




Introduction
Sir2 enzymes, or sirtuins, are NAD+ dependent protein deacetylases and mono-ADP ribosyltransferases which regulate lifespan in S. cerevisiae [1], C. elegans [2] and D. melanogaster [3]. In each of these systems, overexpression or hyperactivation of Sir2 or its homologs extends lifespan. In yeast, this lifespan extension is achieved by promoting genomic stability [4], regulating gene expression [5, 6] and suppressing the formation of extra-ribosomal circles [1, 7]. In the roundworm sir-2.1 promotes longevity by modulating daf-16 (FOXO) signaling [2] and regulating the proteostasis stress response [8]; Sir2 extends lifespan in the fruit fly by coordinating a different stress response - the dietary restriction pathway [3]. Mammalian genomes encode seven Sir2 homologs (SIRT1-7); while it is unclear whether overexpression or hyperactivation of any of these sirtuins can extend lifespan, there is evidence that these genes can protect against several age-related pathologies, including neurodegeneration [9], hearing loss [10], diet induced obesity [11, 12] and cancer [13-15].
Of the mammalian sirtuins, SIRT6 recapitulates many of the biological functions of Sir2 and its homologs in fly and worm. At the molecular level, SIRT6 regulates the expression of a large number of stress-responsive and metabolism related genes [16-18], promotes genomic stability [19, 20] and stimulates base excision [19] and double strand break DNA repair [21-23]. Consistent with these molecular functions, SIRT6 knock-out mice develop a degenerative disorder that in many ways resembles premature aging [19]. These mice exhibit kyphosis, cachexia, greying of the fur, decreased bone mineral density, hypoglycemia, chronic inflammation and a severely shortened lifespan. This progeria is completely penetrant in multiple genetic backgrounds [24].
To date, most of the molecular functions of SIRT6 have been ascribed to the protein's deacetylase activity; SIRT6 is known to deacetylate H3K9 [20], H3K56 [25, 26] and CtIP [22]in vivo. Very little had been known, however, about the biological significance of the protein's mono-ADP ribosyltransferase activity. Recently we reported that SIRT6 mono-ADP ribosylates PARP1 to stimulate DNA repair in response to oxidative stress [23]. In this research perspective, we will review the role of SIRT6 in DNA repair and discuss the emerging implications for sirtuin directed mono-ADP ribosylation in the context of aging and age-related disease.
SIRT6 and double strand break repair
Maintenance of genomic stability is a challenge faced by all organisms. The most grievous challenge to genomic stability comes in the form of DNA damage, and, in particular, lesions which cause double strand breaks (DSBs) [27]. Unrepaired, DSBs can lead to a host of adverse cellular phenotypes including irregular gene expression, permanent cell cycle arrest, cell death and malignant transformation. For this reason, eukaryotes have evolved two independent pathways for repairing this dangerous form of damage: homologous recombination (HR) [28] and non-homologous end joining (NHEJ) [29]. Efficient DSB repair is often a limiting factor for longevity, and mutations in the core DSB repair enzymes frequently result in a variety of disease states including premature-aging and a predisposition for cancer, underscoring the importance of DSB repair in the context of health and aging [30, 31].
The first evidence that SIRT6 may impact on DSB repair came from the observation of genomic instability in SIRT6 knock-out mice; cells from these mice exhibit a high incidence of chromosomal rearrangement and breakage as well as a hypersensitivity to γ-irradiation [19]. Two subsequent studies noted that knockdown of SIRT6 in human cells similarly sensitized the cells to chemically induced DSBs, and also observed that SIRT6 is recruited to break sites after DNA damage [21, 22]. The first of these studies suggested that SIRT6 functions in NHEJ by stabilizing the NHEJ haloenzyme, DNA-PK, at the site of DSBs, and the second study revealed that SIRT6 promotes HR by deacetylating, and thereby activating, the end resection protein, CtIP. These studies provided the first mechanistic insight into how SIRT6 functions in DSB repair.
Our group recently published a report that further clarifies the role of SIRT6 in DNA repair and underscores the dynamic role that SIRT6 plays in promoting genomic stability, especially in response to oxidative stress [23]. Whereas previous studies had depleted SIRT6 to assess its role in DNA repair, we noted that overexpression of SIRT6 stimulated DSB repair through both the HR and NHEJ pathways by approximately 3-fold. Strikingly, when cells were pretreated with oxidative stressors prior to overexpression of SIRT6, DSB repair efficiency was massively stimulated by up to 16-fold, suggesting that SIRT6 specifically integrates stress signaling to prime the DNA repair machinery in response to oxidative stress. Consistent with this hypothesis, we observed that while SIRT6 is normally recruited to DSB sites relatively late, approximately 8 hours after induction of the DSB, pretreatment with paraquat resulted in an early wave of SIRT6 recruitment to the breakage points, within 30 minutes of inducing the break.
To assess which biochemical activity of SIRT6 mediated this stress response, we synthesized SIRT6 mutants which could catalyze only deacetylation or only mono-ADP ribosylation reactions, thereby uncoupling these two enzymatic activities. Intriguingly, both of these activities were required to stimulate DSB repair, indicating that SIRT6 played a role in DSB repair beyond interacting with DNA-PKcs and deacetylating CtIP. Consistent with this hypothesis, we found that SIRT6 mono-ADP ribosylates the upstream DSB repair factor, PARP1, at lysine 521, thereby stimulating its poly-ADP ribosylation activity. Importantly, this modification was required for SIRT6-mediated stimulation of DSB repair. Mechanistically, PARP1 enables several crucial interactions in the early stages of DSB repair by poly-ADP ribosylating itself and other protein substrates. Notably, PARP1 facilitates the recruitment of the MRN complex to DSBs [32], plays a role in the activation of ATM [33] and helps to direct the choice between the NHEJ and HR repair pathways. Additionally, PARP1 is required to promote a non-canonical form of NHEJ (Alt-NHEJ) [34]; consistent with this, SIRT6 can stimulate NHEJ in the absence of DNA-PK, an essential canonical-NHEJ enzyme, suggesting that SIRT6 can stimulate NHEJ through the alternative pathway.
The interaction with PARP1 explains the early recruitment of SIRT6 to sites of DSBs under stressed conditions. Intriguingly, we also observed that H3K9 is globally deacetylated at a late, 8 hour post-break, time point (Figure 1). Previous studies have indicated that this deacetylation is dependent on SIRT6 [21]. Whereas the early interaction between SIRT6 and PARP1 likely functions to stimulate the repair process, it is possible that this later wave of histone deacetylation functions to restore the original chromatin structure of the locus or to condense chromatin to prevent further damage. Another intriguing possibility is that SIRT6 relocalization in response to DNA damage may affect gene expression. Consistent with this possibility, another mammalian sirtuin, SIRT1, relocalizes to DSB sites in response to damage; this relocalization is concomitant with deregulation of SIRT1-regulated genes and may contribute to several aging phenotypes [35]. It will be interesting to see if changes in gene expression are associated with SIRT6 relocalization in response to DNA damage, and what effect these changes may have on the cell.
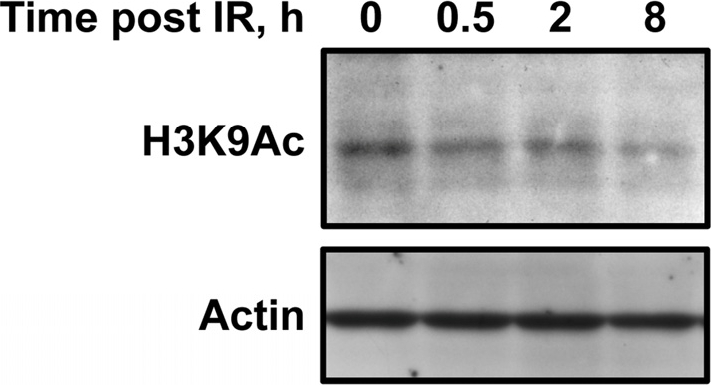
Figure 1. The second wave of SIRT6 recruitment to DSBs is concomitant with global deacetylation of H3K9 Human fibroblasts were exposed to γ-irradiation. Immunoblotting revealed that H3K9-Ac levels are reduced 8 hours following irradiation, concurrent with a second wave of SIRT6 recruitment to DSBs.
The finding that SIRT6 primes the DSB repair machinery in response to stress suggests an energetically pragmatic paradigm, wherein certain DNA repair enzymes exist in a basal state under normal conditions, but can become activated in response to oxidative stress. Such a system would allow cells to stimulate DNA repair under conditions in which they are most likely to sustain DNA damage. This might be beneficial because it would allow the cell to conserve energy and curtail deleterious side-effects associated with chronic activation of DSB repair enzymes which can include oncogenic hyper-recombination and cell death. Constitutive activation of PARP1, for example, promotes cell death [36, 37]; SIRT6 mediated activation of PARP1 specifically in response to stress may represent a mechanism for controlling this response.
This adaptive response, wherein SIRT6 is mobilized following mild doses of oxidative stress to activate DNA repair machinery, in many ways resembles a hormetic response. Hormesis predicts that exposure to low levels of stress can result in favorable biological outcomes such as increased resistance to stress or extended lifespan; caloric restriction is a well characterized example of this phenomenon [38, 39]. At the cellular level, several studies have indicated that low levels of oxidative stress are beneficial to the cell [40, 41]. It is possible that SIRT6 mediates this hormetic response, providing a link between stress sensing pathways and/or NAD+ levels and DNA repair machinery. This raises the intriguing possibility of using pharmacological activators to stimulate SIRT6 activity as a means of slowing and attenuating the onset of age-related pathologies.
While SIRT6 has emerged as an important mediator of genome stability (Figure 2), there are still several questions with regards to the role that SIRT6 plays in DNA repair. In response to oxidative stress, SIRT6 is recruited to chromatin and mono-ADP-ribosylates PARP1, but what triggers this reaction? ATM, NF-κB and the MAPKs are stress responsive kinases which have been implicated in DSB repair or shown to interact with other sirtuins [42-44] - it is possible one or more of these proteins transduces stress signaling to SIRT6. Another intriguing question involves base excision repair (BER) - SIRT6-/- mice exhibit a defect in BER, although the etiology of this deficiency remains unclear [45]. In response to single strand breaks, PARP1 binds to DNA and facilitates the recruitment of BER factors to instigate repair [46]. Could SIRT6 also promote BER through PARP1, and if so, would this response also be heightened in response to oxidative stress? Answering these and other related questions will provide a clearer understanding of the important role that SIRT6 plays in genome maintenance.
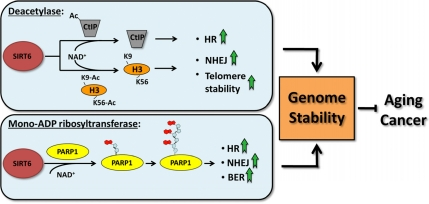
Figure 2. SIRT6 regulates genomic stability SIRT6 promotes genome stability by regulating DNA single-strand and double strand break repair pathways and by facilitating telomere maintenance. The deacetylase and the mono-ADP ribosyltransferase activities of SIRT6 both contribute to this function.
Mono-ADP ribosylation
Although sirtuins are best understood in the context of protein deacetylation, the founding member of this gene family, Sir2, was first described as a mono-ADP ribosyltransferase [47]. This form of post-translational modification was first identified as a feature of several bacterial toxins, including diphtheria, cholera and pertussis. Subsequent studies revealed the presence of eukaryotic mono-ADP ribosyltransferases which function in multiple biological pathways, including signal transduction, gene expression and cellular differentiation [48, 49]. These transferases can broadly be sorted into distinct categories on the basis of homology and localization and include: classical mono-ADP ribosyltransferases (ARTs), certain members of the PARP family and sirtuins [48, 50].
The role of sirtuins as protein deacetylases has been well characterized. Briefly, sirtuins couple NAD+ hydrolysis with lysine deacetylation to generate deacetylated protein, and the metabolites O-acetyl-ADP-ribose and nicotinamide [51]. Sirtuin mediated deacetylation regulates a large array of biological processes. Many sirtuins, however, also possess a less characterized enzymatic activity, mono-ADP ribosylation [52]. In this context, sirtuins transfer the ADP-ribose moiety of NAD+ to acceptor proteins. Ribosylation activities have been reported for sirtuins in a wide range of organisms. For example the protozoan T. brucei Sir2 homolog mono-ADP ribosylates H2A and H2B in response to DNA damage [53]; yeast Sir2 catalyzes the transfer of ADP ribose to itself and histones [47]; and the mammalian sirtuin, SIRT4 ribosylates glutamate dehydrogenase to suppress insulin signaling in pancreatic β-cells [54]. Several studies have also suggested that many other sirtuins, including E. coli CobB, as well as mammalian SIRT1 and SIRT3, possess mono-ADP ribosyltransferase activity, although the biological significance of this activity remains unclear because no in vivo substrates have been identified for this reaction [52, 55]. Akin to Sir2, SIRT6 was first reported as an auto mono-ADP ribosyltransferase [56], and only later discovered to possess protein deacetylase activity [20].
Several recent reports have provided insight into the biochemistry of SIRT6-mediated mono-ADP ribosylation and have suggested that there are additional, as yet uncharacterized, substrates for this reaction. Crystallography of SIRT6 revealed several unique features, including the absence of a helix bundle that typically connects the Rossmann and zinc binding domains in other sirtuins [57]. This distinct structure favors the binding of NAD+ even in the absence of acetylated substrate, and as a result may facilitate mono-ADP ribosyltransfer reactions. Consistent with the notion that SIRT6 is well suited to catalyze mono-ADP ribosylation we recently reported that overexpression of SIRT6 selectively induces massive apoptosis in cancer cells but not non-cancerous cells and that this cytotoxicity is dependent on the mono-ADP ribosylation activity of SIRT6 [15]. As yet it is unclear exactly how SIRT6 mono-ADP ribosylation promotes death in cancer cells, but it appears to be independent of PARP1, suggesting that there are additional targets for SIRT6-mediated mono-ADP ribosylation in vivo.
In consideration of the dual enzymatic activities of sirtuins, early models predicted that the deacetylase activity of sirtuins may function to regulate gene expression, whereas the mono-ADP ribosylation activity may mediate DNA repair [58]. While the discovery of non-histone substrates for deacetylation, and the observation that SIRT4 ribosylates a metabolic enzyme suggests that this model may be simplistic, there is evidence that mono-ADP ribosylation is an important feature of DNA repair. Multiple studies have observed that mono-ADP ribose is transferred to histones in response to DNA damage [59-61], although it is unclear if this response is entirely mediated by sirtuins. The protozoan Sir2 homolog, TbSIR2RP1 ribosylates histones in response to DNA damage [53], and we recently demonstrated that SIRT6 mono-ADP ribosylates PARP1 to promote DNA repair in response to oxidative stress. Future studies will be required to reveal the full importance of sirtuin-mediated mono-ADP ribosylation reactions.
Concluding remarks and prospectus
It is becoming clear that SIRT6 functions in multiple pathways related to aging by facilitating DNA repair, promoting telomere stability, attenuating NF-κB activity and regulating metabolism. Intriguingly, destabilization of any of these pathways can lead to the accumulation of aging related phenotypes [62-65]. It will be interesting to see whether SIRT6 overexpression or hyperactivity can ameliorate or delay the onset of age-related pathologies, possibly by stimulating hormetic response pathways. In this context we have shown that SIRT6 overexpression can improve the efficiency of NHEJ and HR, perhaps by mimicking an endogenous response to oxidative stress. In a separate study, we observed that SIRT6 overexpression induces massive apoptosis in cancer cells, but not non-cancerous cells. Finally, another group has indicated that SIRT6 overexpression protects against diet induced obesity in mice [12]. Collectively, these studies provide support for the hypothesis that SIRT6 may protect against aging or age-associated pathologies, although more evidence will be required to confirm this. It is worth noting that several studies have indicated that it may be possible to modulate SIRT6 activity using physiological or pharmacological interventions [66, 67]. It will be interesting to further assess whether modulating SIRT6 levels and activity can yield desirable clinical outcomes.
Acknowledgments
The authors would like to thank Jorge Azpura, Xiao Tian and Amita Vaidya for critical comments and helpful conversations while composing this research perspective.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & development. 1999; 13: 2570 -2580. [PubMed] .
- 2. Tissenbaum HA and Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001; 410: 227 -230. [PubMed] .
- 3. Rogina B and Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101: 15998 -16003. [PubMed] .
- 4. Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999; 97: 621 -633. [PubMed] .
- 5. Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990; 63: 751 -762. [PubMed] .
- 6. Rine J and Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987; 116: 9 -22. [PubMed] .
- 7. Gottlieb S and Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell. 1989; 56: 771 -776. [PubMed] .
- 8. Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Developmental cell. 2005; 9: 605 -615. [PubMed] .
- 9. Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. The EMBO journal. 2007; 26: 3169 -3179. [PubMed] .
- 10. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010; 143: 802 -812. [PubMed] .
- 11. Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105: 9793 -9798. [PubMed] .
- 12. Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, Cohen HY. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging cell. 2010; 9: 162 -173. [PubMed] .
- 13. Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PloS one. 2008; 3: e2020 [PubMed] .
- 14. Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer cell. 2010; 17: 41 -52. [PubMed] .
- 15. Van Meter M, Mao Z, Gorbunova V, Seluanov A. Sirt6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle. 2011; 10: in press. .
- 16. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009; 136: 62 -74. [PubMed] .
- 17. Kawahara TL, Rapicavoli NA, Wu AR, Qu K, Quake SR, Chang HY. Dynamic chromatin localization of Sirt6 shapes stress- and aging-related transcriptional networks. PLoS genetics. 2011; 7: e1002153 [PubMed] .
- 18. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010; 140: 280 -293. [PubMed] .
- 19. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006; 124: 315 -329. [PubMed] .
- 20. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008; 452: 492 -496. [PubMed] .
- 21. McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 2009; 1: 109 -121. [PubMed] .
- 22. Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010; 329: 1348 -1353. [PubMed] .
- 23. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011; 332: 1443 -1446. [PubMed] .
- 24. Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O, Jou W, Gius D, Deng CX. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. The Journal of biological chemistry. 2010; 285: 36776 -36784. [PubMed] .
- 25. Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009; 8: 2664 -2666. [PubMed] .
- 26. Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009; 8: 2662 -2663. [PubMed] .
- 27. Gorbunova V and Seluanov A. Making ends meet in old age: DSB repair and aging. Mechanisms of ageing and development. 2005; 126: 621 -628. [PubMed] .
- 28. Li X and Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell research. 2008; 18: 99 -113. [PubMed] .
- 29. Weterings E and Chen DJ. The endless tale of non-homologous end-joining. Cell research. 2008; 18: 114 -124. [PubMed] .
- 30. O'Driscoll M and Jeggo PA. The role of double-strand break repair - insights from human genetics. Nature reviews Genetics. 2006; 7: 45 -54. .
- 31. Seviour EG and Lin SY. The DNA damage response: Balancing the scale between cancer and ageing. Aging. 2010; 2: 900 -907. [PubMed] .
- 32. Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. The Journal of biological chemistry. 2008; 283: 1197 -1208. [PubMed] .
- 33. Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. The Journal of biological chemistry. 2007; 282: 16441 -16453. [PubMed] .
- 34. Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic acids research. 2006; 34: 6170 -6182. [PubMed] .
- 35. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008; 135: 907 -918. [PubMed] .
- 36. Chiarugi A and Moskowitz MA. Cell biology. PARP-1–a perpetrator of apoptotic cell death? Science. 2002; 297: 200 -201. [PubMed] .
- 37. Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, Herceg Z, Wang ZQ, Schulze-Osthoff K. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002; 13: 978 -988. [PubMed] .
- 38. Calabrese EJ, Bachmann KA, Bailer AJ, Bolger PM, Borak J, Cai L, Cedergreen N, Cherian MG, Chiueh CC, Clarkson TW, et al. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicology and Applied Pharmacology. 2007; 222: 122 -128. [PubMed] .
- 39. Gems D and Partridge L. Stress-response hormesis and aging. “that which does not kill us makes us stronger”. Cell Metab. 2008; 7: 200 -203. [PubMed] .
- 40. Feinendegen LE. Evidence for beneficial low level radiation effects and radiation hormesis. The British journal of radiology. 2005; 78: 3 -7. [PubMed] .
- 41. Azzam EI, de Toledo SM, Raaphorst GP, Mitchel RE. Low-dose ionizing radiation decreases the frequency of neoplastic transformation to a level below the spontaneous rate in C3H 10T1/2 cells. Radiation research. 1996; 146: 369 -373. [PubMed] .
- 42. Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010; 330: 517 -521. [PubMed] .
- 43. Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochemical pharmacology. 2006; 72: 1493 -1505. [PubMed] .
- 44. Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PloS one. 2009; 4: e8414 [PubMed] .
- 45. Lombard DB. Sirtuins at the breaking point. SIRT6 in DNA repair. Aging. 2009; 1: 12 -16. [PubMed] .
- 46. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell research. 2008; 18: 27 -47. [PubMed] .
- 47. Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999; 99: 735 -745. [PubMed] .
- 48. Corda D and Di Girolamo M. Functional aspects of protein mono-ADP-ribosylation. The EMBO journal. 2003; 22: 1953 -1958. [PubMed] .
- 49. Di Girolamo M, Dani N, Stilla A, Corda D. Physiological relevance of the endogenous mono(ADP-ribosyl)ation of cellular proteins. The FEBS journal. 2005; 272: 4565 -4575. [PubMed] .
- 50. Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Luscher B. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Molecular cell. 2008; 32: 57 -69. [PubMed] .
- 51. Blander G and Guarente L. The Sir2 family of protein deacetylases. Annual review of biochemistry. 2004; 73: 417 -435. .
- 52. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochemical and biophysical research communications. 1999; 260: 273 -279. [PubMed] .
- 53. Garcia-Salcedo JA, Gijon P, Nolan DP, Tebabi P, Pays E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. The EMBO journal. 2003; 22: 5851 -5862. [PubMed] .
- 54. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006; 126: 941 -954. [PubMed] .
- 55. Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. The Journal of biological chemistry. 2005; 280: 13560 -13567. [PubMed] .
- 56. Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. The Journal of biological chemistry. 2005; 280: 21313 -21320. [PubMed] .
- 57. Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. The Journal of biological chemistry. 2011; 286: 14575 -14587. [PubMed] .
- 58. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000; 403: 795 -800. [PubMed] .
- 59. Kreimeyer A, Wielckens K, Adamietz P, Hilz H. DNA repair-associated ADP-ribosylation in vivo. Modification of histone H1 differs from that of the principal acceptor proteins. J Biol Chem. 1984; 259: 890 -896. [PubMed] .
- 60. Adamietz P and Rudolph A. ADP-ribosylation of nuclear proteins in vivo. Identification of histone H2B as a major acceptor for mono-and poly(ADP-ribose) in dimethyl sulfate-treated hepatoma AH 7974 cells. The Journal of biological chemistry. 1984; 259: 6841 -6846. [PubMed] .
- 61. Meyer T and Hilz H. Production of anti-(ADP-ribose) antibodies with the aid of a dinucleotide-pyrophosphatase-resistant hapten and their application for the detection of mono(ADP-ribosyl)ated polypeptides. European journal of biochemistry / FEBS. 1986; 155: 157 -165. [PubMed] .
- 62. Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005; 120: 497 -512. [PubMed] .
- 63. Blasco MA. Telomere length, stem cells and aging. Nature chemical biology. 2007; 3: 640 -649. .
- 64. Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NFκB, reduced IκBα, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell. 2008; 7: 805 -812. [PubMed] .
- 65. Anderson RM, Colman RJ, Weindruch R. Exploring Mechanisms of Aging Retardation by Caloric Restriction: Studies in Model Organisms and Mammals. The Comparative Biology of Agingy. .
- 66. Kanfi Y, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, Lerrer B, Moazed D, Marine JC, de Cabo R, et al. Regulation of SIRT6 protein levels by nutrient availability. FEBS letters. 2008; 582: 543 -548. [PubMed] .
- 67. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004; 430: 686 -689. [PubMed] .