA-3'), miR-28, F (5'-AAGGAGCTCACAGTCTATTGAG-3'); miR-34a, F (5'-TGGCAGTGT CTTAGCTGGTTGT-3'); miR-290, F (5'-gctaatcttctctgtatcgttccaa-3'); U6, F (5'-CGCAAGGATGACACGCAAATTC-3').
Western Blot analysis
Equivalent amounts of proteins were resolved on 10% SDS-PAGE gels and transferred to Hybond-C extra membranes by electro blotting. The resulting blots were blocked with 5% nonfat dry milk solution. Anti-α-tubulin (1:1000), anti-p53 (1:500), anti-p21 (1:1000), anti-ASF/SF2 (1:2000) were used. Incubation was performed overnight at 4°C and bands were revealed after incubation with the recommended secondary antibody coupled to peroxidase using ECL. Scanned images were quantified using scion Image software and normalized to α-tubulin.
Cell cycle
Samples of 5x105 of cells under test were fixed with 95% ethanol, stained with 50 μg/ml propidium iodide (PI), incubated over night at 4°C and cell cycle analyzed using a FACScalibur cytofluorimeter. The Kolmogorov-Smirnov Statistic was used to represent the DNA content profile of MEF at the various passages.
Statistical analysis
Data were analyzed using GraphPad Prism (GraphPad Sofware, Inc., San Diego, CA). Comparisons were evaluated by unpaired t-test. A value of p<0.05 (*) was considered statistically significant.
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro, AIRC [project no. 4753] and by Istituto Superiore di Sanità, ISS [project no. 527/A/3A/4].
References
- 1. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5: 741 -7. [PubMed] .
- 2. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005; 120: 513 -22. [PubMed] .
- 3. vom Brocke J, Schmeiser HH, Reinbold M, Hollstein M. MEF immortalization to investigate the ins and outs of mutagenesis. Carcinogenesis. 2006; 27: 2141 -7. [PubMed] .
- 4. Pitto L, Rizzo M, Simili M, Colligiani D, Evangelista M, Mercatanti A, Mariani L, Cremisi F, Rainaldi G. miR-290 acts as a physiological effector of senescence in mouse embryo fibroblasts. Physiol Genomics. 2009; 39: 210 -8. [PubMed] .
- 5. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593 -602. [PubMed] .
- 6. Maeda T, Hobbs RM, Merghoub T, Guernah I, Zelent A, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP. Role of the proto-oncogene Pokemon in cellular transformation and ARF repression. Nature. 2005; 433: 278 -85. [PubMed] .
- 7. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d'Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006; 444: 638 -42. [PubMed] .
- 8. Yogev O, Anzi S, Inoue K, Shaulian E. Induction of transcriptionally active Jun proteins regulates drug-induced senescence. J Biol Chem. 2006; 281: 34475 -83. [PubMed] .
- 9. Chen JH, Hales CN, Ozanne SE. DNA damage, cellular senescence and organismal ageing: causal or correlative? Nucleic Acids Res. 2007; 35: 7417 -28. [PubMed] .
- 10. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY). 2009; 1: 402 -11. [PubMed] .
- 11. Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany NY). 2010; 2: 333 -43. [PubMed] .
- 12. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995; 83: 993 -1000. [PubMed] .
- 13. Carnero A, Hudson JD, Hannon GJ, Beach DH. Loss-of-function genetics in mammalian cells: the p53 tumor suppressor model. Nucleic Acids Res. 2000; 28: 2234 -41. [PubMed] .
- 14. Busuttil RA, Rubio M, Dolle ME, Campisi J, Vijg J. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2003; 2: 287 -94. [PubMed] .
- 15. Odell A, Askham J, Whibley C, Hollstein M. How to become immortal: let MEFs count the ways. Aging (Albany NY). 2010; 2: 160 -5. [PubMed] .
- 16. Borel F, Lohez OD, Lacroix FB, Margolis RL. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc Natl Acad Sci U S A. 2002; 99: 9819 -24. [PubMed] .
- 17. Vitale I, Galluzzi L, Senovilla L, Criollo A, Jemaa M, Castedo M, Kroemer G. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2010; .
- 18. Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello-Varona S, Manic G, Metivier D, Vivet S, Tajeddine N, Joza N, Valent A, Castedo M, Kroemer G. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010; 29: 1272 -84. [PubMed] .
- 19. Poliseno L, Pitto L, Simili M, Mariani L, Riccardi L, Ciucci A, Rizzo M, Evangelista M, Mercatanti A, Pandolfi PP, Rainaldi G. The proto-oncogene LRF is under post-transcriptional control of MiR-20a: implications for senescence. PLoS One. 2008; 3: e2542 [PubMed] .
- 20. Verduci L, Simili M, Rizzo M, Mercatanti A, Evangelista M, Mariani L, Rainaldi G, Pitto L. MicroRNA (miRNA)-mediated interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J Biol Chem. 2010; 285: 39551 -63. [PubMed] .
- 21. Yamakuchi M and Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009; 8: 712 -5. [PubMed] .
- 22. Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007; 104: 15472 -7. [PubMed] .
- 23. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -4. [PubMed] .
- 24. Vigneron A and Vousden KH. p53, ROS and senescence in the control of aging. Aging (Albany NY). 2010; 2: 471 -4. [PubMed] .
- 25. Kim H, You S, Farris J, Kong BW, Christman SA, Foster LK, Foster DN. Expression profiles of p53-, p16(INK4a)-, and telomere-regulating genes in replicative senescent primary human, mouse, and chicken fibroblast cells. Exp Cell Res. 2002; 272: 199 -208. [PubMed] .
- 26. Lall S, Grun D, Krek A, Chen K, Wang YL, Dewey CN, Sood P, Colombo T, Bray N, Macmenamin P, Kao HL, Gunsalus KC, Pachter L, Piano F, Rajewsky N. A genome-wide map of conserved microRNA targets in C. elegans. Curr Biol. 2006; 16: 460 -71. [PubMed] .
- 27. Krainer AR, Conway GC, Kozak D. The essential pre-mRNA splicing factor SF2 influences 5' splice site selection by activating proximal sites. Cell. 1990; 62: 35 -42. [PubMed] .
- 28. Krainer AR, Conway GC, Kozak D. Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 1990; 4: 1158 -71. [PubMed] .
- 29. Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, Xiang D, Desano JT, Bommer GT, Fan D, Fearon ER, Lawrence TS, Xu L. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009; 4: e6816 [PubMed] .
- 30. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011; 17: 211 -5. [PubMed] .
- 31. Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010; 17: 193 -9. [PubMed] .
- 32. Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR, Ji H, Vojtesek B, Bourdon JC, Lane DP, Harris CC. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat Cell Biol. 2009; 11: 1135 -42. [PubMed] .
- 33. Rizzo M, Mariani L, Pitto L, Rainaldi G, Simili M. miR-20a and miR-290, multi-faceted players with a role in tumourigenesis and senescence. J Cell Mol Med. 2010; 14: 2633 -40. [PubMed] .