




Introduction
Mitochondria are essential eukaryotic organelles that participate in processes such as cellular energy production, cell signaling and apoptosis [1-3]. The production of cellular energy by mitochondria produces as a byproduct reactive oxygen species (ROS). While the effect of ROS on the accumulation of mutations in the mitochondrial genome is still under debate, ROS have been shown to promote protein oxidation and consequently misfolding and/or unfolding of these proteins inside mitochondria [4, 5]. The resulting proteotoxic stress has been shown to increase with age and to participate in several age-related disorders, such as neurodegenerative diseases [6]. To maintain mitochondrial proteostasis and, hence, mitochondrial function, it is important that damaged proteins are eliminated by mitochondrial proteases and that mitochondrial chaperones assist in the folding of nascent proteins. Another challenge that mitochondria face is the fact that the assembly of complexes of the Electron Transport Chain (ETC) in the inner mitochondrial membrane (IMM) requires a proper stoichiometric ratio of their subunits. Hence, misfolding of one subunit of an ETC complex results in the failure to assemble the entire complex. Consequently, the other subunits will accumulate in the mitochondrial matrix and thereby compromise mitochondrial proteostasis. Finally, ETC components are encoded by the mitochondrial and the nuclear genome. In order to maintain their respective stoichiometric ratios, the expression of mitochondrial and nuclear encoded proteins therefore has to be properly coordinated [7].
Several mitochondrial stress response pathways that maintain mitochondrial function have been described. One or more of these pathways are activated, depending on the extent to which mitochondrial proteostasis is compromised or mitochondria are damaged. For example, severely damaged mitochondria, which have irremediably lost their membrane potential, are eliminated from the functional mitochondrial network by mitochondrial-specific autophagy (mitophagy) (see for review [8]). The accumulation of unfolded or misfolded mitochondrial proteins activates the mitochondrial unfolded protein response (UPRmt). This response leads to the increased production and import into mitochondria of chaperones (such as HSP60 and HSP70) and proteases (such as ClpP), which help misfolded proteins to fold properly or cause their degradation, respectively (see for review [9]). Finally, the mitochondrial hyperfusion response has recently been described in mammalian cells and in C. elegans. Various forms of stress, such as a decrease in complex IV activity, the inhibition of cytosolic protein synthesis or starvation, induce mitochondrial hyperfusion in order to maintain mitochondrial ATP production [10-12].
The human LRPPRC gene encodes a leucine-rich pentatricopeptide repeat containing protein that is imported into mitochondria and that is mutated in patients with French Canadian Leigh Syndrome, a neurodegenerative disorder associated with complex IV deficiency [13]. Inside the mitochondrial matrix, the LRPPRC protein is part of a ribonucleoprotein complex that post-transcriptionally controls the expression of specific mitochondrial mRNAs such as the mRNA coding for COX I, a component of complex IV [14, 15]. We have previously shown that reducing LRPPRC function in mammalian tissue culture cells or reducing the function of the LRPPRC-like gene mma-1 (mma, mitochondrial morphology-abnormal) in C. elegans leads to a decrease in the level of COX I and, consequently, a decrease in complex IV activity [11]. This decrease in complex IV activity is compensated by an evolutionarily conserved mitochondrial hyperfusion response [11]. COX I is one of three mitochondria-encoded subunits of complex IV. The remaining 11 subunits in mammals and 6 subunits in C. elegans are nuclear-encoded [16, 17]. Therefore, we hypothesized that the reduction of COX I protein level upon LRPPRC mma-1 knock-down might cause an imbalance between nuclear- and mitochondria-encoded subunits and, hence, trigger UPRmt. Here we report that in addition to triggering a mitochondrial hyperfusion response, LRPPRC mma-1 knock-down also triggers UPRmt in both mammalian cell cultures and C. elegans. We propose that these two responses act together to maintain and restore mitochondrial function, in response to decreased complex IV activity.
Results
LRPPRC siRNA leads to an imbalance between mitochondria-encoded and nuclear-encoded sub-units of complex IV and triggers UPRmt
Inactivation of LRPPRC results in a decrease in the production of mitochondria-encoded subunits of complex IV [11, 14, 15]. We reasoned that this decrease may lead to an imbalance between mitochondria-encoded and nuclear-encoded subunits of this complex and thereby trigger UPRmt. To test this hypothesis, we inactivated LRPPRC in SH-SY5Y cells using small interfering RNA (siRNA) and quantified the level of COX I protein (a mitochondria-encoded subunit of complex IV) and the level of COX IV protein (a nuclear-encoded subunit of complex IV). As shown in Figure 1A, the level of COX I protein decreases after 3 days of LRPPRC siRNA (down to 50% of the level in control siRNA cells). In contrast, the level of COX IV protein remains stable, resulting in an imbalance between COX I and COX IV subunits with ~1.8 times more COX IV subunits than COX I subunits upon LRPPRC siRNA.
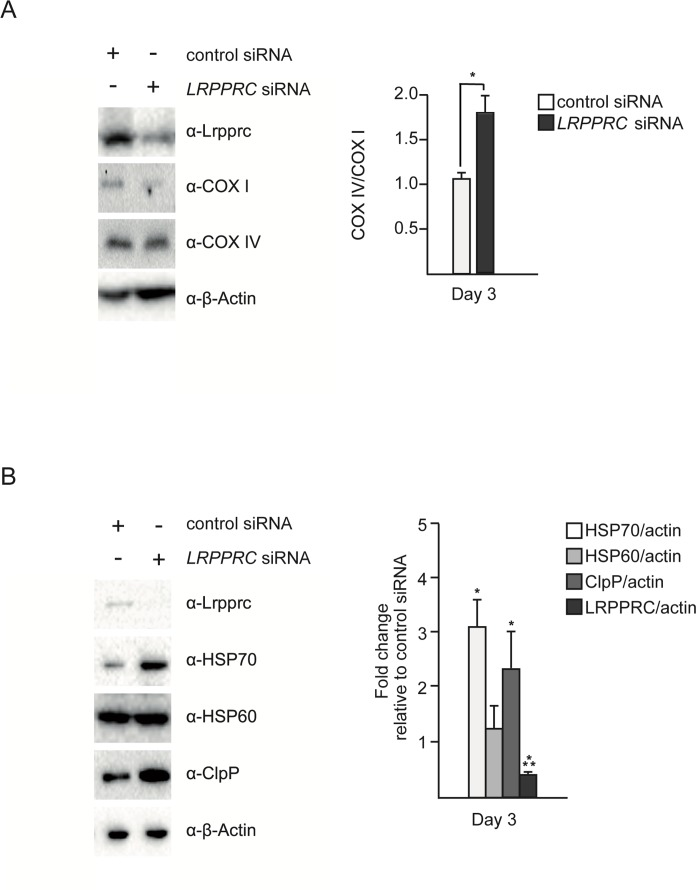
Figure 1. Silencing of LRPPRC leads to an imbalance between mitochondria-encoded and nuclear-encoded subunits of complex IV and triggers UPRmt SH-SY5Y cells were treated with control or LRPPRC siRNA for three days and transferred for 24 hours to low glucose medium (5 mM) to enhance oxidative phosphorylation. (A) Total protein extracts were analyzed by Western using anti-LRPPRC, anti-COX I, anti-COX IV and anti-β-Actin antibodies. COX IV/COX I ratios are indicated. (B) The same protein extracts as in panel A were also analyzed by Western using anti-LRPPRC, anti-HSP70, anti-HSP60, anti-ClpP and anti-β-Actin antibodies. Ratios relative to the control siRNA are indicated. (For all panels, quantifications are based on data from three independent experiments; average values are shown and error bars indicate s.d.; *p ≤ 0.05, ***p ≤ 0.001 by one way ANOVA for panel A and one sample t-test for panel B).
In order to test whether this imbalance triggers UPRmt, we measured the levels of the mitochondrial chaperones HSP60 and HSP70 as well as the mitochondrial protease ClpP. We observed that 3 days of LRPPRC siRNA triggers a ~3-fold increase in the level of endogenous HSP70 protein (Figure 1B; n = 3; p = 0.0473 by one sample t-test), a ~1.3-fold increase in the level of endogenous HSP60 protein (Figure 1B; n = 3; p = 0.178 by one sample t-test) and a ~2.3-fold increase in the level of the endogenous mitochondrial protease ClpP (Figure 1B; n = 3; p = 0.0202 by one sample t-test). Similar results were observed using HEK293T cells (Figure S1). Based on these results, we conclude that the imbalance between nuclear- and mitochondria-encoded subunits of complex IV caused by LRPPRC siRNA triggers UPRmt.
The transient activation of UPRmt by LRPPRC siRNA correlates with the restoration of the balance between complex IV subunits
In order to test the effect of the induction of UPRmt upon LRPPRC siRNA on the balance between nuclear- and mitochondria-encoded complex IV subunits, we performed a time course experiment. The imbalance between COX I and COX IV is first observed after 3 days of LRPPRC siRNA (Figure 2A). This time point also corresponds to the highest level of induction of HSP70 and ClpP (Figure 2B). After 5 days of LRPPRC siRNA, the level of COX IV protein decreases (down to 50% of the level in control siRNA cells), restoring the balance between COX I and COX IV subunits (Figure 2A). This time point coincides with a decrease of the levels of HSP70 and ClpP back to the levels measured in control cells (Figure 2B). A similar transient activation of UPRmt was observed in HEK293T cells (Figure S1). Therefore, the transient activation of UPRmt correlates with the restoration of mitochondrial proteostasis in the context of LRPPRC siRNA.
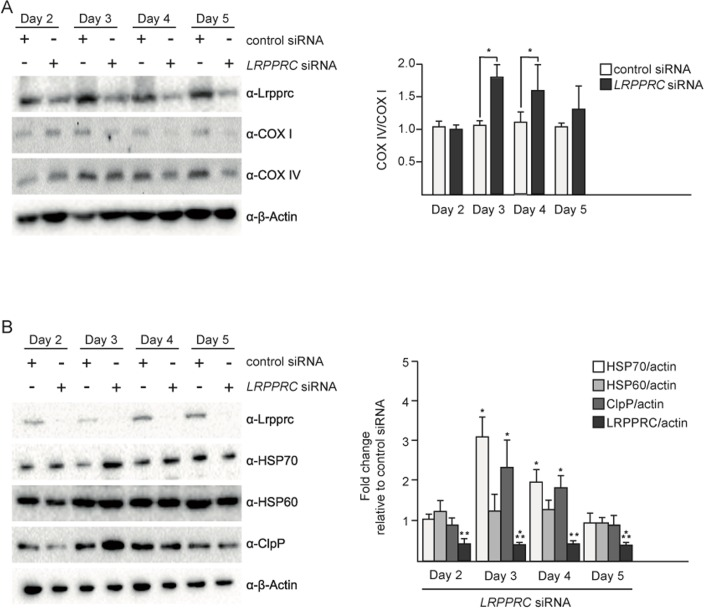
Figure 2. Upon LRPPRC siRNA-induced UPRmt, mitochondrial proteostasis is restored UPRmt in SH-SY5Y cells treated with control or LRPPRC siRNA for 2, 3, 4 or 5 days and transferred for 24 hours in low glucose medium. (A) Total protein extracts were analyzed by Western using anti-LRPPRC, anti-COX I, anti-COX IV and anti-β-Actin antibodies. COX IV/COX I ratios are indicated. (B) The same protein extracts as in panel A were also analyzed by Western using anti-LRPPRC, anti-HSP70, anti-HSP60, anti-ClpP and anti-β-Actin antibodies. Ratios relative to the control siRNA are indicated. (For all panels, quantifications are based on data from three independent experiments; average values are shown and error bars indicate s.d.; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by one way ANOVA for panel A and one sample t-test for panel B).
UPRmt and mitochondrial hyperfusion are transient responses to LRPPRC siRNA that follow similar kinetics
We have previously shown that mitochondrial hyper-fusion is a transient response, which peaks at 3 days of LRPPRC siRNA (Figure S2; day 3: 50% of cells have hyperfused mitochondria, 32% have tubular mitochondria and 18% have fragmented mitochondria). After 4 or 5 days of inactivation, the population of cells that display hyperfused mitochondria decreases while the population of cells that display fragmented mitochondria increases (Figure S2; day 5: 35% of cells have hyper-fused mitochondria, 45% have tubular mitochondria and 28% have fragmented mitochondria). Similarly, we observed that the induction of UPRmt is at its maximum at 3 days of LRPPRC siRNA, in both SH-SY5Y cells and HEK293T cells (Figure 2B, Figure S1). After 4 and 5 days of inactivation, the UPRmt decreases in both cell lines (Figure 2B, Figure S1B). Hence, in mammalian cell culture, UPRmt and the mitochondrial hyperfusion response both are transient and follow similar kinetics.
Inactivation of the LRPPRC-like gene mma-1 in C. elegans also induces UPRmt
In order to address whether the induction of UPRmt in response to LRPPRC siRNA is conserved, we inactivated the LRPPRC-like gene mma-1 in C. elegans. We used the transcriptional reporters Phsp-6GFP, Phsp-60GFP and Phsp-4GFP to monitor the expression of the genes encoding the mitochondrial HSP70 chaperone (hsp-6), the mitochondrial HSP60 chaperone (hsp-60) or the endoplasmic reticulum (ER) chaperone BiP (hsp-4), respectively [18]. We reduced mma-1 function using RNA-mediated interference (RNAi) by feeding [19]. Specifically, we grew L4 larvae of the reporter strains on RNAi plates seeded with an Escherichia coli strain expressing double-strand RNA (dsRNA) of mma-1. The plates were seeded with mma-1(RNAi) bacteria diluted 1:5 (v/v) with mock(RNAi) bacteria; hereafter referred to as ‘mma-1(RNAi) 1:5 dil.′. The expression of the reporters was analyzed four days later in the F1 generation. Using these conditions, the level of MMA-1 protein is reduced by ~40% (see below, Figure S4). As shown in Figure 3, the Phsp-6GFP reporter is strongly up-regulated in response to mma-1(RNAi) (on average ~200-fold compared to mock(RNAi); Figure 3A; n = 5; p = 0.037 by one sample t-test). Phsp-60GFP expression was also increased in mma-1(RNAi) animals compared to mock(RNAi) animals, albeit to a lesser extent (Figure 3B; n = 9; p = 0.011 by one sample t-test). Finally, mma-1 knock-down did not affect the expression of the ER chaperone hsp-4 (Figure 3C; n = 5; p = 0.36 by one sample t-test). We also tested the effect of reducing the activity of the C. elegans homolog of paraplegin spg-7, which has previously been shown to induce UPRmt [18]. Using the RNAi conditions described above, the inactivation of spg-7 induces a strong up-regulation of the Phsp-6GFP reporter and a weak up-regulation of the Phsp-60GFP reporter but has no effect on the expression of the Phsp-4GFP reporter (Figure 3).
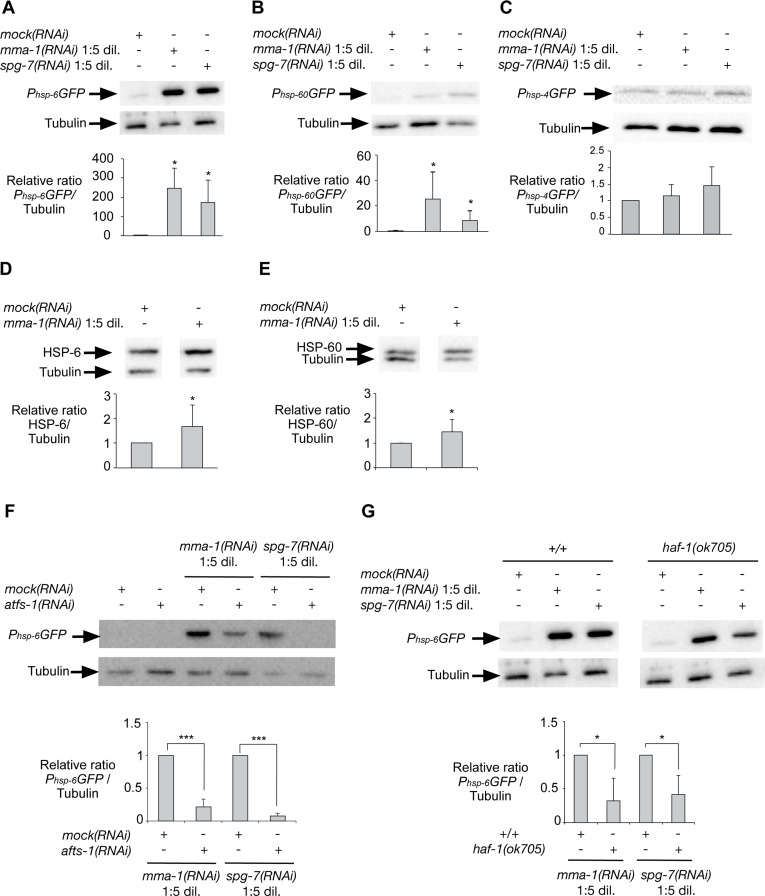
Figure 3. Inactivation of mma-1 by RNAi in C. elegans induces ATFS-1-dependent UPRmt Western analysis of (A) Phsp-6GFP (mitochondrial Hsp70), (B) Phsp-60GFP (mitochondrial Hsp60) or (C) Phsp-4GFP (ER BiP) reporter strains treated with mock(RNAi), mma-1(RNAi) 1:5 dil. (diluted 1:5 (v/v) with mock(RNAi)) or spg-7(RNAi) 1:5 dil. (diluted 1:5 (v/v) with mock(RNAi)). Ratios of Phsp-6GFP/Tubulin, Phsp-60GFP/Tubulin and Phsp-4GFP/Tubulin relative to the mock(RNAi) treated animals are indicated (n = 5 for Phsp-6GFP + mma-1(RNAi); n = 7 for Phsp-6GFP + spg-7(RNAi); n = 6 for Phsp-60GFP + mma-1(RNAi); n = 9 for Phsp-60GFP + spg-1(RNAi); n = 5 for Phsp-4GFP). Western analysis of the effect of mma-1(RNAi) on endogenous (D) HSP-6 or (E) HSP-60 protein level. Ratios of HSP-6/Tubulin and HSP-60/Tubulin relative to the mock(RNAi) treated animals are indicated (n = 8 for HSP-6 and n = 10 for HSP-60). (F) Western analysis of the effect of ATFS-1 on mma-1(RNAi)-induced UPRmt. mma-1(RNAi) or spg-7(RNAi) were diluted either with mock(RNAi) or atfs-1(RNAi). Relative ratios of Phsp-6GFP/Tubulin are indicated (n = 7 for mma-1(RNAi) and n = 10 for spg-7(RNAi)). (G) Western analysis of the effect of HAF-1 on mma-1(RNAi)-induced UPRmt. Wild-type Phsp-6GFP reporter strain (+/+) or Phsp-6GFP reporter strain carrying the haf-1(ok705) loss-of-function mutation were analyzed. The relative ratios of Phsp-6GFP/Tubulin are indicated (n = 5 for mma-1(RNAi) and n = 6 for spg-7(RNAi)) (For all panels, average values are shown and error bars indicate s.d.; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by one sample t-test).
In order to confirm the effect of mma-1(RNAi) on the expression of hsp-6 and hsp-60 at the levels of endogenous HSP-6 and HSP-60 protein, we used a mouse monoclonal anti-HSP-60 antibody [20] and generated rabbit polyclonal anti-HSP-6 antibodies. We confirmed that the anti-HSP-6 antibodies specifically recognize HSP-6 (Figure S3). Using these antibodies we found that the inactivation of mma-1 by RNAi leads to a ~1.7-fold increase in the level of endogenous HSP-6 protein (Figure 3D; n = 8; p = 0.024 by one sample t-test). In addition, we found that mma-1(RNAi) induces a ~1.4-fold increase in the level of endogenous HSP-60 protein (Figure 3E; n = 10; p = 0.022 by one sample t-test). The discrepancy between the level of induction for endogenous proteins (~1.4-fold and ~1.7-fold for HSP-60 and HSP-6, respectively) and for transcriptional reporters (~20-fold and ~200-fold for Phsp-60GFP and Phsp-6GFP, respectively) is most likely due to the copy number of the Phsp-6GFP and Phsp-60GFP transgenes integrated in the genome of the reporter strains; however, we cannot exclude that this discrepancy is also caused by post-transcriptional regulations of hsp-6 and/or hsp-60 expression. Based on these results we conclude that mma-1(RNAi) induces UPRmt and the transcriptional up-regulation of the hsp-6 and hsp-60 genes, which results in a significant increase in the level of endogenous HSP-6 and HSP-60 protein.
mma-1(RNAi)-induced up-regulation of hsp-6 transcription is dependent on ATFS-1 and HAF-1
In C. elegans, UPRmt is dependent on the peptide exporter HAF-1 as well as the bZip transcription factor ATFS-1 [21, 22]. Consistent with previous studies [22], we confirmed that the inactivation of atfs-1 by RNAi suppresses the up-regulation of the Phsp-6GFP reporter induced by spg-7(RNAi) (Figure 3F; n = 7; p = 0.0001 by one sample t-test). Furthermore, we found that mma-1(RNAi)-induced UPRmt is suppressed by atfs-1(RNAi) as well (Figure 3F; n = 10; p = 0.0001 by one sample t-test). To rule out that the effects observed are due to differences in RNAi efficiency, we quantified the knock-down of mma-1 using anti-MMA-1 antibodies [11]. As shown in Figure S4A, the efficiency of mma-1(RNAi) in atfs-1(RNAi) animals is similar to that in wild-type animals (reduction by ~40% on average). Hence, the mma-1(RNAi)-induced up-regulation of hsp-6 transcription is dependent on ATFS-1. In contrast, the haf-1 loss-of-function mutation ok705 only partially suppresses the up-regulation of Phsp-6GFP in response to mma-1(RNAi) (Figure 3G; n = 5; p = 0.0217 by one sample t-test). We observed a similar result for spg-7(RNAi)-induced Phsp-6GFP up-regulation (Figure 3G; n = 6; p = 0.017 by one sample t-test). We confirmed that the effects observed were not due to differences in the efficiency of mma-1(RNAi) (Figure S4B). Therefore, mma-1(RNAi) induces UPRmt in a manner that is dependent on the bZip transcription factor ATFS-1 and that is partially dependent on the peptide exporter HAF-1.
Strong depletion of MMA-1 protein leads to both mitochondrial hyperfusion and UPRmt induction whereas mild depletion of MMA-1 protein only leads to UPRmt
When growing animals on ‘non-diluted’ mma-1(RNAi) plates, we achieved on average an ~80% reduction in the level of MMA-1 protein (Figure 4A; mma-1(RNAi)). Under these conditions, as previously reported, animals exhibit hyperfused mitochondria in body wall muscle cells (76%; Figure 4A; n = 17; two independent experiments) [11]. These animals also show a stronger UPRmt as reflected by the up-regulation of the Phsp-6GFP reporter (on average ~600-fold compared to mock(RNAi) animals; Figure 4A). When reducing the level of MMA-1 protein by only 40% (mma-1(RNAi) 1:5 dil.), the Phsp-6GFP reporter is still up-regulated (on average 200-fold compared to mock(RNAi) animals; Figure 4A); however, these animals do not exhibit mitochondrial hyperfusion (6%; Figure 4A; n = 16; two independent experiments). Hence, mild depletion of MMA-1 protein leads to the activation of the UPRmt pathway and strong depletion of MMA-1 protein additionally leads to the activation of the mitochondrial hyperfusion response pathway.
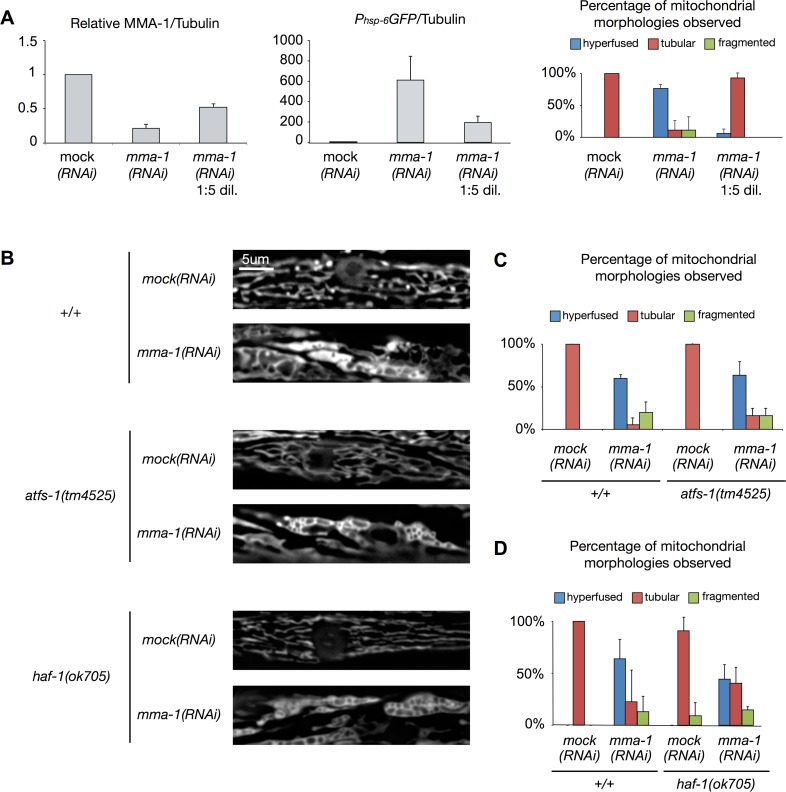
Figure 4. mma-1(RNAi)-induced mitochondrial hyperfusion is not dependent on ATFS-1 or HAF-1 (A) Strong depletion of MMA-1 protein (mma-1(RNAi)) leads to both mitochondrial hyperfusion and UPRmt response whereas mild depletion of MMA-1 protein (mma-1(RNAi) 1:5 dil.) only leads to UPRmt. Average relative ratios of MMA-1/Tubulin and Phsp-6GFP/Tubulin are indicated. Mitochondrial morphology in body wall muscles was assessed in L4 larvae of the F1 generation. Percentage of animals showing fragmented, tubular or hyperfused mitochondria are indicated (n >10 for each condition; average values are shown and error bars indicate s.d.). (B) Fluorescence microscopy analysis of wild-type Pmyo-3mitoGFP reporter strain (+/+) or Pmyo-3mitoGFP reporter strain carrying the haf-1(ok705) mutation or atfs-1(tm4525) mutation. (C-D) Quantification of the different mitochondrial morphologies observed (at least 15 animals were analyzed for each condition; two independent experiments were performed for panel C; three independent experiments were performed for panel D; average values are shown and error bars indicate s.d.).
mma-1(RNAi)-induced mitochondrial hyperfusion is independent of HAF-1 and ATFS-1
In order to test whether the mitochondrial hyperfusion response induced by the inactivation of mma-1 is dependent on HAF-1 and ATFS-1, we used a transgene (bcIs78 [Pmyo-3mitoGFP]) that expresses mitochondrial matrix-targeted GFP in body wall muscles, which allows us to monitor steady-state mitochondrial morphology in these cells [11]. In an otherwise wild-type background, mma-1(RNAi) causes mitochondrial hyperfusion (64%, n = 30, Figure 4B-D, three independent experiments) [11]. Similarly, mma-1(RNAi) causes mitochondrial hyperfusion in animals homozygous for the atfs-1 loss-of-function mutation tm4525 (67%, Figure 4B-C; n = 19, two independent experiments). Finally, mma-1(RNAi) also causes mitochondrial hyperfusion in animals homozygous for the haf-1 loss-of-function mutation ok705 (45%; Figure 4B-D; n = 33; three independent experiments). Compared to the response in the wild-type and atfs-1(tm4525) background, the response in the haf-1(ok705) background is slightly reduced; however, the difference is not statistically significant (p = 0.22 by Student t-test). Based on these results we conclude that mma-1(RNAi)-induced mitochondrial hyperfusion is independent of ATFS-1 and HAF-1.
ATFS-1-dependent UPRmt is not essential for viability in response to mma-1(RNAi)
We previously showed that mma-1(RNAi) causes synthetic embryonic lethality and synthetic midlarval arrest in the fzo-1(tm1133) loss-of-function mutant background, indicating that mitochondrial hyperfusion is essential for viability in response to mma-1(RNAi) [11]. To test whether UPRmt is also essential for viability in response to mma-1(RNAi), we inactivated mma-1 in animals carrying a loss-of-function mutation in the atfs-1 gene (atfs-1(tm4525)) [22]. We found that mma-1(RNAi) does not cause any obvious embryonic lethality or midlarval arrest in atfs-1(tm4525) mutant animals (Figure S5A). We also performed the experiment using double RNAi (afts-1(RNAi)+mma-1(RNAi)) and did not observe any obvious synthetic lethality or arrest (Figure S5B). This indicates that in response to mma-1(RNAi), mitochondrial hyperfusion is essential for viability while ATFS-1-dependent UPRmt is not.
Discussion
Depending on the type and the severity of the stress to which mitochondria are exposed, different mito-chondrial stress responses (such as UPRmt, mitochondrial hyperfusion or mitophagy) can be activated in order to protect mitochondrial function [23]. Here, we show that reducing LRPPRC or mma-1 function leads to an evolutionary conserved co-activation of UPRmt and mitochondrial hyperfusion. Depletion of LRPPRC or MMA-1 protein results in a decrease in the production of mitochondria-encoded subunits of complex IV and thereby causes a reduction of complex IV activity [11, 14, 15]. Depletion of LRPPRC or MMA-1 protein also leads to an imbalance between nuclear-encoded and mitochondria-encoded subunits of this complex. We show that the activation of the UPRmt occurs when this balance and, hence mitochondrial proteostasis, is disrupted (Figure 2; day 3 of LRPPRC siRNA). This finding suggests that the accumulation of unassembled complex IV subunits is most likely the signal that triggers UPRmt. Furthermore, our data indicate that UPRmt is transiently activated until mitochondrial proteostasis is restored (Figure 2; day 5 of LRPPRC siRNA). We, therefore, propose that the transient activation of UPRmt participates in the restoration of mitochondrial proteostasis by either proteolytic degradation of unassembled subunits by ClpP or assisted folding of nascent subunits by HSP70. In addition, the response may also involve the down-regulation of the genes encoding nuclear-encoded subunits of complex IV.
Whereas UPRmt and mitochondrial hyperfusion in response to LRPPRC mma-1 RNAi follow similar kinetics, genetic experiments in C. elegans indicate that the pathways that mediate these two responses are distinct. Specifically, LRPPRC mma-1 RNAi-induced UPRmt is dependent on the previously described HAF-1, ATFS-1 pathway, whereas LRPPRC mma-1 RNAi-induced mitochondrial hyperfusion is not. This notion is furthermore supported by the fact that in C. elegans, a mild depletion of MMA-1 protein activates only the UPRmt pathway whereas a strong depletion of MMA-1 activates both pathways. The pathway through which a reduction in LRPPRC mma-1 function induces mitochondrial hyperfusion remains to be elucidated. Similarly, the mechanism through which mitochondrial hyperfusion and UPRmt are coordinated is currently unknown.
We propose that the mitochondrial hyperfusion response and UPRmt act together to maintain and restore mitochondrial function (Figure 5). We have previously shown that mitochondrial hyperfusion helps to maintain cellular ATP levels despite a reduction in complex IV activity caused by LRPPRC mma-1 RNAi [11]. We now propose that this transient compensation enables the cell to restore the balance between mitochondria-encoded and nuclear-encoded subunits of complex IV. However, as previously shown, prolonged inactivation of LRPPRC mma-1 eventually leads to mitochondrial fragmentation, a drop in ATP level and the loss of cellular and organismal functions [11]. The two mitochondrial stress response pathways we unraveled are therefore likely to be important for the maintenance of mitochondrial function in response to moderate and short-term changes in mitochondrial proteostasis. Interestingly, UPRmt as well as an ‘elongation’ of mitochondria are also induced in response to the infection of C. elegans by Pseudomonas aeruginosa [24]; however, how these two stress responses are induced in this context remains to be determined. In contrast, UPRmt and mitochondrial hyperfusion may not protect against more drastic and long-term changes in mitochondrial proteostasis, such as in C. elegans in which mma-1 has been chronically inactivated or in French Canadian Leigh Syndrome patients carrying mutations in the LRPPRC gene. In cell lines derived from these patients, mitochondria are fragmented [15], which indicates that the mitochondrial hyperfusion response has already failed. Whether UPRmt is activated in these cell lines is currently unknown. In case it is activated, it will be interesting to determine whether this pathway contributes to the pathophysiology of this neurodegenerative disorder, which has not been completely elucidated yet.
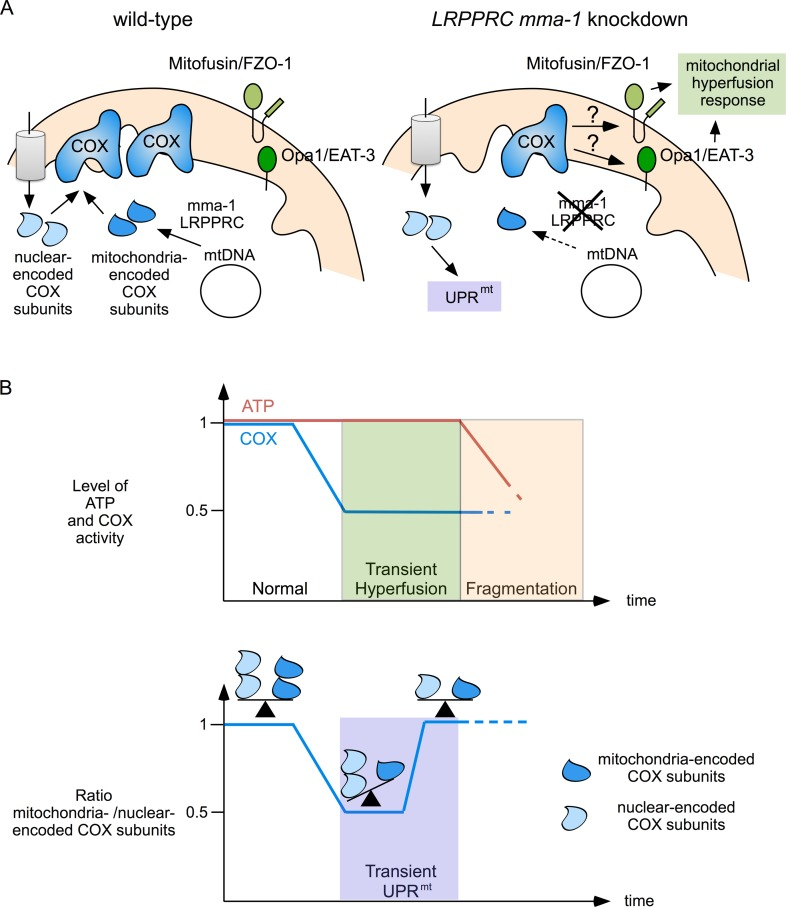
Figure 5. Mitochondrial hyperfusion and UPRmt act together to protect mitochondrial function (A) Complex IV of the ETC is composed of mitochondria-encoded and nuclear-encoded subunits. Upon LRPPRC mma-1 RNAi, the level of mitochondria-encoded subunits decreases, leading to a reduction in complex IV activity, which triggers the mitochondrial hyperfusion response and an accumulation of unassembled nuclear-encoded subunits, which triggers UPRmt. (B) While mitochondrial hyperfusion helps to maintain ATP level in response to a reduction in complex IV activity [11], UPRmt helps to restore the balance between mitochondria-encoded and nuclear-encoded subunits of this complex. Mitochondrial hyperfusion can only compensate for reduced complex IV activity for a limited time. Prolonged inactivation of LRPPRC mma-1 eventually leads to mitochondrial fragmentation and drop in ATP level as previously shown [11].
We previously showed that the mitochondrial hyperfusion response is essential for viability in response to mma-1(RNAi) [11]. In contrast, we present evidence that ATFS-1-dependent UPRmt is not essential for viability upon inactivation of mma-1. Furthermore, we show that ATFS-1-dependent UPRmt is also not essential for viability in response to spg-7(RNAi). One possible explanation for this finding is the existence of an ATFS-1-independent UPRmt pathway. Indeed, Haynes and co-workers have shown that while spg-7(RNAi) induces the up-regulation of 685 genes, 294 of these genes are still up-regulated in atfs-1(tm4525) mutant animals [22]. Hence, in the context of mma-1 inactivation, the ATFS-1-independent UPRmt in combination with the mitochondrial hyperfusion response may be sufficient for the completion of embryonic and larval development.
The effect of the activation of UPRmt on aging remains controversial. While Houtkooper et al reported an extension of lifespan in response to the activation of UPRmt [7], Bennett et al observed no clear correlation between UPRmt and lifespan [25]. We and others have previously shown in C. elegans and in rat that mitochondrial fragmentation increases with age and that this fragmentation is accompanied with a loss of mitochondrial volume [26, 27]. Interestingly, a recent study reports that fibroblasts isolated from long-lived human individuals exhibit hyperfused mitochondria [28]. While the genetic determinants resulting in this change in mitochondrial morphology remain unknown, the mitochondrial hyperfusion in the cells of these individuals has been shown to increase mitochondrial mass by preventing mitophagy [28]. Consequently, while the activity of their individual mitochondria decreases with age, the overall mitochondrial function of their cells is maintained, implicating a beneficial role of mitochondrial hyperfusion in aging [28]. In conclusion, a better understanding of UPRmt and the mitochondrial hyperfusion response has not only the potential to increase our knowledge of how cells respond to mitochondrial stress under physiological and pathophysiogical conditions (i.e. French Canadian Leigh Syndrome) but also during aging.
Materials and Methods
General C. elegans methods and strains
C. elegans strains were cultured as previously described [29]. Bristol N2 was used as the wild-type strain. Mutations used in this study were described by Riddle and co-workers [30] except: (LG IV) haf-1(ok705) (OMRF Knockout Group); (LG V) atfs-1(tm4525) (National BioResource Project). A summary of the transgenic lines is provided in Table S1.
C. elegans RNA interference
RNAi by feeding was performed as previously described [11] with the following modifications. RNAi plates containing 1 mM IPTG were inoculated with 50 μl of 0.5 OD600nm/ml of concentrated mma-1(RNAi) or spg-7(RNAi) bacterial clones from the Ahringer library [31] or diluted 1:5 (v/v) with mock(RNAi) bacteria (HT115 bacteria transformed with the empty RNAi feeding vector pPD129.36) or atfs-1(RNAi) bacteria. 24 hours later, L4 larvae were inoculated on the RNAi plates and incubated at 20°C for 4 days. Mitochondrial morphology in L4 larvae of the F1 generation was analyzed by fluorescent microscopy as described [32]. For protein analyses, mixed-stage populations of worms were harvested and analyzed as described below. For the analysis of the effect of RNAi on C. elegans development, three wild-type (+/+), three atfs-1(tm4525) and 15 fzo-1(tm1133) L4 animals (15 animals of the fzo-1(tm1133) genotype were used since the broodsize of these animals is smaller) were inoculated on mock(RNAi), mma-1(RNAi) or spg-7(RNAi) plates and incubated at 20°C for 18 hours. For the double RNAi experiment, three wild-type L4 larvae were inoculated on mock(RNAi), atfs-1(RNAi), mma-1(RNAi) (diluted 1:5 with mock(RNAi) or atfs-1(RNAi)) or spg-7(RNAi) (diluted 1:5 with mock(RNAi) or atfs-1(RNAi)) plates and incubated at 20°C for 18 hours. The adults were then transferred onto freshly seeded RNAi plates and incubated for an additional three hours at 20°C. After removing the adults, the embryos laid during these three hours were counted. 24 hours later, the number of unhatched embryos was counted to determine the embryonic lethality. The number of animals that reached adulthood by day 5 and the number of animals that underwent a midlarval arrest were also counted.
Mammalian cell culture and transfection
SH-SY5Y cells were cultivated in DMEM:F12 supplemented with 15% FBS (Sigma), 1% non essential amino acids and 1% penicillin/streptomycin (Invitrogen); HEK293T cells were cultivated in DMEM supplemented with 10% FBS (Sigma) and 1% penicillin/streptomycin (Invitrogen); both cell lines were passaged twice a week and kept at 37°C with 5% CO2. For RNA interference, cells were transfected reversely with control stealth siRNA medium GC or stealth siRNA oligos targeting LRPPRC using Lipofectamine RNAiMax in OPTI-MEM (Invitrogen). 24 hours after transfection, fresh culture medium (25 mM glucose) was added. For the analysis of mitochondrial morphology, cells were kept in normal culture medium until harvested. For the analysis of UPRmt, the normal culture medium was replaced with medium containing only 5 mM glucose 24 hours before analysis.
Protein analysis
Rabbit polyclonal anti–HSP-6 antibodies were generated using the antigenic peptide C472QEAKTAEEPKKEQN (cysteine plus HSP-6 C-terminal sequence) by Thermo Fisher Scientific, as previously described [33] and used at 1:5000 for Western analysis. Worm protein extracts were subjected to SDS/PAGE and Western. To detect HSP-60, GFP and α-Tubulin, we used a mouse monoclonal antibody developed by Nonet and co-workers and available at DHSB [20] (1:2000), rabbit polyclonal antibodies from Abcam ab290 (1:2000) and a mouse monoclonal antibody from Abcam ab7291 (1:2000), respectively. Images were quantified using the ChemiDoc XRS+ System (Bio-Rad). The data presented are ratios relative to the mock(RNAi). Only samples for which mma-1 knock-down was at least 20% were used for the statistically analysis. Statistical analysis was performed by using one sample t-test. Normality of the data was assessed by Shapiro-Wilk Normality test.
SH-SY5Y and HEK293T cells were harvested with pre-warmed 1x Laemmli-sample buffer and boiled for 10 minutes at 95°C and then subjected to SDS/PAGE and Western. The following antibodies were used: mouse monoclonal anti-β-Actin (Sigma AC-15, 1:5000), mouse monoclonal anti-LRP130 (sc-166178, 1:1000), goat polyclonal anti-HSP60 (sc-1052, 1:2000), rabbit polyclonal anti mortalin/mitochondrial HSP70 (sc-13967, 1:1000), rabbit polyclonal anti-ClpP (sc-134496, 1:1000), goat polyclonal anti-COXI (sc-48143, 1:500) and goat polyclonal anti-COXIV (sc-69359, 1:1000) (With the exception of the anti-β-Actin, all the antibodies were provided from Santa Cruz Biotechnology).
Analysis of mitochondrial morphology in SH-SY5Y cells
Mitochondrial morphology was analyzed as previously described with the following modifications [11]. Cells grown on glass coverslips were fixed with PBS 3.7% PFA at 20°C for 10 minutes and permeabilized with PBS 0.2% Triton X-100 for 10 minutes at 20°C. The coverslips were blocked with PBS 5% BSA for 1 hour at 4°C and incubated overnight at 4°C in PBS 5% BSA with polyclonal anti-TOM20 antibodies (1:2000, Santa Cruz Biotechnology). After three washes with PBS, the coverslips were incubated with an Alexa555 goat anti-rabbit antibody (1:2000, Molecular Probes) in PBS 5% BSA for 2 hours at 20°C. After three washes with PBS, three washes with PBS 0.2% Tween and three washes with PBS, the glass coverslips were mounted on glass slides with MOWIOL 4–88 containing DAPI (Sigma). At least 300 cells per coverslip were counted in a blinded manner. Results are based on three independent experiments.
Supplementary Materials
Acknowledgments
We thank Eric Lambie, Nikola Wagener, Tamara Mikeladze-Dvali, Nadin Memar and members of the B.C. laboratory for comments on the manuscript; N. Lebedeva, L. Jocham, and M. Schwarz for excellent technical support; S. Mitani (National BioResource Project, Tokyo, Japan) for atfs-1(tm4525); and the Caenorhabditis elegans Genetics Center (supported by the National Institutes of Health National Center for Research Resources) for strains. The anti-HSP-60 antibody developed by Nonet, M.L., Hadwiger, G. and Dour, S was obtained from the Developmental Studies Hybridoma Bank (DSHB), created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Funding
This work was funded by NIH grant GM076651 and CIPSM (to BC).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- 1. Blackstone NW. The impact of mitochondrial endosymbiosis on the evolution of calcium signaling. Cell calcium. 2015; 57: 133 -139. [PubMed] .
- 2. Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999; 283: 1488 -1493. [PubMed] .
- 3. Wang C and Youle RJ. The role of mitochondria in apoptosis. Annual review of genetics. 2009; 43: 95 -118. .
- 4. Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annual review of biochemistry. 2010; 79: 683 -706. .
- 5. Niforou K, Cheimonidou C, Trougakos IP. Molecular chaperones and proteostasis regulation during redox imbalance. Redox biology. 2014; 2: 323 -332. [PubMed] .
- 6. Jensen MB and Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell metabolism. 2014; 20: 214 -225. [PubMed] .
- 7. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013; 497: 451 -457. [PubMed] .
- 8. Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harbor perspectives in biology. 2012; 4: a011338 [PubMed] .
- 9. Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochimica et biophysica acta. 2013; 1833: 410 -416. [PubMed] .
- 10. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature cell biology. 2011; 13: 589 -598. [PubMed] .
- 11. Rolland SG, Motori E, Memar N, Hench J, Frank S, Winklhofer KF, Conradt B. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: E2967 -2976. [PubMed] .
- 12. Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. The EMBO journal. 2009; 28: 1589 -1600. [PubMed] .
- 13. Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100: 605 -610. [PubMed] .
- 14. Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Camara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K, Erdjument-Bromage H, Tempst P, Brandt U, et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. The EMBO journal. 2012; 31: 443 -456. [PubMed] .
- 15. Sasarman F, Brunel-Guitton C, Antonicka H, Wai T, Shoubridge EA, Consortium L. LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Molecular biology of the cell. 2010; 21: 1315 -1323. [PubMed] .
- 16. Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landazuri MO, Enriquez JA. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell metabolism. 2012; 16: 378 -386. [PubMed] .
- 17. Lemire B. Mitochondrial genetics. biology. 2005; 1 -10. [PubMed] .
- 18. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of cell science. 2004; 117: 4055 -4066. [PubMed] .
- 19. Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001; 263: 103 -112. [PubMed] .
- 20. Hadwiger G, Dour S, Arur S, Fox P, Nonet ML. A monoclonal antibody toolkit for C. elegans. PloS one. 2010; 5: e10161 [PubMed] .
- 21. Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular cell. 2010; 37: 529 -540. [PubMed] .
- 22. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012; 337: 587 -590. [PubMed] .
- 23. Friedman JR and Nunnari J. Mitochondrial form and function. Nature. 2014; 505: 335 -343. [PubMed] .
- 24. Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014; 516: 414 -417. [PubMed] .
- 25. Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, Pineda VV, Kaeberlein M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nature communications. 2014; 5: 3483 .
- 26. Regmi SG, Rolland SG, Conradt B. Age-dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging. 2014; 6: 118 -130. [PubMed] .
- 27. Iqbal S, Ostojic O, Singh K, Joseph AM, Hood DA. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle & nerve. 2013; 48: 963 -970. [PubMed] .
- 28. Sgarbi G, Matarrese P, Pinti M, Lanzarini C, Ascione B, Gibellini L, Dika E, Patrizi A, Tommasino C, Capri M, Cossarizza A, Baracca A, Lenaz G, et al. Mitochondria hyperfusion and elevated autophagic activity are key mechanisms for cellular bioenergetic preservation in centenarians. Aging. 2014; 6: 296 -310. [PubMed] .
- 29. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974; 77: 71 -94. [PubMed] .
- 30. Riddle DL, Blumenthal T, Meyer BJ, Priess JR. . in . Riddle D.L., Blumenthal T., Meyer B.J., Priess J.R.. Cold Spring Harbor (NY) 1997; .
- 31. Kamath RS and Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003; 30: 313 -321. [PubMed] .
- 32. Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,- and EAT-3/Opa1-dependent mitochondrial fusion. The Journal of cell biology. 2009; 186: 525 -540. [PubMed] .
- 33. Yokoyama K, Fukumoto K, Murakami T, Harada S, Hosono R, Wadhwa R, Mitsui Y, Ohkuma S. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS letters. 2002; 516: 53 -57. [PubMed] .