



The ability of our cells to maintain genomic integrity is fundamental for adequate protection from developmental defects, cancer, and aging. Central to this process is the ability of cells to accurately recognize and correctly repair DNA damage in a timely manner to then allow regulated and orderly progression through the cell cycle. By linking genome stability surveillance, cell cycle, and energy metabolism, the ~370 kDa Serine (Ser)/Threonine (Thr) kinase ataxia telangiectasia mutated (ATM) has begun to emerge as a central DNA damage checkpoint that connects cellular bioenergetics with cancer predisposition and aging [1-4]. ATM is a member of the phosphoinositide 3-kinase-related protein kinase (PIKK) family to which the master regulator of cell growth and metabolism mammalian target of Rapamycin (mTOR) also belongs [5]. ATM is a well-known primary regulator of the cellular response to DNA double-strand breaks (DSBs). A number of studies have recently established that oxidative stress also activates ATM, even in the absence of DSBs. In response to oxidative stress, ATM is phosphorylated at Ser-1981, which results in phosphorylation of its substrates, including p53, the master controller of DNA metabolic stresses, and AMP-activated protein kinase-α (AMPKα), the key sensor of fuel and energy status [6, 7].
ATM: Connecting the energy restriction mimetic metformin to its metabolic target AMPK
Energetic stress due to glucose restriction increases the AMP/ATP ratio. Treatments with drugs that increase the AMP/ATP ratio, including the AMP analog 5-aminoimidazole-4-carboxamide-1-β-ribofuranoside (AICAR) or the anti-diabetic biguanide metformin, activate AMPKα through phosphorylation of Thr-172 and also increase the levels of the AMPKα protein. Although several proteins can phosphorylate AMPKα (e.g., the master upstream Ser/Thr kinase 11 (STK11)/Liver Kinase B1 [LKB1]), it should be noted that activating phosphorylation of AMPKα in response to energetic stress takes place in an ATM-dependent and STK11/LKB1-independent manner [7]. Accordingly, the selective ATM inhibitor KU-55933 markedly reduces the AMPKα-activating effects of metformin in rat hepatoma cells, functionally supporting the first genome-wide association study that unexpectedly found the ATM gene as the causal modulator of glycemic responsiveness to metformin among type 2 diabetic patients [8]. Indeed, treatment with the ATM inhibitor KU-55933 is sufficient to prevent metformin-induced phosphorylation of AMPKα and of the AMPKα downstream target Acetyl-CoA Carboxylase (ACC), concluding that ATM works upstream of AMPKα and that ATM is required for a full response to metformin [8]. Although these results support and extend previous reports of ATM involvement in the activation of AMPKα by stimuli other than metformin [7, 9, 10], metformin's ability to function as a general activator of the ATM-dependent DDR pathway remains to be explored to prove a causal link between the metformin-induced activation of ATM and the diminished risk of developing cancer in individuals taking this drug [11].
We have recently added metformin to the growing list of agents that may have potent cancer-preventive properties by activating the ATM-regulated DDR pathway [12]. The treatment of cultured tumor cells with millimolar concentrations of metformin was found to promote significant activation of ATM, as determined by immunofluorescence microscopy using a monoclonal antibody directed against Ser-1981-phosphorylated ATM. Because cellular DNA damage and particularly the induction of DSBs result in activating phosphorylation of ATM at Ser-1981 and Histone H2AX at Ser-139, we also explored whether the Ser-139 Histone H2AX phosphorylation was altered in response to metformin. Metformin-induced induction of phospho-γH2AXSer139 foci was not accompanied by the expected incorporation of 53BP1 to nuclear repair foci, and metformin-induced Ser-1981 ATM phosphorylation displayed a uniform, nuclear signal that failed to colocalize with phospho-γH2AXSer139 foci. Thus, we termed these metformin-triggered events "pseudo-DDR" [13] to distinguish them from a bona fide DDR triggered in response to true DNA damage. Importantly, “metformin-induced pseudo-DDR” was accompanied by the activation of functional elements typically involved in ATM-regulated genomic stress. First, metformin treatment greatly enhanced phosphorylation of Chk2 at Thr-68, an ATM kinase-dependent event that mediates the response of the ATM pathway following DNA damage [14, 15]. Second, metformin exposure notably enhanced phosphorylation of cAMP-Response Element Binding protein (CREB) at Ser-133, an ATM kinase-regulated event in response to oxidative DNA damage and DNA replication stress [16, 17].
Metformin and ATM-sensed energy metabolism: Reactivating oxidative phosphorylation biogenesis to impede glycolytic cancer cell growth
As metformin is thought to activate AMPK by inhibiting oxidative phosphorylation [18, 19] and because phosphorylation of CREB at Ser-133 can be observed in cultured cells that have been incubated with oxidative phosphorylation inhibitors [20], it could be argued that metformin-induced CREB activation might merely reflect metformin's ability to impair mitochondrial activity in tumor cells. However, CREB phosphorylation is pivotal in mediating peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α)-stimulated mitochondrial biogenesis [21-24]. Therefore, metformin-stimulated phosphorylation of CREB at Ser-133, which activates the promoter of PGC-1α and increases PGC-α mRNA and protein expression [25, 26], can also be viewed as part of the mechanism through which metformin may control mitochondrial biogenesis in tumor cells. Tumor cells are dependent on glycolysis to support their metabolic requirements; even under aerobic conditions, tumor cells continue to rely on glycolysis rather than oxidative phosphorylation (Warburg effect), resulting in high glucose requirements to generate energy and biosynthetic precursors because of the increased availability of glycolytic intermediates [27-30]. As such, metformin-induced reactivation of oxidative phosphorylation biogenesis may contribute to the growth arrest of cancer cells. A recently developed high-throughput respirometric assay for mitochondrial biogenesis used the Seahorse Bioscience analyzer to measure mitochondrial function in real time. In adapted primary cultures of non-glycolytic renal proximal tubular cells, metformin augmented mitochondrial biogenesis [31]. Recent experiments from our own laboratory have established that culturing human cancer cells in the presence of metformin significantly enhances the expression of cytochrome c oxidase I (COX-1) and mitochondrial succinate dehydrogenase (SDH-A), which are encoded by mitochondrial and nuclear genomes, respectively (Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Menendez OJ, Martin-Castillo B, Joven J, Menendez JA. Metformin rescues cell surface major histocompatibility complex class I deficiency caused by oncogenic transformation. Submitted for publication). Using cancer cell lines, non-cancer cells, embryonic cells and Rho(0) cells (i.e., cells depleted of mitochondrial DNA), Jose et al. [32] recently confirmed that the AMPK agonist AICAR exhibits a strong and cancer-specific growth effect that depends on the bioenergetic signature of the cells and involves upregulation of oxidative phosphorylation. In fact, the sensitivity to pharmacological activation of AMPK is higher when cells display a high proliferation rate accompanied by a low steady-state content of ATP. Although it remains to be established if AMPKα-related induction of mitochondrial biogenesis to increase oxidative phosphorylation is instrumental and possibly required for the anti-cancer/anti-aging effects of metformin [33, 34], it is becoming clear that some health-promoting capabilities of metformin may rely on its ability to function as a bona fide glucose-starvation mimetic. Accordingly, recent experiments from our own laboratory have confirmed that, when added to glucose-free medium, where growth is highly oxidative phosphorylation-dependent, metformin drastically increases apoptotic cell death in glucose-addicted cancer cell cultures. Because transformed human cell types appear to be more sensitive to glucose deprivation-induced cytotoxicity and metabolic oxidative stress than non-transformed human cell types, we suggest that a rational use of metformin in combination with fasting could significantly potentiate the effects of chemotherapy in cancer while protecting normal cells, thus further increasing the therapeutic window [35-38] (Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Menendez OJ, Joven J, Martin-Castillo B, Menendez JA. Glucose deprivation enhances metformin-induced apoptosis in a breast cancer cell type-dependent manner: Implications for cyclotherapy. Manuscript in preparation).
In the last issue of Aging, Halicka et al. [39] reported that treatment of normal mitogenically stimulated lymphocytes or tumor cell lines treated with metformin attenuated ATM activation and constitutive H2AX phosphorylation. Their observation that cells treated with metformin have reduced expression of Ser-1981-phosphorylated ATM and Ser-139-phosphorylated Histone H2AX is in contrast not only to data previously reported by our own group in Cell Cycle [12] but also to those presented by Dian et al. [40] in a recent issue of PLoS One. Using human diploid fibroblasts (HDFs), these authors reported that treatment with pharmacological agents increasing the AMP/ATP ratio (i.e., AICAR and metformin) is molecularly equivalent to the effects of glucose restriction in terms of activation of the ATM/AMPK pathway. On one hand, glucose restriction-induced activation of AMPK-driven intracellular signaling was found to be an ATM-dependent process. Thus, the ability of glucose restriction to increase the activating phosphorylation of AMPKα cannot be observed in ataxia-telangiectasia (A-T) cells. On the other hand, treatment of HDFs with the AMPK agonists AICAR or metformin activates ATM at Ser-1981, increases the overall levels of ATM protein and activates AMPK [40]. These findings together indicate that the energetic stress that is induced by glucose restriction or metformin treatment can activate the ATM/AMPKα pathway to induce autophagy and likely cellular senescence. These observations are consistent with the idea that disruption of an energetic stress-induced checkpoint through the loss of ATM function may provide a growth advantage to cells under energetic stress but exacerbate cytotoxic responses to metformin [41].
We recently hypothesized that the unexpected ability of metformin to promote the activation of ATM may be due to the short (24 or 48 h) time courses of most published studies on metformin-induced energetic stress and human cancer cell death in vitro. To test this hypothesis and to simulate patients receiving metformin on a daily basis, we maintained A431 epidermoid cancer cells in long-term uninterrupted subculture with metformin concentrations as high as 10 mmol/L for longer than 4 months before starting any experimental procedure. Metformin-induced loss of proliferative potential, as measured by the absence of immuno-reactive Histone H3 phosphorylated at Ser-10 (Figure 1A), was accompanied by chronic activation of autophagy, as measured by confocal imaging of the recruitment of ATG8/LC3 to autophagic vesicles (“LC3 puncta”) and loss of the specific autophagy receptor p62/SQSTM1, a protein that is selectively degraded by autophagy (Figure 1B) [42]. Of note, growth retardation and subsequent arrest of A431 tumor cells in response to the chronic energetic stress imposed by continuous exposure to metformin drastically up-regulated ATM activity and ATM protein accumulation. Indeed, fluorescence microscopic analyses revealed a massive accumulation of a uniform, nuclear signal of both total ATM (Figure 2A) and Ser-1981 phosphorylated ATM (Figure 2B). Furthermore, A431 cells chronically treated with metformin displayed flattened, giant, polynucleated morphology (Figure 2C). These findings not only reaffirm our earlier results and those reported by Duan et al. [40] on metformin-induced activation of the ATM/AMPKα pathway, but they additionally suggest that metformin-mimicked glucose restriction appears to reactivate the senescence program in cancer cells (Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Metformin lowers the threshold for stress-induced cellular senescence. Manuscript in preparation).
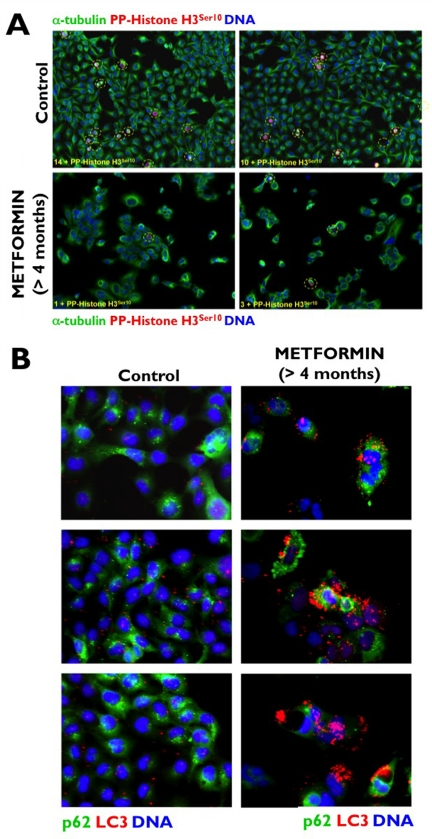
Figure 1. Chronic exposure to metformin suppresses the proliferative activity of human [A431] cancer cells (A) to enter a form of permanent cell-cycle arrest accompanied by the induction of “self-digesting” autophagy (B).
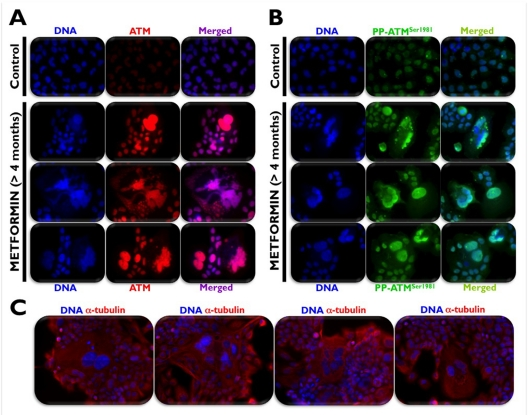
Figure 2. Chronic exposure to metformin activates the DNA damage sensor kinase ATM in poly-morphic, polyploidy [A431] cancer cells that contained giant nuclei and/or multiple varied size nuclei, including micronuclei (A, B). Chronically-treated cells demonstrated a large, spreading and flattening morphology, typical of senescent cells (C).
The above mentioned data suggest an attractive complementary strategy by which we might prevent or halt cancer that is functionally compatible with activation of the ATM pathway: the senescence process is dependent on ATM signaling, and senescence can be bypassed or suppressed by microinjection of kinase-dead constructs of ATM or by treatment with ATM inhibitors [42-46]. Metformin-treated A431 cell cultures typically revealed senescent cells with a damaged nucleus that, in some cases, appeared to evolve through progressive encircling of the nucleus by refringent components. Numerous dense particles (probably damaged components) and several autophagic vacuole-like structures of different sizes were observed within large cells displaying multiple nuclei, micronucleation, or lobulated nuclei. Chronic exposure of cancer cells to metformin might provoke permanent senescent-cell growth arrest as a result of a high macroautophagic activity that continuously targets many vital metabolic cancer cell components. This supports earlier studies demonstrating that, in contrast to the widely accepted antioxidant properties of the anti-aging polyphenol resveratrol, chronic culture of cancer cells with resveratrol initiates replication stress via activation of the ATM pathway and induces senescence associated with mitochondria-increased reactive oxygen species (ROS) levels [47]. This report and our current findings appear to functionally link the cell cycle with pro-oxidant/pro-senescent effects of anti-aging compounds in cancer cells [39]. In contrast, Halicka et al. found that the anti-oxidant activity of metformin is functionally linked to enhanced genomic stability, the pivotal mode of action underlying the anti-aging effects of metformin. At present, we cannot explain the apparent discrepancy of our results [12] and those of Duan et al. [40] with the data presented by Halicka et al. [39]. These contradictory hypotheses must be tested adequately before concluding the ultimate mechanism by which metformin exerts anti-cancer and/or anti-aging effects.
Metformin accelerates the onset of cellular senescence in human diploid fibroblasts (HDFs)
The onset of cellular senescence is thought to protect against the initiation of tumor formation in response to certain cellular stresses, including genotoxic and energetic stresses [48]. Environmental factors that place oxidative stress on cells promote the early onset of cellular senescence by significantly increasing the AMP/ATP ratio and activating stress pathways involving AMPK [49]. AMP/ATP ratios are significantly higher in senescent fibroblasts compared with young fibroblasts and, accordingly, in vitro senescence is accompanied by a marked elevation of AMPK activity. Indeed, ATP depletion in senescent fibroblasts is due to the dysregulation of glycolytic enzymes and a failure to maintain ATP levels, which finally leads to a drastic increase in cellular AMP. This, in turn, acts as a growth-suppressive signal that induces premature senescence [50]. Within this model, escaping fromcellular senescence and becoming immortal constitutes a crucial step in oncogenesis that most tumors require for ongoing proliferation [51].
The cumulative oxidative damage induced by growth in conditions that are hyperoxic (by the standard of living tissues) leads to the onset of senescence in HDFs and mouse embryo fibroblasts (MEFs). Indeed, when HDFs/MEFs are propagated in hypoxic conditions (1-3%) rather than the commonly used 20% oxygen, HDFs/MEFs avoid senescence; when grown in 20% oxygen, HDFs/MEFs rapidly accumulate DNA damage and eventually initiate a positive feedback loop of oxidative damage and growth arrest that masquerades as cellular senescence [51, 52]. Immortalized MEFs and mouse/human embryonic stem cells display higher glycolytic flux with reduced oxygen consumption and therefore present more resistance to oxidative damage than senescent cells. As such, they demonstrate the Warburg effect (enhanced glycolysis), which plays a causative role in cell immortality by protecting cells from senescence induced by oxidative damage [53-56]. Thus, it can be speculated that exogenous supplementation with metformin should increase the population-doubling potential of cultured HDFs and MEFs by preventing the accumulation of ROS and oxidative damage as suggested by Halicka et al. [39].
Conversely, recent experiments conducted in our laboratory have concluded that chronic exposure to millimolar concentrations of metformin (1 and 10 mmol/L) drastically reduces the lifespan of non-transformed HDFs by accelerating replicative cellular senescence (Figure 3A). Indeed, metformin exposure reduced cumulative population doublings by up to 70% in HDFs (Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Metformin lowers the threshold for stress-induced cellular senescence. Manuscript in preparation). Metformin's ability to accelerate the onset of replicative senescence was more significant in WI-38 fetal lung HDFs, which are highly sensitive to stress-induced pre- mature senescence [57]. BJ-1 fibroblasts required longer exposures to higher concentrations of metformin, as they are extremely resistant to hyperoxia and H2O2 [58, 59]. Although we did not explore the activation status of ATM in HDFs chronically exposed to metformin, it is reasonable to conclude that the metformin-lowered threshold for stress-induced senescence must be explained in terms of metformin-augmented oxidative damage in HDFs. In other words, metformin-accelerated replicative senescence might mostly rely on metformin's ability to establish a stronger DDR-dependent cell cycle arrest because exogenous supplementation with metformin appears to synergistically enhance hyperoxic culture-induced DNA damage and cellular senescence in cultured HDFs.
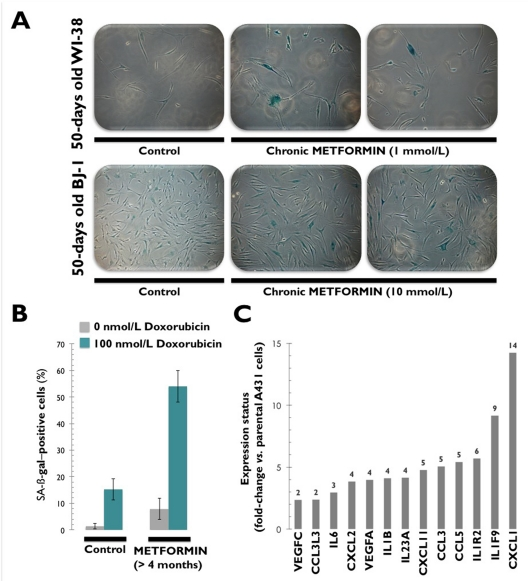
Figure 3. Chronic exposure to metformin accelerates the onset of replicative senescence in human [WI-38 and BJ-1] fibroblast cultures confirmed by senescence-associated β-galactosidase (SA-β-gal) staining (A). Chronic exposure to metformin sensitizes [MCF-7] cancer cells to the senescence program activated by the DNA-damaging drug doxorubicin (B). Chronic exposure to metformin transcriptionally activates a senescence-associated secretory phenotype (SASP) or senescence messaging secretome (SMS) involving the production of factors that reinforce the senescence arrest, alter the surrounding microenvironment, and trigger immune surveillance of the senescent cells.
2. Metformin sensitizes HDFs and cancer cells to DSB-induced cellular senescence
Doxorubicin is an anthracycline that indirectly causes DSBs, activates ATM-dependent signaling, and induces cell senescence at concentrations significantly lower than those required to induce apoptotic cell death. Treatment of cells with doxorubicin leads to the phosphorylation of Histone H2AX on Ser-139, with dependence on ATM for the initial response [60]. Treatment with doxorubicin also stimulates ATM autophosphorylation on Ser-1981 and the ATM-dependent phosphorylation of numerous effectors in the ATM-signaling pathway, including Chk2, in a ROS-dependent manner [60]. Because free radical scavengers have been shown to attenuate the accelerated senescence response triggered by treatment with a low concentration of doxorubicin in MCF-7 breast cancer cells [61-63], Halicka's hypothesis that metformin acts as an anti-oxidant that enhances genome stability via ATM inhibition [39] would dictate that metformin treatment must efficiently block doxorubicin-induced senescence [64]. We recently assessed whether metformin treatment can regulate the senescence-like growth arrest induced by doxorubicin in primary MEFs from wild-type (p53+/+) mice. Of note, exposure of MEFs to millimolar concentrations of metformin (1 and 10 mmol/L) augmented baseline senescence in doxorubicin-untreated control cultures and notably potentiated cell senescence triggered by doxorubicin-induced DNA damage (Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Metformin lowers the threshold for stress-induced cellular senescence. Manuscript in preparation). Furthermore, we maintained MCF-7 breast cancer cells (wild-type p53) in long-term uninterrupted subculture with metformin concentrations as high as 10 mmol/L for longer than 4 months and then challenged them with a senescence-inducing concentration of doxorubicin. Interestingly, the pre-conditioned MCF-7 cells became sensitized to senescence induction by low doses of doxorubicin (Figure 3B). We observed that sequential incubation with metformin, followed by 100 nmol/L of doxorubicin, produced a drastic change in the cellular response program. In response to doxorubicin-induced stress, wild-type MCF-7 cells showed low levels of SA-β-gal positive cells (~15%), and MCF-7/Metformin cells showed very high levels (~54%). This indicated a senescent-like phenotype without signs of apoptotic cell death. By activating AMPK, metformin treatment appears to induce a sensitizing stress that creates a metabolic cellular imbalance in favor of the pro-senescent effects induced by DNA damaging agents. Metformin's ability to accelerate the onset of cellular senescence in HDFs and enhance DNA damage-induced senescence might provide a rational approach to sensitizing pre-malignant and cancer cells to further stress induced by oncogenic stimuli.
3. Metformin impedes nuclear reprogramming of somatic cells to induced Pluripotent Stem Cells (iPSCs)
Somatic cells can be reprogrammed by the expression of four factors associated with pluripotency, the so-called “Yamanaka factors” OSKM (O = OCT4, S = SOX2, K = KLF4, M = and c-MYC) [65]. Several groups have observed that a DDR compatible with DNA replication-induced DNA damage is mounted upon the expression of the OSKM reprogramming factors [66-68]. This appears to be similar to what occurs during oncogene-induced senescence (OIS), when cell proliferation and transformation induced by oncogene activation in early tumorigenesis is restrained by cellular senescence, which results from the ATM-mediated DDR triggered by oncogene-induced DNA hyper-replication [69, 70]. However, it should be noted that expression of the four Yamanaka factors has been shown to result in the accumulation of 8-oxoguanine adducts in human fibroblasts, which are commonly the result of oxidative stress. Furthermore, c-MYC overexpression induces DNA damage in a mainly ROS-dependent rather than DNA replication-dependent manner [71, 72]. Therefore, the DNA damage occurring upon reprogramming may be caused not only by OSKM-driven aberrant replication but also through the generation of ROS, which can explain why reprogramming is significantly more efficient under either low oxygen conditions or in the presence of anti-oxidants such as vitamin C [73-76]. Vitamin C efficiently alleviates reprogramming-induced sense-cence (RIS) [66, 75-77], suggesting that antioxidants or other compounds that transiently inhibit senescence could be used to improve reprogramming efficiency. As such, the interplay between the expression of reprogramming factors and the activation of a p53-mediated [68, 78] DDR due to increased DNA replication and/or ROS creates a model in which to test the anti-oxidant (Halicka's findings [39]) or pro-senescent (Vazquez-Martin's findings [12]) effects of metformin in terms of enhanced or repressed reprogramming efficiency, respectively. Because reprogramming in the presence of pre-existing, but tolerated, DNA damage is aborted by the activation of DDR- and p53-dependent apoptosis [68], metformin's ability to reduce ATM activity should attenuate the p53 response to DNA damage (as in some preneoplastic lesions [79, 80]), resulting in accelerated somatic reprogramming. Using MEFs or mouse adult fibroblasts (MAFs), we recently tested the effect of metformin in reprogramming experiments. We found that treatment with metformin dose-dependently inhibited somatic cell reprogramming induced by the OSK stemness factors in MEFs. At 10 mmol/L metformin, iPSC formation was virtually undetectable in MEFs and in MAFs (Vazquez-Martin, Vellon L, Cufi S, Oliveras-Ferraros C, Quirós PM, Lopez-Otin C, Javier A. Menendez. Metformin impedes reprogramming of somatic cells into stem cells. Manuscript in preparation). Parallel experiments performed with human BJ-1 fibroblasts transduced with OSKM reprogramming factors produced effects similar to metformin in drastically inhibiting reprogramming (Vazquez-Martin, Vellon L, Cufi S, Oliveras-Ferraros C, Quirós PM, Lopez-Otin C, Javier A. Menendez. Metformin impedes reprogramming of somatic cells into stem cells). Manuscript in preparation). Because p53-mediated DDR limits reprogramming to ensure iPSC genomic integrity [68], it could be argued that these findings are consistent with a genome-protective effect of metformin, which in turn can reduce DNA replication stress [39]. However, metformin exposure was found to abolish highly efficient reprogramming upon abrogation of p53 in MAFs. Importantly, the observed effects on reprogramming efficiencies were not due to metformin-induced cell death of the starting somatic population but rather to the metabolic, pro-senescent effects exerted via AMPK activation [81, 82]. When metformin, which indirectly activates AMPK through effects on the mitochondria, was replaced with the small-molecule A-769662, which directly activates AMPK by mimicking both effects of AMP, including allosteric activation and inhibition of dephosphorylation [83-86], reprogramming efficiency was also drastically reduced.
Recent studies have indicated that somatic cells convert from an oxidative to glycolytic state when they are reprogrammed [87-90] and that the bioenergetic states of somatic cells appear to correlate with their reprogramming efficiencies. Furthermore, manipulating these bioenergetic changes can affect reprogramming, as the glycolysis inhibitor 2-deoxy-D-glucose [2-DG] decreases reprogramming, whereas the glycolysis stimulator D-fructose-6-phosphate (F6P) increases reprogramming of fibroblasts [89, 90]. Although further studies aimed at dissecting the exact mechanism of metformin action in regulating somatic cell reprogramming are needed, it is reasonable to suggest that impaired reprogramming following metformin treatment might result from compromised glycolysis and energy crisis, leading to the sustained activation of AMPK and the establishment of a senescent phenotype, a crucial roadblock for reprogramming [66, 75].
Metformin and cancer: Hastening the onset of stress-induced senescence to enhance protection against cancer
Understanding the metabolic changes associated with somatic cell reprogramming might shed light on the “metabolic transformation” that is required to support not only the increased biosynthetic needs of the tumor cell but also to enable the acquisition of stemness properties in cancer stem cells (CSCs). Many of the changes in cell metabolism that have been identified to be important in regulating somatic cell reprogramming and induced pluripotency also play roles in oncogenesis. On the other hand, reprogramming to a more dedifferentiated state occurs during tumor progression (i.e., the activation of an embryonic stem cell-like transcriptional program in differentiated adult cells may induce pathologic self-renewal/stemness characteristics of CSCs [91]) and might be favored by alterations in crucial tumor suppressors. Indeed, the stress mechanisms triggered by expression of the Yamanaka stemness factors, which ultimately lead to reprogramming, elicit the tumor suppressor pathways that naturally protect cells against the uncontrolled growth that occurs during tumorigenesis. Unsurprisingly, the cellular response to expression of the reprogramming factors or stem-cell-specific genes molecularly mimics the senescence response observed during OIS, thus emphasizing the parallels between RIS and OIS. In this scenario, if metformin treatment appears to reinforce RIS in somatic reprogramming experiments, we can then infer that metformin treatment should improve cells' ability to establish a more efficient senescence response in pre-malignant and malignant tissues. Many tumor cells appear to have developed mechanisms to reduce AMPK activation and therefore escape from its growth-arrest and tumor-suppressor effects. In fact, more aggressive tumors exhibit reduced signaling via the AMPK pathway, and an inverse relationship exists between the AMPK activation status with histological grade and metastasis [92-94]. As such, metformin-induced energy crisis and, therefore, AMPK (re-)activation, may functionally disrupt the deleterious connection between pluripotency and oncogenic transformation. In fact, Struhl's team discovered that metformin treatment can selectively kill the chemotherapy-resistant subpopulation of CSCs in genetically distinct types of breast cancer cell lines [95, 96]. Our own group has confirmed that treatment with metformin can suppress the self-renewal and proliferation of cancer stem/progenitor cells in HER2 gene-amplified breast carcinomas cells refractory to HER2-targeted drugs [97, 98]. The central mechanisms through which metformin exposure blocks the ontogenesis of the CSC molecular signature have begun to be elucidated in cultured cancer cells, including alterations of epithelial-to-mesenchymal transition drivers/effectors, tumor-suppressor miRNAs and oncomiRs [99-101]. Metformin's ability to oppose reprogramming of cell energy metabolism from oxidative mitochondria toward an alternative ATP-generating glycolytic metabotype may be sufficient to inhibit the network required for the establishment and maintenance of stem cell pluripotency and self-renewal imposed by certain oncogenic stimuli in the right cellular context [102].
Recently, evidence has emerged that the DDR is one of the earliest events that impedes the multistep progression of human epithelial carcinomas to invasive malignancy. DNA damage can be due to a variety of factors, such as telomere dysfunction or oncogene-induced replication stress [103-105]. Consequently, there is a strong selective pressure for mutation in DDR components because DNA damage checkpoints act as native blockades against the acquisition of invasive/metastatic properties during malignant transformation [103, 105]. Epithelial cells within pre-malignant lesions with markers of senescence maintain an intact response to cellular stress and are less likely to develop subsequent tumors. Accordingly, the presence of functional pro-senescence mechanisms is the most accurate predictor of recurrence and progression of premalignant lesions in situ (e.g., in ductal carcinoma in situ [DCIS] of the breast) to biologically aggressive invasive carcinomas (e.g., basal-like breast carcinomas) [106]. Because the DDR is the major innate tumor suppressor barrier in early human tumorigenesis, selective activation of DDR surveillance mechanisms may therefore directly contribute to metformin's cancer preventive effects. Proliferative invasive cancer cells with activated oncogenes acquire mechanisms to suppress senescence in the early stages of cancer pathogenesis (e.g., in situ lesions). Organisms in which cells fail to undergo senescence die prematurely of cancer [107]. Therefore, activating the program of senescence in tumor cells is an attractive approach to cancer treatment [108, 109] and may help to explain the differential impact of metformin on cancer incidence in non-prone and cancer-prone animal models and perhaps also in cancer-prone individuals. It remains to be clearly defined whether metformin's ability to strongly activate the ATM-regulated DDR checkpoint is the critical event that prevents neoplastic epithelium from progressing unimpeded into invasive cancer in individuals without type 2 diabetes. However, reduced cancer risk in type 2 diabetic patients taking metformin can be explained in terms of metformin's ability to activate DNA damage-like signaling that induces specific senescence-like growth inhibition of pre-malignant or malignant cells without altering the normal function of non-neoplastic tissues. We are currently developing a pre-clinical framework for pro-senescence, metformin-based anti-cancer therapies by evaluating metformin's effects on DCIS xenografts during their spontaneous transition to invasive cancer lesions [110-112]. It would also be relevant to evaluate whether metformin facilitates the “accelerated senescence” triggered in normal cells by the expression of mutated, transforming versions of oncogenes (e.g., Ras or Raf) and by some other forms of supraphysiological mitogenic signaling irrespective of senescence-inhibiting adaptations (e.g., inactivation of p53) [69, 113, 114]. In a clinical scenario, it would be interesting to test whether metformin can significantly increase senescence in premalignant lesions of the skin, the lung, the pancreas, the liver or the breast. Additionally, forthcoming studies should evaluate metformin's effects in clinical scenarios in which senescence has been recognized to have positive effects on organ maintenance. Senescence limits pathological responses to either acute forms of injury such as fibrotic scarring in response to chemically induced liver injury [98, 115, 116] or to chronic viral infections such as hepatitis C virus (HCV) with or without concomitant human immunodeficiency virus (HIV) infection. Indeed, HCV infection increases rates of hepatocellular carcinoma via the accumulation of senescent hepatocytes in human liver [117].
Metformin: Lowering the threshold for stress-induced senescence to limit cancer development and delay aging-associated disorders
Metformin's ability to enhance senescence in established premalignant disease or in fully malignant disease is a largely unexplored mechanism that may explain why reductions in cancer mortality related to metformin use are similar in magnitude to reductions in cancer incidence. This suggests that the anti-cancer effects of metformin largely depend on (or are restricted to) its preventive effects [118]. The most widely accepted interpretation for the biological function of cellular senescence is that it serves as a mechanism for restricting cancer progression. Based on this, escaping from cellular senescence and becoming immortal constitute a required additional step in the progression of oncogenesis [51, 52]. Recent studies have suggested that the accumulation of ROS and oxidative damage are commonly involved in culture stress- or oncogene-induced cellular senescence. Increasing accumulation of ROS is observed during replicative senescence, and the replicative potential of MEFs and HDFs is significantly higher under low oxygen conditions. As such, the ability of immortalized cells, including embryonic stem cells (ESCs), iPSCs and CSCs, to buffer oxidative stress may be pivotal for explaining their immortality [53-56; 87-90]. Enhanced glycolysis actively protects cells from senescence induced by oxidative stress [53-56], a metabolic protection that appears to causally contribute to the maintenance of the self-renewal capacity of stem cells [87-90]. In fact, the enhanced glycolysis of the Warburg effect is a crucial metabolic feature that helps cancer cells bypass senescence, and this may provide indirect evidence that metformin's primary target is the immortalizing step during tumorigenesis. In other words, if enhanced glycolysis is necessary and sufficient to enable indefinite proliferation (i.e., immortalization) very early during multi-step carcinogenesis in vivo, then metformin's ability to inhibit glucose flux while simultaneously stimulating lactate/pyruvate flux and mitochondrial biogenesis must cause ATP depletion accompanied by a drastic increase in cellular AMP, which is expected to induce premature senescence [50]. Many tumor cells retain the ability to senesce in response to DNA-damaging drugs in culture and in vivo. Because of this, metformin-accelerated replicative senescence due to a stronger DDR-dependent cell cycle arrest may underlie metformin's ability to increase the rate of pathological complete response (pCR) in neoadjuvant chemotherapy in diabetic patients with breast cancer [119] and to promote tumor regression and prevent relapse when combined with suboptimal doses of chemotherapy in animal models [96].
Senescent cells accumulate in various tissues and organs with aging and have been hypothesized to disrupt tissue structure and function [120-122]. Cellular senescence halts the proliferation of damaged or dysfunctional cells, thus functioning as a pivotal mechanism to constrain the malignant progression of tumor cells [123, 124]. Indeed, upon the aberrant activation of oncogenes, normal cells can enter the cellular senescence program, a state of stable cell-cycle arrest that represents an important barrier against tumor development in vivo [125]. Senescent cells communicate with their environment by secreting various cytokines and growth factors, and this “secretory phenotype” has been reported to have pro- as well as anti-tumorigenic effects [126-130]. In this regard, it is remarkable that metformin-induced chronic activation of ATM signaling in A431 epidermoid cancer cells is accompanied by the increased expression of a wide variety of cyto-/chemokines (e.g., IL6, IL1B, CCL3, CCL5, IL1F9 or CXCL11), as measured by Agilent's whole human genome arrays (Figure 3C) (Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Metformin lowers the threshold for stress-induced cellular senescence. Manuscript in preparation). Cellular senescence is often the result of ATM sensing of nuclear DNA damage fueling chronic DDR, and upstream elements of the DDR signaling cascade are necessary to successfully establish a SASP [131]. Thus, it might be tempting to suggest that an AMPK-induced energy crisis imposed by continuous exposure to metformin is a novel regulator of the DDR-SASP axis that reinforces the senescent phenotype.
Two recent landmark studies [117, 132] help clarify the apparently counterintuitive pro-senescent activity of the anti-aging biguanide metformin [133-138]. Kang et al. [117] have confirmed that pre-malignant senescent hepatocytes secrete chemo- and cytokines and are subject to immune-mediated clearance (“senescence surveillance”) that strictly depends on an intact CD4+ T-cell-mediated adaptive immune response. Accordingly, impaired immune surveillance of pre-malignant senescent hepatocytes results in the development of hepatocellular carcinomas, thus showing that senescence surveillance is important for tumor suppression in vivo by mounting specific immune responses against antigens expressed in pre-malignant senescent cells [117]. The molecular mechanisms presented here suggest that metformin's use of the senescence program may significantly diminish the number of cells that can escape senescence. By activating the chemopreventive ATM-dependent DDR, metformin-treated cells might be sensitized for damage signaling, and metformin treatment might mimic the precancerous stimulus that generates a barrier against carcinogenesis. Because energy metabolism is being recognized as a/the critical pathway regulating the immortalization-to-senescence transition, metformin-based therapies based on the induction of senescence would prevent the generation of CSCs. Concurrently, metformin treatment could lead to a significant alteration of “cancer immunoediting” (e.g., via restoration of MHC-related pathways) to impede tumor cell escape from immunosurveillance. The net effect of metformin treatment should be, therefore, a significant decrease in the accumulation of dysfunctional, pre-malignant cells in tissues, including those with the ability to initiate tumors (i.e., CSCs).
Whether senescent cells are causally implicated in age-related dysfunction and whether their removal is beneficial have remained unknown until now. Baker et al. [132] have nicely answered this pivotal question by designing a transgenic strategy for the clearance of senescent cells in mice. Using a BubR1 progeroid mouse background designed for inducible elimination of p16Ink4a-positive senescent cells, the authors demonstrate that in tissues in which p16Ink4a contributes to the acquisition of age-related pathologies (i.e., adipose tissue, skeletal muscle and eye), life-long removal of p16Ink4a-expressing cells significantly delays onset of these pathologies. Furthermore, late-life clearance notably attenuates progression of already established age-related disorders [132]. If metformin's ability to prevent escape from the senescence anti-tumor barrier is accompanied by an enhanced clearance of pre-malignant/tumor senescent cells, including those exhibiting tumor-initiating ability (i.e., CSCs), the net effect of the tissue sweeper functioning of metformin will be to halt the malignant/metastatic progression of pre-malignant/tumor senescent cells while preventing or delaying tissue dysfunction, thus extending the human lifespan.
Note
In addition to SA-β-gal activity, senescence has been previously linked to induction of ROS production, which is believed to be necessary for maintenance of the senescence phenotype [139, 140]. ROS also play a pivotal role in promoting SA-β-gal activity following radiation-induced DNA damage. Accordingly, inhibition of ROS using N-acetyl cysteine (NAC) following radiation treatment drastically decreases senescence in tumor cells with normally high levels of radiation-induced SA-β-gal. In accordance with our current observations and taking advantage of earlier studies suggesting that metformin treatment induces ROS in certain cellular backgrounds [141], Skinner et al [142] have recently confirmed that enhanced rather than reduced ROS and SA-β-gal activity occurred when metformin was concurrently added to radiation in p53-deficient tumor cells. Skinner's findings that metformin treatment can overcome locoregional treatment failure in head and neck carcinomas by reinforcing the radiation-induced cellular senescence, which clinically translates into a significantly improved survival in patients taking metformin, strongly support our hypothesis that metformin's ability to reinforce the establishment of accelerated senescence may function as an effective barrier to tumor growth and disease recurrence.
Acknowledgments
Work in the laboratory of Javier A. Menendez is supported by the Instituto de Salud Carlos III (Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain, Grants CP05-00090, PI06-0778 and RD06-0020-0028), the Fundación Científica de la Asociación Española Contra el Cáncer (AECC, Spain) and by the Ministerio de Ciencia e Innovación (SAF2009-11579, Plan Nacional de I+D+ I, MICINN, Spain). Alejandro Vazquez-Martin is the recipient of a Sara Borrell post-doctoral contract (CD08/00283, Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria -FIS-, Spain). Sílvia Cufí is the recipient of a Research Fellowship (Formación de Personal Investigador, FPI) from the Ministerio de Ciencia e Innovación (MICINN, Spain).
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Cheema AK, Timofeeva O, Varghese R, Dimtchev A, Shiekh K, Shulaev V, Suy S, Collins S, Ressom H, Jung M, Dritschilo A. Integrated analysis of ATM mediated gene and protein expression impacting cellular metabolism. J Proteome Res. 2011; 10: 2651 -2657. [PubMed] .
- 2. Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011; 30: 546 -55. [PubMed] .
- 3. Krüger A and Ralser M. ATM is a redox sensor linking genome stability and carbon metabolism. Sci Signal. 2011; 4: pe17 [PubMed] .
- 4. Perry JJ and Tainer JA. All stressed out without ATM kinase. Sci Signal. 2011; 4: pe18 [PubMed] .
- 5. Lovejoy CA and Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst). 2009; 8: 1004 -1008. [PubMed] .
- 6. Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signaling and cancer. Nat Rev Mol Cell Biol. 2008; 9: 759 -769. [PubMed] .
- 7. Sun Y, Connors KE, Yang DQ. AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol Cell Biochem. 2007; 306: 239 -245. [PubMed] .
- 8. GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group, Wellcome Trust Case Control Consortium 2, Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ, Burch L, Carr F, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011; 43: 117 -120. [PubMed] .
- 9. Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One. 2008; 3: e2009 [PubMed] .
- 10. Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J, Tsakiridis T. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010; 78: 221 -229. [PubMed] .
- 11. Birnbaum MJ and Shaw RJ. Genomics: Drugs, diabetes and cancer. Nature. 2011; 470: 338 -339. [PubMed] .
- 12. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Martin-Castillo B, Menendez JA. Metformin activates an Ataxia Telangiectasia Mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011; 10: 1499 -501. [PubMed] .
- 13. Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNAdamage response in senescent cells. Cell Cycle. 2009; 8: 4112 -4118. [PubMed] .
- 14. Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000; 97: 10389 -10394. [PubMed] .
- 15. Shi Y, Venkataraman SL, Dodson GE, Mabb AM, LeBlanc S, Tibbetts RS. Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc Natl Acad Sci U S A. 2004; 101: 5898 -5903. [PubMed] .
- 16. Dodson GE and Tibbetts RS. DNA replication stress-induced phosphorylation of cyclic AMP response element-binding protein mediated by ATM. J Biol Chem. 2006; 281: 1692 -1697. [PubMed] .
- 17. Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010; 9: 1057 -64. [PubMed] .
- 18. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Martin-Castillo B, Menendez JA. Metformin and energy metabolism in breast cancer: from insulin physiology to tumour-initiating stem cells. Curr Mol Med. 2010; 10: 674 -691. [PubMed] .
- 19. Arnould T, Vankoningsloo S, Renard P, Houbion A, Ninane N, Demazy C, Remacle J, Raes M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002; 21: 53 -63. [PubMed] .
- 20. Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J Biol Chem. 2010; 285: 142 -152. [PubMed] .
- 21. De Rasmo D, Signorile A, Papa F, Roca E, Papa S. cAMP/Ca2+ response element-binding protein plays a central role in the biogenesis of respiratory chain proteins in mammalian cells. IUBMB Life. 2010; 62: 447 -52. [PubMed] .
- 22. Than TA, Lou H, Ji C, Win S, Kaplowitz N. Role of cAMP-responsive element-binding protein (CREB)-regulated transcription coactivator 3 (CRTC3) in the initiation of mitochondrial biogenesis and stress response in liver cells. J Biol Chem. 2011; 286: 22047 -22054. [PubMed] .
- 23. López-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol. 2008; 43: 813 -819. [PubMed] .
- 24. Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006; 55: 120 -127. [PubMed] .
- 25. Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol. 2006; 101: 1685 -1692. [PubMed] .
- 26. Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK, Shapiro CL, Chen CS. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer cells by a novel AMPK activator. J Biol Chem. 2011; Sep 14. [Epub ahead of print] .
- 27. De Berardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7: 11 -20. [PubMed] .
- 28. Jones RG and Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009; 23: 537 -548. [PubMed] .
- 29. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324: 1029 -1033. [PubMed] .
- 30. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646 -674. [PubMed] .
- 31. Beeson CC, Beeson GC, Schnellmann RG. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem. 2010; 404: 75 -81. [PubMed] .
- 32. Jose C, Hébert-Chatelain E, Bellance N, Larendra A, Su M, Nouette-Gaulain K, Rossignol R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim Biophys Acta. 2011; 1807: 707 -718. [PubMed] .
- 33. Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase: development of the energy sensor concept. J Physiol. 2006; 574: 7 -15. [PubMed] .
- 34. Ristow M and Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010; 45: 410 -418. [PubMed] .
- 35. Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C, Cohen P, Longo VD. Fasting and cancer treatment in humans: A case series report. Aging. 2009; 1: 988 -1007. [PubMed] .
- 36. Raffaghello L, Safdie F, Bianchi G, Dorff T, Fontana L, Longo VD. Fasting and differential chemotherapy protection in patients. Cell Cycle. 2010; 9: 4474 -4476. [PubMed] .
- 37. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011; 2: 222 -233. [PubMed] .
- 38. Lee C and Longo VD. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene. 2011; 30: 3305 -3316. [PubMed] .
- 39. Halicka HD, Zhao H, Li J, Traganos F, Zhang S, Lee M, Darzynkiewicz Z. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging. 2011; 3: 1028 -1038. [PubMed] .
- 40. Duan X, Ponomareva L, Veeranki S, Choubey D. IFI16 induction by glucose restriction in human fibroblasts contributes to autophagy through activation of the ATM/AMPK/p53 pathway. PLoS One. 2011; 6: e19532 [PubMed] .
- 41. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007; 67: 6745 -6752. [PubMed] .
- 42. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One. 2009; 4: e6251 [PubMed] .
- 43. Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999; 283: 1321 -1325. [PubMed] .
- 44. d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003; 426: 194 -198. [PubMed] .
- 45. Crescenzi E, Palumbo G, de Boer J, Brady HJ. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: implications for chemotherapy. Clin Cancer Res. 2008; 14: 1877 -1887. [PubMed] .
- 46. Zajkowicz A and Rusin M. The activation of the p53 pathway by the AMP mimetic AICAR is reduced by inhibitors of the ATM or mTOR kinases. Mech Ageing Dev. 2011; Sep 21. [Epub ahead of print] .
- 47. Heiss EH, Schilder YD, Dirsch VM. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J Biol Chem. 2007; 282: 26759 -26766. [PubMed] .
- 48. Campisi J. Senescent cells, tumor supression, and organismal aging: good citizens, bad neighbors. Cell. 2005; 120: 513 -522. [PubMed] .
- 49. Wang W, Yang X, López de Silanes I, Carling D, Gorospe M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem. 2003; 278: 27016 -27023. [PubMed] .
- 50. Zwerschke W, Mazurek S, Stöckl P, Hütter E, Eigenbrodt E, Jansen-Dürr P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J. 2003; 376: 403 -411. [PubMed] .
- 51. Wright WE and Shay JW. Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr Opin Genet Develop. 2011; 11: 98 -103. .
- 52. Ben-Porath I and Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004; 113: 8 -13. [PubMed] .
- 53. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005; 65: 177 -185. [PubMed] .
- 54. Kondoh H, Lleonart ME, Bernard D, Gil J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol Histopathol. 2007; 22: 85 -90. [PubMed] .
- 55. Kondoh H, Lleonart ME, Nakashima Y, Maruyama T, Yokode M, Tanaka M, Bernard D, Gil J. A common metabolic profile shared between murine ES cells and primary cells bypassing senescence. Med Hypotheses Res. 2008; 4: 29 -36. .
- 56. Kondoh H. Cellular life span and the Warburg effect. Exp Cell Res. 2008; 314: 1923 -1928. [PubMed] .
- 57. Toussaint O, Dumont P, Remacle J, Dierick JF, Pascal T, Frippiat C, Magalhaes JP, Zdanov S, Chainiaux F. Stress-induced premature senescence or stress-induced senescence-like phenotype: one in vivo reality, two possible definitions? Scientific World Journal. 2002; 2: 230 -247. [PubMed] .
- 58. Lorenz M, Saretzki G, Sitte N, Metzkow S, von Zglinicki T. BJ fibroblasts display high antioxidant capacity and slow telomere shortening independent of hTERT transfection. Free Radic Biol Med. 2001; 31: 824 -831. [PubMed] .
- 59. de Magalhães JP, Chainiaux F, Remacle J, Toussaint O. Stress-induced premature senescence in BJ and hTERT-BJ1 human foreskin fibroblasts. FEBS Lett. 2002; 523: 157 -162. [PubMed] .
- 60. Kurz EU, Douglas P, Lees-Miller SP. 2004 Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004; 279: 53272 -53281. [PubMed] .
- 61. Elmore LW, Rehder CW, Di X, McChesney PA, Jackson-Cook CK, Gewirtz DA, Holt SE. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002; 277: 35509 -35515. [PubMed] .
- 62. Song YS, Lee BY, Hwang ES. Dinstinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech Ageing Dev. 2005; 126: 580 -590. [PubMed] .
- 63. Di X, Shiu RP, Newsham IF, Gewirtz DA. Apoptosis, autophagy, accelerated senescence and reactive oxygen in the response of human breast tumor cells to adriamycin. Biochem Pharmacol. 2009; 77: 1139 -1150. [PubMed] .
- 64. Sliwinska MA, Mosieniak G, Wolanin K, Babik A, Piwocka K, Magalska A, Szczepanowska J, Fronk J, Sikora E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech Ageing Dev. 2009; 130: 24 -32. [PubMed] .
- 65. Takahashi K and Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126: 663 -676. [PubMed] .
- 66. Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, Vallier L, Gil J. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009; 23: 2134 -2139. [PubMed] .
- 67. Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Belmonte JC. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009; 460: 1140 -1144. [PubMed] .
- 68. Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009; 460: 1149 -1153. [PubMed] .
- 69. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre' M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d'Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006; 444: 638 -642. [PubMed] .
- 70. Reddy JP, Peddibhotla S, Bu W, Zhao J, Haricharan S, Du YC, Podsypanina K, Rosen JM, Donehower LA, Li Y. Defining the ATM-mediated barrier to tumorigenesis in somatic mammary cells following ErbB2 activation. Proc Natl Acad Sci U S A. 2010; 107: 3728 -3733. [PubMed] .
- 71. Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002; 9: 1031 -1044. [PubMed] .
- 72. Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005; 24: 8038 -8050. [PubMed] .
- 73. Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009; 460: 1145 -1148. [PubMed] .
- 74. Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009; 5: 237 -241. [PubMed] .
- 75. Banito A and Gil J. Induced pluripotent stem cells and senescence: learning the biology to improve the technology. EMBO Rep. 2010; 11: 353 -359. [PubMed] .
- 76. Esteban MA, Wang T, Qin B, Yang J, Qin D, Cai J, Li W, Weng Z, Chen J, Ni S, Chen K, Li Y, Liu X, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 2010; 6: 71 -79. [PubMed] .
- 77. Menendez JA, Vellon L, Oliveras-Ferraros C, Cufí S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle. 2011; 10: 3658 -3677. [PubMed] .
- 78. Choi YJ, Lin CP, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Bennett MJ, Chen C, Ozturk A, Hicks GG, Hannon GJ, He L. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol. 2011; 13: 1353 -1360. [PubMed] .
- 79. Silins I, Finnberg N, Ståhl A, Högberg J, Stenius U. Reduced ATM kinase activity and an attenuatedp53 response to DNA damage in carcinogen-induced preneoplastic hepatic lesions in the rat. Carcinogenesis. 2001; 22: 2023 -2031. [PubMed] .
- 80. Cao L, Kim S, Xiao C, Wang RH, Coumoul X, Wang X, Li WM, Xu XL, De Soto JA, Takai H, Mai S, Elledge SJ, Motoyama N, Deng CX. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006; 25: 2167 -2177. [PubMed] .
- 81. Phadke M, Krynetskaia N, Mishra A, Krynetskiy E. Accelerated cellular senescence phenotype of GAPDH-depleted human lung carcinoma cells. Biochem Biophys Res Commun. 2011; 411: 409 -415. [PubMed] .
- 82. Sung JY, Woo CH, Kang YJ, Lee KY, Choi HC. AMPK induces vascular smooth muscle cell senescence via LKB1 dependent pathway. Biochem Biophys Res Commun. 2011; 413: 143 -148. [PubMed] .
- 83. Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006; 3: 403 -416. [PubMed] .
- 84. Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007; 282: 32539 -32548. [PubMed] .
- 85. Göransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007; 282: 32549 -32560. [PubMed] .
- 86. Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008; 412: 211 -221. [PubMed] .
- 87. Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010; 28: 721 -733. [PubMed] .
- 88. Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA 4th, Ramalho-Santos J, Van Houten B, Schatten G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One. 2011; 6: e20914 [PubMed] .
- 89. Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011; 14: 264 -271. [PubMed] .
- 90. Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, Berggren WT, Zhang K, Evans RM, Siuzdak G, Belmonte JC. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2011; Nov 8. doi: 10.1038/cr.2011.177. [Epub ahead of print] .
- 91. Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008; 2: 333 -344. [PubMed] .
- 92. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011; 25: 1895 -1908. [PubMed] .
- 93. Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, Fleming S, Thompson AM. Histological evaluation of AMPKsignalling in primarybreast cancer. BMC Cancer. 2009; 9: 307 [PubMed] .
- 94. Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009; 8: 3679 -3683. [PubMed] .
- 95. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009; 69: 7507 -7511. [PubMed] .
- 96. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011; 71: 3196 -3201. [PubMed] .
- 97. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drugmetforminsuppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2011; 126: 355 -364. [PubMed] .
- 98. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metforminagainst TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010; 9: 4461 -4468. [PubMed] .
- 99. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metforminregulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010; 9: 3807 -3814. [PubMed] .
- 100. Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, Menendez JA. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFβ-induced oncomiR miRNA-181a. Cell Cycle. 2011; 10: 1144 -1151. [PubMed] .
- 101. Vazquez-Martin A, López-Bonetc E, Cufí S, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. Repositioning chloroquine and metformin to eliminate cancer stem cell traits in pre-malignant lesions. Drug Resist Updat. 2011; 14: 212 -223. [PubMed] .
- 102. Abollo-Jiménez F, Jiménez R, Cobaleda C. Physiological cellular reprogramming and cancer. Semin Cancer Biol. 2010; 20: 98 -106. [PubMed] .
- 103. Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Ørntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434: 864 -870. [PubMed] .
- 104. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005; 434: 907 -913. [PubMed] .
- 105. Collado M and Serrano M. The senescent side of tumor suppression. Cell Cycle. 2005; 4: 1722 -1724. [PubMed] .
- 106. Gauthier ML, Berman HK, Miller C, Kozakeiwicz K, Chew K, Moore D, Rabban J, Chen YY, Kerlikowske K, Tlsty TD. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007; 12: 479 -491. [PubMed] .
- 107. Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res. 2007; 35: 7475 -7484. [PubMed] .
- 108. Lleonart ME, Artero-Castro A, Kondoh H. Senescence induction; a possible cancer therapy. Mol Cancer. 2009; 8: 3 [PubMed] .
- 109. Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011; 11: 503 -511. [PubMed] .
- 110. Tait LR, Pauley RJ, Santner SJ, Heppner GH, Heng HH, Rak JW, Miller FR. Dynamic stromal-epithelial interactions during progression of MCF10DCIS.com xenografts. Int J Cancer. 2007; 120: 2127 -2134. [PubMed] .
- 111. Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, Bauerlein EL, Hahn WC, Gelman RS, Allred C, Bissell MJ, Schnitt S, Polyak K. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008; 13: 394 -406. [PubMed] .
- 112. Behbod F, Kittrell FS, LaMarca H, Edwards D, Kerbawy S, Heestand JC, Young E, Mukhopadhyay P, Yeh HW, Allred DC, Hu M, Polyak K, Rosen JM, Medina D. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 2009; 11: R66 [PubMed] .
- 113. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593 -602. [PubMed] .
- 114. Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007; 21: 43 -48. [PubMed] .
- 115. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008; 134: 657 -667. [PubMed] .
- 116. Zender L and Rudolph KL. Keeping your senescent cells under control. Aging. 2009; 1: 438 -441. [PubMed] .
- 117. Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011; doi:10.1038/nature10599 .
- 118. Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev Res (Phila). 2010; 3: 1060 -1065. [PubMed] .
- 119. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009; 27: 3297 -3302. [PubMed] .
- 120. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005; 120: 513 -522. [PubMed] .
- 121. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6: 2853 -2868. [PubMed] .
- 122. Rodier F and Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192: 547 -556. [PubMed] .
- 123. Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev. 2011; 21: 107 -112. [PubMed] .
- 124. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010; 2463 -2479. [PubMed] .
- 125. Narita M and Lowe SW. Senescence comes of age. Nature Med. 2005; 11: 920 -922. [PubMed] .
- 126. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001; 98: 12072 -12077. [PubMed] .
- 127. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445: 656 -660. [PubMed] .
- 128. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008; 133: 1006 -1018. [PubMed] .
- 129. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008; 133: 1019 -1031. [PubMed] .
- 130. Kuilman T and Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009; 9: 81 -94. [PubMed] .
- 131. Fumagalli M and d'Adda di Fagagna F. SASPense and DDRama in cancer and ageing. Nat Cell Biol. 2009; 11: 921 -923. [PubMed] .
- 132. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479: 232 -236. doi: 10.1038/nature10600. [PubMed] .
- 133. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect ofmetforminon life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005; 40: 685 -693. [PubMed] .
- 134. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, Semenchenko AV. Metforminslows down aging and extends life span of female SHR mice. Cell Cycle. 2008; 7: 2769 -2773. [PubMed] .
- 135. Anisimov VN. Metforminfor aging and cancer prevention. Aging. 2010; 2: 760 -774. [PubMed] .
- 136. Anisimov VN, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Egormin PA, Yurova MV, Rosenfeld SV, Semenchenko AV, Kovalenko IG, Poroshina TE, Berstein LM. Gender differences inmetformineffect on aging, life span and spontaneous tumorigenesis in 129/Sv mice. Aging. 2010; 2: 945 -958. [PubMed] .
- 137. Anisimov VN, Berstein LM, Popovich IG, Zabezhinski MA, Egormin PA, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Kovalenko IG, Poroshina TE. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging. 2011; 3: 148 -157. [PubMed] .
- 138. Menendez JA, Cufí S, Oliveras-Ferraros C, Vellon L, Joven J, Vazquez-Martin A. Gerosuppressantmetformin: less is more. Aging. 2011; 3: 348 -362. [PubMed] .
- 139. Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, Aaronson SA. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002; 21: 2180 -2188. [PubMed] .
- 140. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010; 6: 347 [PubMed] .
- 141. Anedda A, Rial E, González-Barroso MM. Metformin induces oxidative stress in white adipocytes and raises uncoupling protein 2 levels. J Endocrinol. 2008; 199: 33 -40. [PubMed] .
- 142. Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK, Myers JN. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2011; Nov 16. [Epub ahead of print] .