



Introduction
The molecular genetics governing eukaryotic longevity are strongly conserved from yeast to mammals [1]. The genetic malleability of yeast has facilitated discoveries that have determined factors involved in both replicative (divisions of a single cell, mitotic) and chronological (population survival time, post-mitotic) lifespan, with corresponding orthologs in higher eukaryotes subsequently proven to carry out similar roles. Specifically, the technique of yeast replicative lifespan (RLS) follows how many mitotic divisions a yeast cell will go through prior to senescence, whereas chronological lifespan (CLS) measures how long a post-mitotic population of cells will remain metabolically active [2, 3]. These are two very different measures of the yeast life cycle, and are dictated by similar, yet distinct, sets of genes [3]. Intertwined with the genes affecting aging are those related to stress response and adaptive survival. The Anaphase Promoting Complex (APC) and the yeast Forkhead (Fkh) proteins (Fkh1 and Fkh2) interact to play important roles in both lifespan and stress response [4-6].
Stress response genes play a significant role in lifespan in multiple model systems [7-9]. The evolutionarily conserved Fox family of proteins are stress response transcription factors that are critical for properly controlling apoptosis, autophagy, metabolism and cell proliferation [10-12]. This, in turn, improves cell health, survival, and extension of lifespan. In higher eukaryotes including flies, mice, and worms, mutations of various Fox family members results in significant elevations of cancer incidence, supporting their role in apoptosis and proliferation [13, 14]. Higher eukaryotic Fox proteins undergo numerous post-translational modifications including phosphorylation, and ubiquitin-dependent degradation by the proteasome, leading to a variety of molecular outcomes [15, 16]. For example, phosphorylation of FoxO proteins by different upstream kinases, including AKT [15] or AMPK [17], affects FoxO nuclear localization and gene target expression. Here, we demonstrate that the yeast Fox ortholog, Fkh1, is also regulated at the level of protein degradation, highlighting that the Fkh system in yeast is a viable lower eukaryotic model for genetic and molecular investigations into lifespan and stress response mechanisms that will benefit our understanding of human lifespan and disease.
Yeast encode four Fox orthologues: Fkh1, Fkh2, Hcm1 and Fhl1 [18]. The Fkh1 and Fkh2 transcription factors are evolutionarily conserved, and at least Fkh1 and Fkh2 are redundant [5, 10, 19-21]. The Fkhs interact with the transcriptional cofactor Ndd1 in G2 to drive transcription of G2/M genes. In G1, Sir2 is recruited to promoters via the Fkh proteins to effect repression of the same key cell cycle progression genes activated earlier during G2 [19, 22, 23]. Because of their redundancy, genomic deletion of both FKH1 and FKH2 genes is required to assess their inherent function. The fkh1Δ fkh2Δ strain is exquisitely sensitive to oxidative stress and has a significantly shortened lifespan [5]. Conversely, elevated expression of either FKH1 or FKH2 independently enhanced lifespan, and augmented oxidative stress resistance [5].
An interdependence also exists in yeast between the APC and the Fkh proteins that impacts lifespan and stress response [5, 11]. Fkh1 and Fkh2 proteins can both activate the APC under normal growth conditions to coordinate cell cycle progression [5]. The APC is a multi-subunit ubiquitin ligase, or E3, that is predominantly known as being required for cell cycle progression through mitosis and for G1 maintenance, in lower and higher eukaryotes [24, 25]. Cdc20 controls APC function through mitosis, while Cdh1 regulates APC-dependent processes through G1 passage. We have described biological roles affected by the APC that go beyond lifespan, including critical functions in stress response, mitotic chromatin assembly, and mitotic-associated histone modifications [4, 26-29]. We observed that deletion of both FKH genes was necessary to further impair mutant APC phenotypes, such as sensitivity to temperature and oxidative stress, and reduced lifespan, indicating how important this combination of genes is to cell health and adaptive survival.
Activators of the APC, such as Clb2 and Cdc20, are often targeted for ubiquitin-dependent degradation through the E3 activity of the APC itself [30, 31]. Although we had evidence that the Fkh proteins likely activated the APC [5], we did not know if Fkh1 was targeted for degradation like other APC activators. Our hypothesis that Fkh1 served as an APC target grew from our observation that deletion of FKH1 suppressed mutant APC defects. This is based on observations that deletion of APC targets, which accumulate in APC mutants, is predicted to alleviate APC mutant phenotypes [32]. Thus we queried if Fkh1 is also degraded in a cell cycle-dependent manner. We demonstrate here that the regulation of Fkh1 occurs at the onset of mitosis via targeted degradation initiated by the APCCdc20 complex. Mutation of a highly conserved lysine stabilized Fkh1, conferred cell cycle, heat stress, and lifespan defects, but did not impair Fkh1/Apc5 interactions nor recruitment to promoters. These findings of conserved regulation of the Fox family of proteins from yeast to humans demonstrates that yeast provide valuable insight into conserved Fox molecular regulation mechanisms.
Results
Deletion of FKH1 suppresses APC mutant defects
We have extensively used the apc5CA (chromatin assembly) mutant allele for the bulk of our genetic studies to gain insight into APC function [4-6, 26-29, 33]. We discovered the allele in a screen for chromatin assembly mutants [26]. This allele harbors a 2 bp deletion (37AT38), conferring a temperature sensitive phenotype (ts); growth is impaired at 37°C, versus the optimal growth temperature of 30°C. This allele is predicted to encode a short N-terminal portion of Apc5 (the first 12 Apc5 residues followed by 18 novel residues). However, since APC5 is an essential gene and a short N-terminal portion of Apc5 does not rescue the apc5CA ts defect [26, 34, 35], we fused the TAP epitope to the C-terminus of the apc5CA allele and discovered that the ts phenotype is due to an N-terminally truncated protein that likely starts from an internal methionine, and/or undergoes programmed ribosome frameshifting [36] (Fig. S1A). We previously used this allele to show that deletion of both FKH1 and FKH2 was necessary to further impair apc5CA mutant phenotypes [5]. However, here we show that deletion of only FKH1, and not FKH2, improved APC mutant phenotypes (Fig. 1) implicating Fkh1, but not Fkh2, as a potential APC target. This is consistent with other literature noting that deletion of either FKH1 or FKH2 alone has mild, but opposing and independent phenotypes [37, 38].
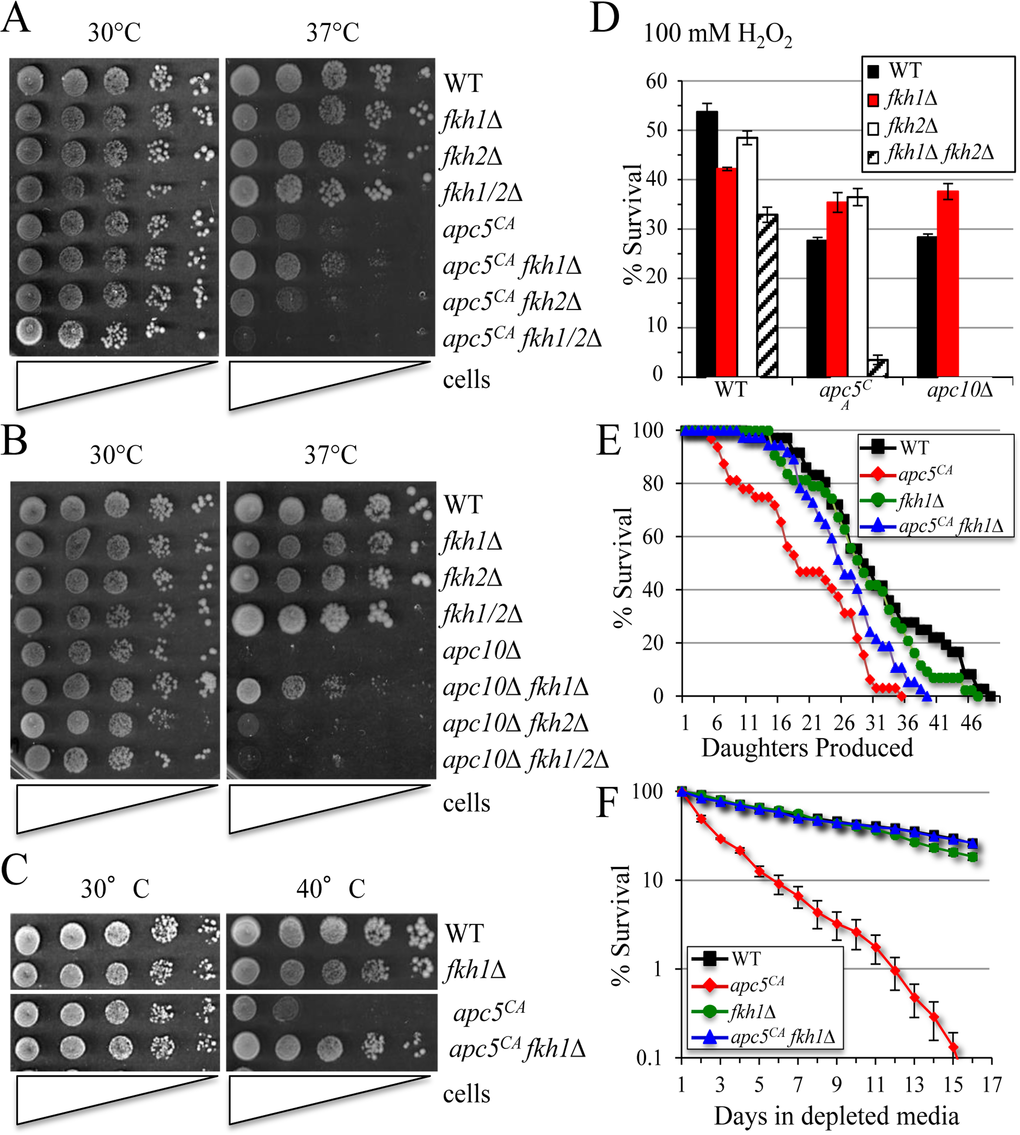
Figure 1. Deletion of FKH1 reverses APC mutant phenotypes (A) The various yeast strains shown were grown overnight at 30°C. The next morning a 10-fold serial dilution series starting with 1 × 107 cells/ml was spotted onto YPD plates and grown at 30°C and 37°C for 3 to 5 days. (B) The strains shown were treated as above. (C) Cells were spot diluted and grown at 30°C and 40°C to accentuate temperature sensitive growth. (D) The cells shown were grown to day 5 of stationary phase then split, with one half treated with 100 mM H2O2 for 1 hour. Equal numbers of cells were then plated onto YDP with all treated cell counts compared to untreated controls. The experiment was done in triplicate with standard error shown. (E) Replicative lifespan was performed with the cells shown. (F) Chronological lifespan of the cells used in (E) was performed in triplicate. Cells were grown in Complete Media (CM) and maintained in the Depleted Media (DM) once stationary phase was reached. Standard error of each timepoint is shown.
We began by examining APC mutant phenotypes, including temperature and oxidative stress sensitivity, and reduced lifespan. Deletion of only FKH1 suppressed ts growth in both apc5CA and in the more severe apc10Δ mutant (Figs. 1A-C, S1B). The ability to improve apc5CA ts growth was more apparent when the cells were grown at 40°C. Sensitivity of apc5CA cells to oxidative stress was also partially alleviated by deletion of FKH1 or FKH2. In contrast, loss of FKH1 in apc10 Δ cells suppressed oxidative stress phenotypes, while deletion of FKH2 was lethal (Fig. 1D). These observations supported our consideration that Fkh1 may serve as an APC target. The remainder of this manuscript will focus on Fkh1-APC interactions in order to understand the molecular mechanisms regulating this interaction.
We had previously observed that overexpression of FKH genes was toxic to cells and induced apoptotic-like cell death [5]. Thus, it is likely that if Fkh1 is accumulating in apc5CA mutant cells, it may likewise be linked to a shorter lifespan. Our results confirm that deletion of FKH1 completely restored apc5CA RLS (Fig. 1E) and CLS (Fig. 1F) to wild type (WT) levels. Thus, by removing Fkh1, stress and lifespan defects are reduced. Taken together, these observations suggest that Fkh1 plays a detrimental role in cells experiencing compromised APC function.
Fkh1 accumulates in apc5CA cells in a ubiquitin- and proteasome-dependent manner
To test the hypothesis that Fkh1 is targeted for ubiquitin-dependent degradation by the APC, we first assessed Fkh1 protein levels in asynchronous WT and apc5CA cells. We integrated Fkh1-TAP into the WT and apc5CA strains, expressed under its endogenous promoter. As a control, we followed the protein levels of a known APC targeted protein, Clb2, which is selected for degradation at the end of mitosis and is at minimal abundance in G1 [39]. Like Clb2, Fkh1 was dramatically reduced in G1 arrested cells and accumulated in apc5CA cells (Figs. 2A, 2B). The accumulation of Fkh1 protein in the apc5CA strain is most likely a post-translational event, as accumulation of Fkh1 protein did not correlate with increased mRNA transcript (Figs. 2C, 2D).
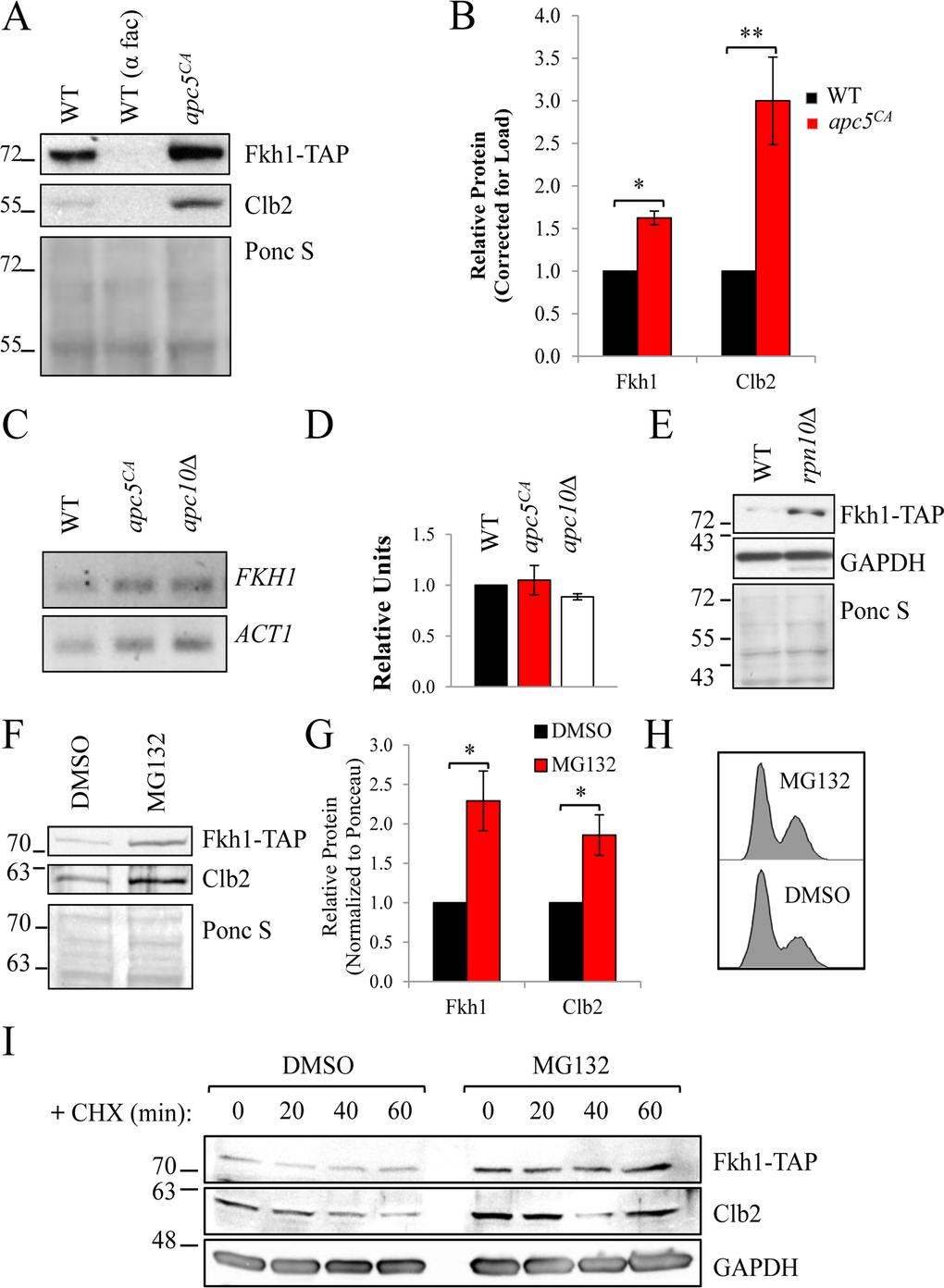
Figure 2. Fkh1 stability depends on the APC and the proteasome (A) WT and apc5CA cells expressing Fkh1-TAP were grown to mid log phase. WT cells were also treated with α-factor to arrest cells in G1. Whole cell lysates were prepared and analyzed by Western analyses with the antibodies shown. (B) The experiment in (A) was repeated 3 times, with all protein bands quantified, normalized to Ponceau S stained gels and plotted. Standard error is shown. * - p≤0.001; ** - p≤0.005. (C) The cells shown were grown to mid log phase, after which total RNA was extracted and used for quantitative reverse transcriptase PCR using primers against FKH1 and ACT1 as a normalization control. (D) The experiment in (C) was repeated 3 times with bands quantified, normalized to ACT1, and plotted. Standard error is shown. (E) WT and rpn10Δ cells expressing endogenous FKH1-TAP were grown to mid log phase, with proteins analyzed by Westerns using antibodies against TAP and GAPDH, with the Ponceau S stained gel confirming equivalency of load. (F) Asynchronous FHK1-TAP cells were incubated with 75 μM MG132 for 3 hours at 30°C prior to preparing cell lysates to inhibit the proteasome. Westerns were then performed using antibodies against TAP and Clb2. (G) The bands from (F) were quantified, normalized to the Ponceau S stained gel and plotted. * - p≤0.05. (H) Cells in (F) were used for flow cytometry to determine cell cycle distribution. (I) Asynchronous FKH1-TAP expressing cells, were treated +/− MG132 as in (F). Cycloheximide (CHX) was then added to inhibit protein synthesis with samples taken every 20 minutes to determine Fkh1-TAP stability.
To determine whether Fkh1 G1 instability was dependent on the proteasome, we expressed endogenous Fkh1-TAP in rpn10Δ cells, which lack the proteasome ubiquitin receptor [40]. In rpn10Δ cells, potentially ubiquitinated proteins accumulate [6, 29], and this was observed for Fkh1-TAP (Fig. 2E). WT Fkh1-TAP expressing cells were next exposed to 75 μM MG132, a proteasome inhibitor, for 3 hours prior to harvesting proteins. In the presence of MG132, both Clb2 and Fkh1-TAP accumulated (Figs. 2F, 2G). This was not cell cycle related, as cells treated with MG312 had the same flow cytometry profile as untreated cells (Fig. 2H). Next, we added cycloheximide (CHX), which halts all protein synthesis, to MG132 treated cells to determine whether MG132 impacts Fkh1 stability or steady state levels. Fkh1-TAP and Clb2 protein levels were both elevated in MG132 treated cells, consistent with stable proteins (Fig. 2I).
If Fkh1 degradation is indeed dependent on the APC E3 activity, then increased APC subunit expression may further decrease Fkh1-TAP levels through enhanced E3 activity. When plasmids expressing copper-inducible promoter (CUP1prom) driven GST-APC5 (Fig 3A) or GST-APC10 (Fig. 3B) were expressed in Fkh1-TAP cells, we indeed observed that Fkh1-TAP levels decreased as compared to empty vector controls. Interestingly, Fkh1-TAP levels increased in response to copper alone, potentially due to induction of Fkh1 by the heavy metal stress. Taken together, these observations are consistent with the idea that Fkh1-TAP instability is APC, ubiquitin- and proteasome-dependent.
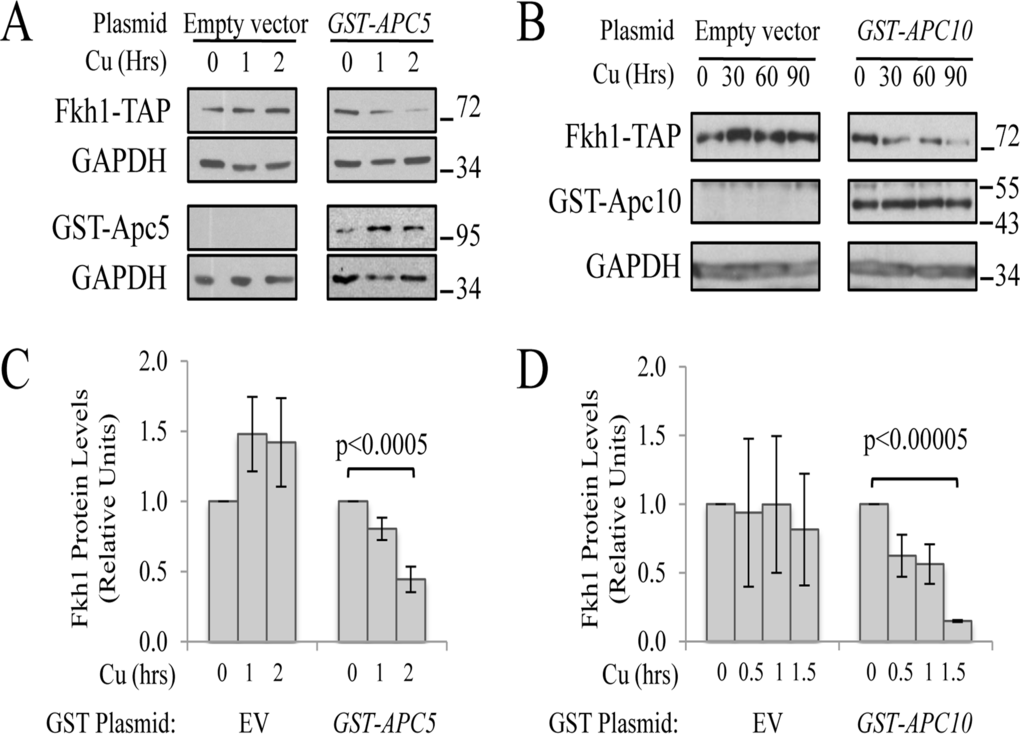
Figure 3. APC subunit content controls Fkh1 protein levels (A) Fkh1-TAP cells expressing plasmid-borne GST-APC5, or the empty vector control, were grown to early log and then exposed to 50 μM CuSO4 for 0, 1 or 2 hours. Cell lysates were prepared and analyzed using Westerns with antibodies against TAP, GST and GAPDH as a load control. (B) The same experiment as in (A) was performed except the plasmid expressed GST-APC10. (C) The bands from 3 repeat experiments in (A) were scanned, quantified, normalized to GAPDH, and plotted. (D) The bands from 2 repeat experiments in (B) were scanned and treated as in (C). Standard error of the mean is shown. Significance was calculated using a Student t-test.
Fkh1 is specifically targeted for degradation at the onset of mitosis by APCCdc20
To determine whether Fkh1 stability is correlated with specific positions within the cell cycle, we arrested Fkh1-TAP expressing cells at various points in the cell cycle. CHX was then added to block further protein synthesis with samples removed every 30 minutes to assess protein stability from the arrest point forward (Figs. 4A-D). We observed that Fkh1-TAP was at its lowest level during G1, but remained stable (Figs. 4B, 4E). In contrast, Fkh1 is unstable during mitosis in the WT strain (Figs 4C, 4E). Stable Fkh1-TAP appears to again accumulate during S phase (Figs. 4D, 4E), consistent with cyclical FKH1 mRNA expression throughout the cell cycle, which peaks during S phase [41]. In apc10Δ cells, Fkh1-TAP protein levels in G1 arrested cells, compared to G1 arrested WT cells, was higher when compared to asynchronous cells (Figs. 4F, S4C). Thus, these results indicate that Fkh1-TAP is specifically targeted for degradation at the onset of mitosis by the APC, which is also active at the onset of mitosis.
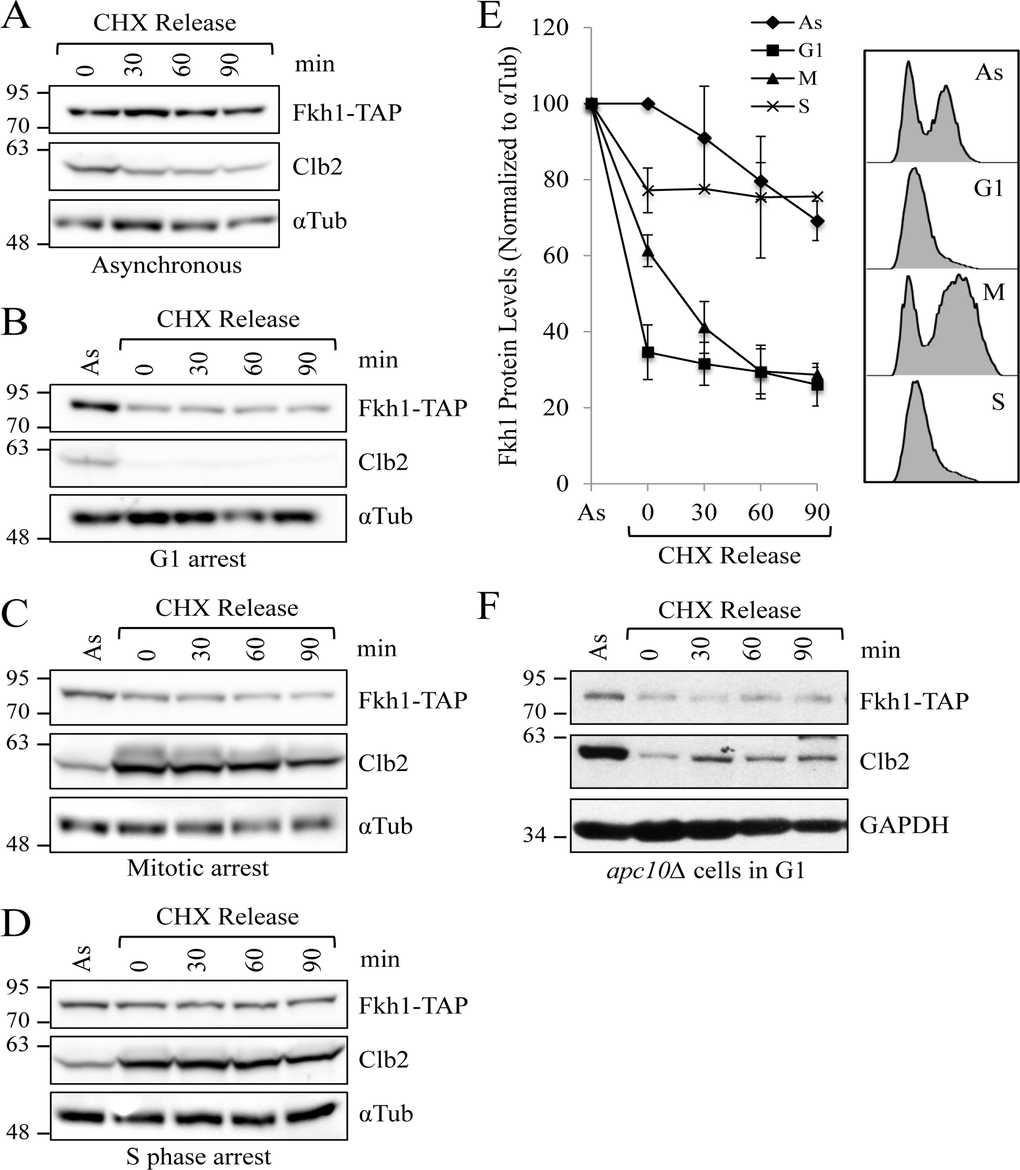
Figure 4. Fkh1 instability occurs specifically in mitosis (A) WT cells expressing endogenous FKH1-TAP were grown to mid log. CHX was added to the asynchronous cells to stop further protein synthesis. Samples were removed every 30 minutes and assessed by Western analyses with the antibodies shown. (B) The experiment described in (A) was performed, except the cells were arrested in G1 using α-factor. (C) The experiment described in (A) was performed, except the cells were arrested in mitosis using nocodazole. (D) The experiment described in (A) was performed, except the cells were arrested in S phase using hydroxyurea. (E) Blots from 3 repeat experiments of (A), (B), and (C), and 2 repeats of (D), were imaged with Fkh1-TAP levels compared over time to visualize Fkh1 stability. Standard error of the mean is shown. Samples were also removed following cell treatment for flow cytometry to verify cell cycle arrest. (F) apc10Δ Fkh1-TAP cells were arrested in G1 using α-factor. CHX was then added with samples removed every 30 minutes for protein analyses.
The APC forms two complexes: a Cdc20-containing complex required for mitotic progression (APCCdc20), and a Cdh1-containing complex that is required for G1 maintenance (APCCdh1) [25, 42]. Cdc20 and Cdh1 are thought to recruit targets to the APC in order to be ubiquitinated [43]. To confirm that Fkh1 is specifically degraded during mitosis, we integrated Fkh1-TAP into the genomes of cdh1Δ and cdc20-1 cells predicting that a cdc20-1 mitotic mutant would disrupt Fkh1 recruitment and degradation in mitosis. cdh1Δ FKH1-TAP cells were chemically arrested in G1, treated with CHX, then assessed for Fkh1-TAP protein stability. Clb2 can be targeted by either APCCdc20 or APCCdh1 complex [44, 45], thus serves as a stability control as it should not be degraded at the specific cell cycle points defined by these mutants. We show that in cdh1Δ mutants, Fkh1-TAP levels remain low in G1, whereas Clb2 levels are markedly elevated, as predicted (Figs. 5A-C). This indicates that Fkh1-TAP is not targeted for degradation by the G1 specific APCCdh1-since it remains low in cdh1Δ cells. On the other hand, in asynchronous cdc20-1 FKH1-TAP cells, both Clb2 and Fkh1-TAP proteins were clearly stabilized, in contrast to their degradation in WT cells (Figs. 5D, 5E). Lastly, we confirmed that the protein levels of Clb2 and Fkh1-TAP predictably rise and fall throughout the cell cycle following synchronous release of S phase-arrested cells back into the cell cycle. Our analysis confirmed that Fkh1-TAP levels began to drop soon after S phase arrest, prior to Clb2 levels, and began to accumulate before Clb2 (Fig. S2). Taken together, the above observations clearly demonstrate that Fkh1 turnover is cell cycle dependent, depends on the APCCdc20 complex, and occurs at the onset of mitosis.
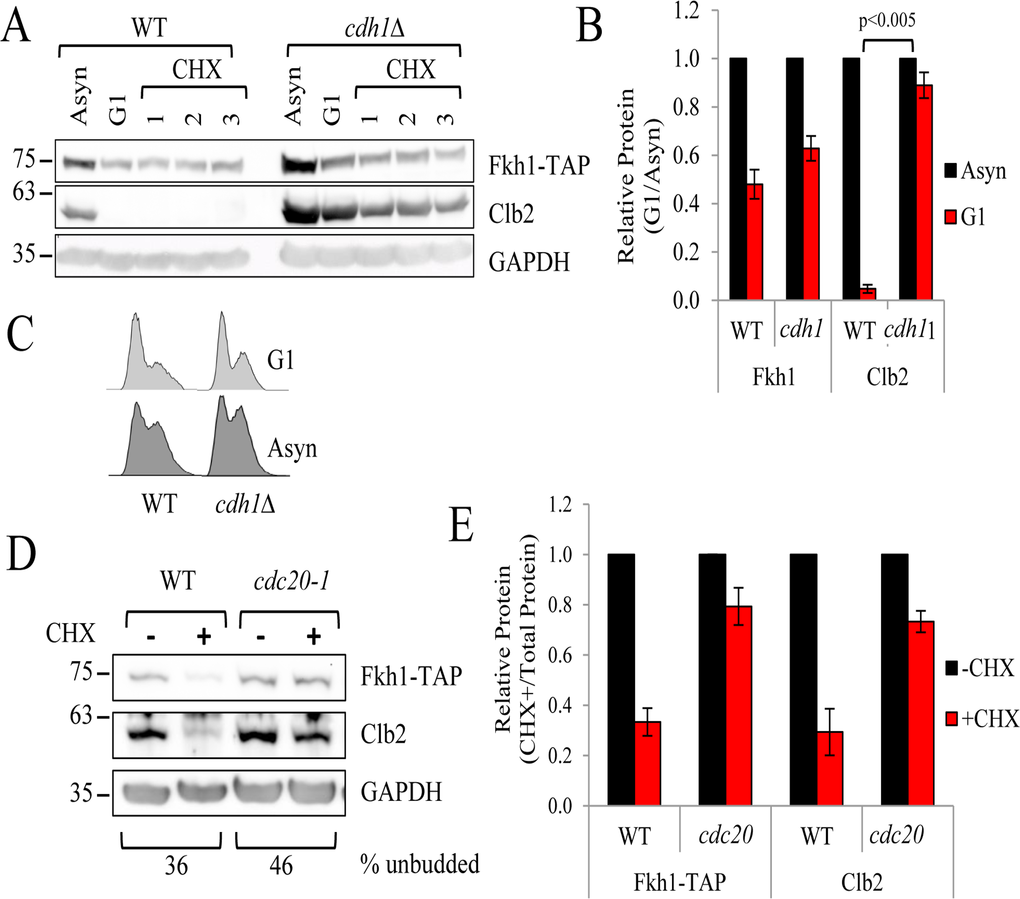
Figure 5. Fkh1 instability is determined by the APCCdc20 complex (A) WT and cdh1Δ FKH1-TAP cells were arrested in G1 using α-factor for 3 hours, followed by the addition of CHX. Proteins were removed every hour to determine Fkh1-TAP stability. Clb2, a known target of the APCCdc20 complex was also monitored as a control. (B) The experiment in (A) was repeated 3 times, with bands quantified, normalized to asynchronous controls, and plotted. Standard error and p-values are shown. (C) Asynchronous and G1 arrested WT and cdh1Δ cells were used for flow cytometry. (D) Asynchronous WT and cdc20-1 FKH1-TAP cells were treated with CHX for 1 hour. Proteins were harvested and Fkh1-TAP and Clb2 levels were determined. (E) Protein bands from three repeats of the experiment shown in (D) were quantified, normalized to - CHX samples and plotted. Standard error is shown.
Mutation of a highly conserved lysine residue stabilizes Fkh1
If Fkh1 is indeed targeted for degradation by the APC, then mutation of a ubiquitinated lysine (K) should abolish ubiquitination, and stabilize the protein. We searched Fkh1 for a conserved K, and found a highly conserved K at position 373 (Fig. 6A). We mutated the highly conserved K373 in the endogenously-TAP tagged FKH1 gene to arginine (R) to generate the Fkh1K-R strain. Since Fkh2 is believed to compensate for the loss of Fkh1 [37], we deleted FKH2 in these cells to examine the isolated effects of the FHK1 mutation. fkh1K-R fkh2Δ cells were flocculent, but not as severely as fkh1Δ fkh2Δ cells (Fig. 6B), indicating that the K373R mutation impaired Fkh1 function, but did not completely disable it. Next, we tested whether the mutation altered Fkh1 mitotic protein instability. We observed that in cells arrested in mitosis, Fkh1 was unstable, whereas Fkh1K-R was stable (Figs. 6C, 6D). Lastly, we determined the cycling profile of the Fkh1K-R protein. The results show that the K373R mutation stabilized Fkh1 throughout the cell cycle (Figs. 6E, S3), indicating that K373 is required for mitotic Fkh1 instability.
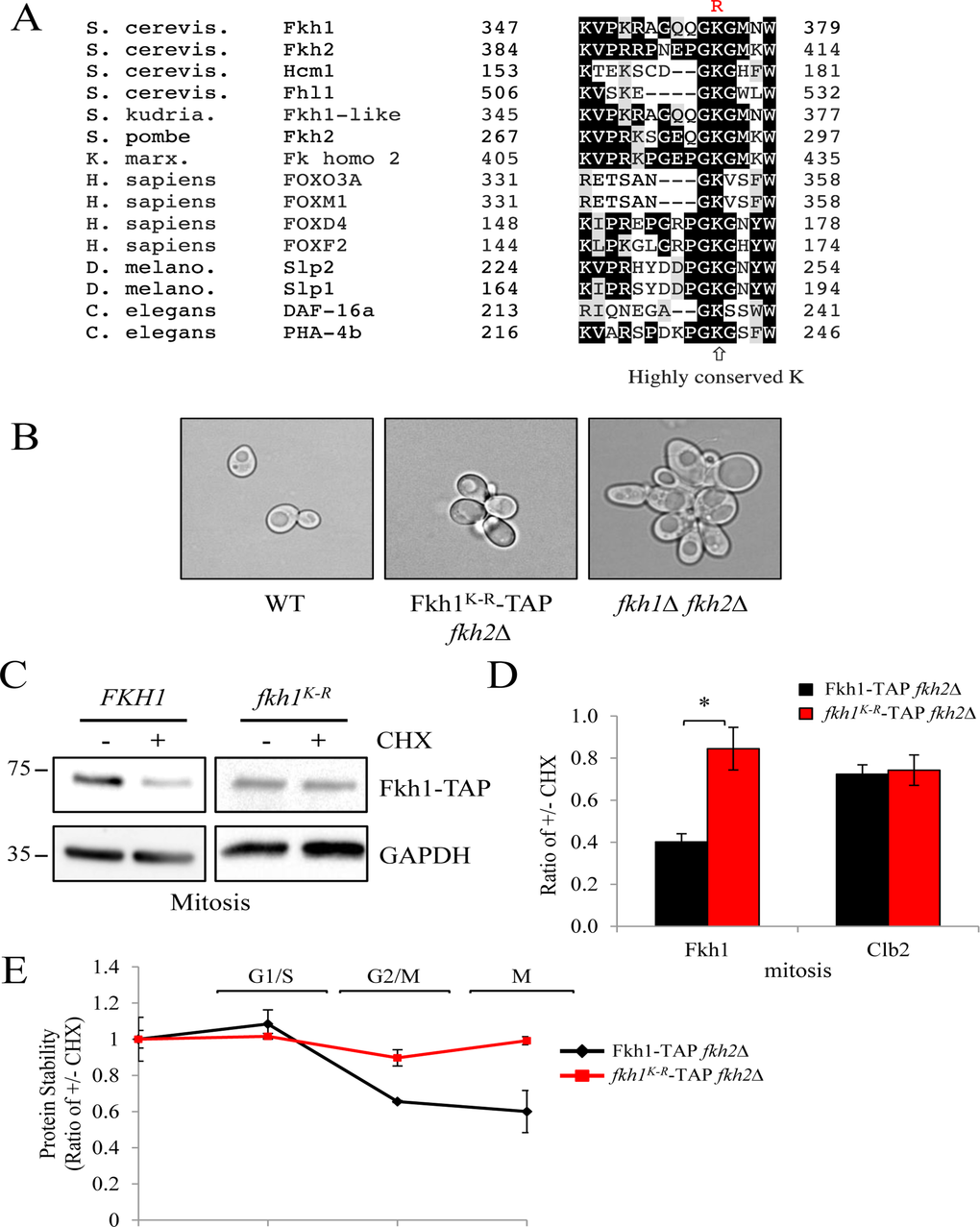
Figure 6. Mutation of the highly conserved K373 in Fkh1 stabilizes the protein in mitosis (A) Sequence alignment of Fkh1-like proteins across evolutionary boundaries highlighting the highly conserved lysine (K373). The mutation made is shown above in red. (B) Morphology of fkh2Δ cells harboring the FKH1K-R mutation shown in (A). (C) fkh2Δ cells expressing FKH1K-R were arrested in mitosis using nocodazole, then treated with CHX for 60 minutes. Proteins were then harvested and examined for Fkh1-TAP, Clb2 and GAPDH content. (D) All bands in (C) were imaged from three separate experiments and the ratio of intensities before and after CHX addition were determined. Standard error of the mean is shown. * - p>0.001. (E) fkh2Δ cells expressing FKH1K-R were arrested in S phase using HU, then released back into the cell cycle. Samples were removed every 30 minutes afterwards, treated +/− CHX for 1 hr, then proteins were recovered for Westerns. Bands from two experiments were scanned and the ratio of bands +/− CHX was plotted. See also Figure S3.
Given the flocculent phenotype of fkh1K-R fkh2Δ cells (Fig. 6B), consistent with a defective Fkh1 protein, we asked if the K373R mutation impaired the core ability of Fkh1: binding promoter DNA. We used chromatin immunoprecipitation (ChIP) in asynchronous cells, with primers designed to amplify the CLB2 and FKH2 promoters, as Fkh1 binds these promoters (Fig. 7) [46]. The high abundance of rDNA, leading to nonspecific interactions with the beads, allows it to be used as a load control. In the bound lanes, Fkh1-TAP was observed at both CLB2 and FKH2 promoters, with Fkh1-TAP abundance clearly increased when FKH2 was deleted (the FKH2 promoter is still present in the FKH2 coding region deletion). Thus, the ability of Fkh1 to compensate for Fkh2 [37] likely involves increased Fkh1 recruitment to promoters in the absence of FKH2. The stable Fkh1K-R protein was bound to both CLB2 and FKH2 promoters at WT levels, demonstrating that the Fkh1K-R defect, leading to flocculence in the fkh2Δ background, is not due to impaired promoter binding, at least at CLB2 and FKH2.
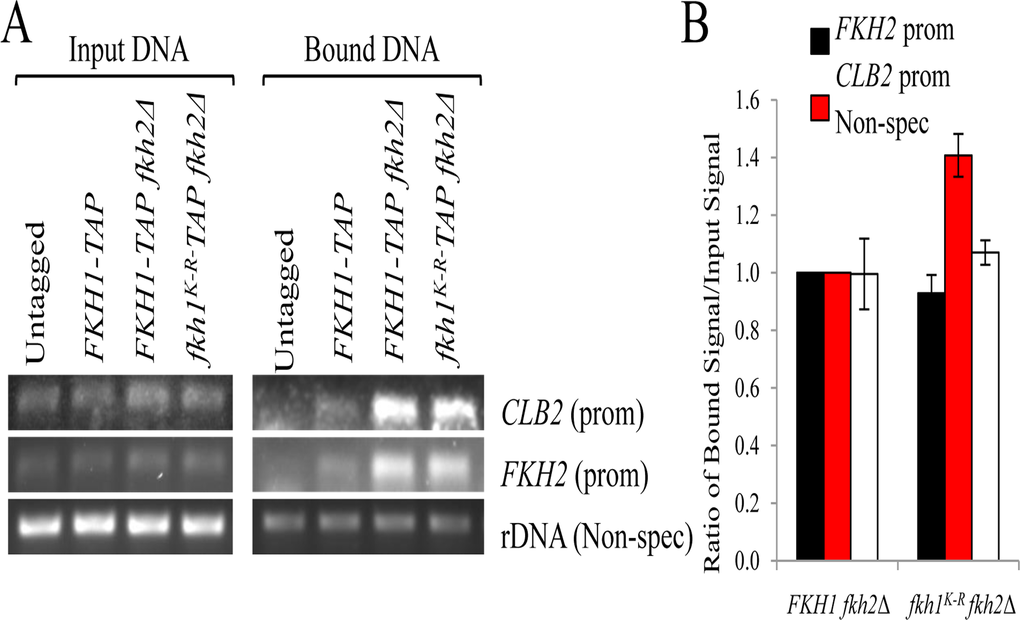
Figure 7. The Fkh1K-R mutation does not impair recruitment of Fkh1 to CLB2 and FKH2 promoters (A) ChIP was performed on the strains shown. DNA was purified and used as template in PCR reactions using primers specific for the loci indicated. 36-38 cycles were determined to be in the linear range for these PCR reactions. DNA extracted prior to IP was used as input controls. (B) The DNA bands in (A) were scanned, quantified, and a ratio of bound:input DNA was plotted.
Fkh1 interacts physically with Apc5
If the APC targets Fkh1 for ubiquitination and degradation, then it is likely that Apc5 and Fkh1 physically interact. Cells expressing endogenous Fkh1-TAP and plasmid-borne GST-Apc5 were harvested after asynchronous log phase growth. Our results demonstrate that Fkh1 and Apc5 do in fact physically interact in asynchronous cells (Fig. 8A). The control using only GST shows that the TAP antibody does not interact with GST (Fig. 8B). Fkh1 protein levels do fluctuate predictably throughout the cell cycle, demonstrated by G1 arrest followed by synchronous release back into the cell cycle (Fig. S4A). In WT cells, Fkh1-TAP levels were lowest in G1, then steadily increased as the cell cycle progressed. These cell cycle-dependent fluctuations of Fkh1-TAP were blunted and minimized in APC mutants, as Fkh1 protein levels were higher in G1 arrested APC mutants (Figs. S4A, S4C).
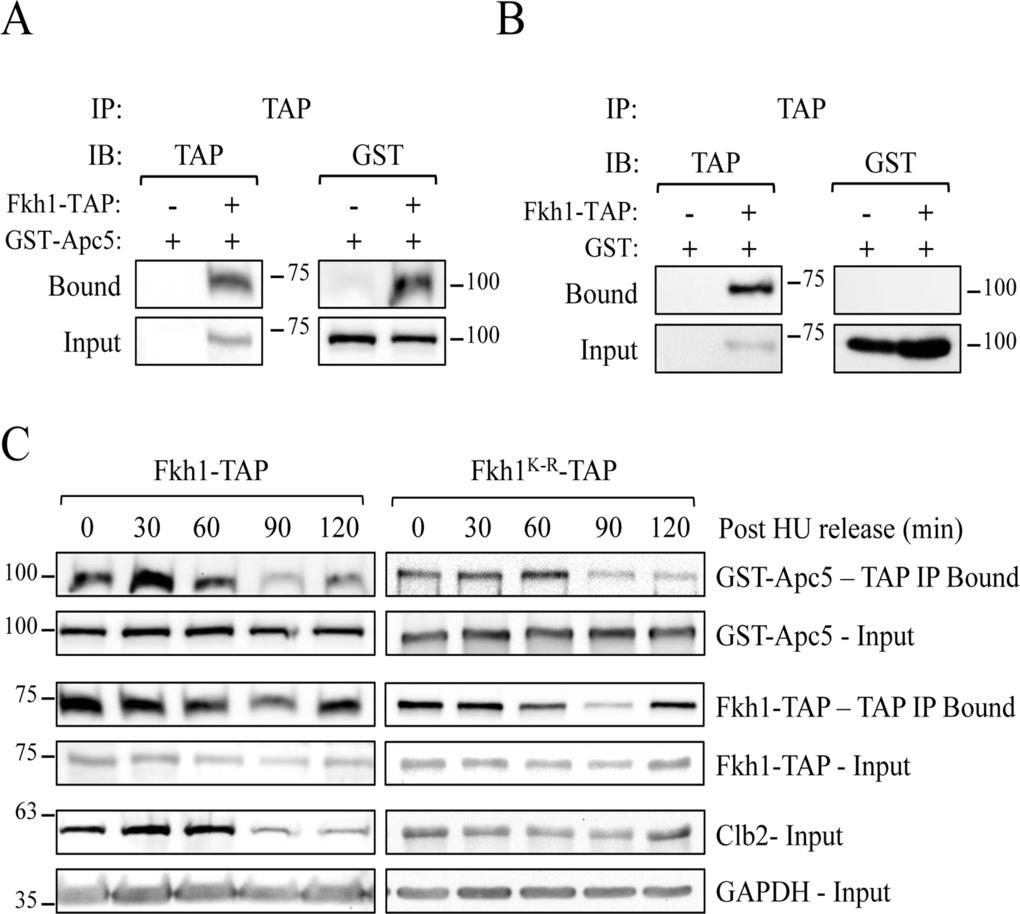
Figure 8. Fkh1 and Apc5 physically associate throughout the cell cycle (A) Fkh1-TAP cells expressing plasmid-borne GST-APC5 were grown overnight to early log phase. Lysates were prepared and IPed using antibodies against the TAP epitope. Westerns using antibodies against TAP and GST were done. (B) Fkh1-TAP cells expressing the empty GST vector were used as described for (A). (C) The various Fkh1-TAP cells expressing plasmid-borne GST-APC5 were arrested in S phase using HU. HU was then washed away, with cells released synchronously back into the cell cycle. Samples removed every 30 minutes were subjected to CoIP using TAP and Clb2 antibodies. Westerns were performed on input and bound samples using antibodies against TAP and GST. GAPDH was used as a load control. See also Figure S4.
We next asked if the Fkh1-Apc5 interaction is cell cycle dependent, perhaps preceding mitosis when Fkh1 is degraded. To determine this, we arrested the Fkh1-TAP cells in S phase, with samples removed for CoIP every 30 minutes after synchronous release back into the cell cycle (Fig. 8C). Apc5 co-immunoprecipitated with Fkh1 throughout the cell cycle, and its levels were proportional to Fkh1. Apc5 likewise continued to be observed in Fkh1K-R precipitates, with levels also proportional to the amount of Fkh1 IP'd. Our observations therefore suggest that Fkh1 and Apc5 are physically bound throughout the cell cycle, even though Fkh1 degradation only occurs during mitosis, and the stabilizing K373R mutation is irrelevant for this interaction.
Degradation of Fkh1 in mitosis is required for normal cell cycle progression through G1
The demonstration that the APC is required to target Fkh1 for degradation leads to the next question: what is the biological significance of this interaction? We hypothesized that Fkh1 degradation in mitosis is necessary to facilitate ordered cell cycle progression into G1. Furthermore, we propose that this progression involves Fkh1-dependent transcriptional control of Clb2, based on a combination of several observations: asynchronous apc5CA mutants exhibit high Clb2 and Fkh1 protein levels (Figs. 2A, 2B) [6, 28]; G1 arrested apc5CA mutants exhibit very low Clb2 levels (Figs. 9A, S4A); and Fkh proteins normally repress CLB2 expression during G1 [19, 23]. Together, this raises the possibility that the higher levels of Fkh1 protein (in G1 arrested APC mutants) could be keeping Clb2 protein levels low through transcriptional repression. To test this, WT, apc5CA and apc5CA fkh1Δ fkh2Δ cells were arrested in G1, then released back into the cell cycle with samples removed to determine Clb2 protein levels (Fig. 9A). Our hypothesis predicts that removal of Fkh1/2 function in the mutant APC background will return Clb2 protein levels to near normal. In apc5CA mutants, Clb2 was not apparent until after 90 minutes, and it took 120 minutes for cells to begin to move into G2/M. However, in apc5CA fkh1Δ fkh2Δ cells, Clb2 expression was clear after 15 minutes, with a synchronous movement of cells into S phase as early as 30 minutes (Figs. 9A, 9C). In WT cells, Clb2 protein levels begin to accumulate again after 60 minutes, which coincided with movement of cells from G1 to G2/M (Figs. 9A, 9B). These experiments, therefore, provide the biological relevance of Fkh1 degradation in mitosis as being essential for normal cell cycle progression.
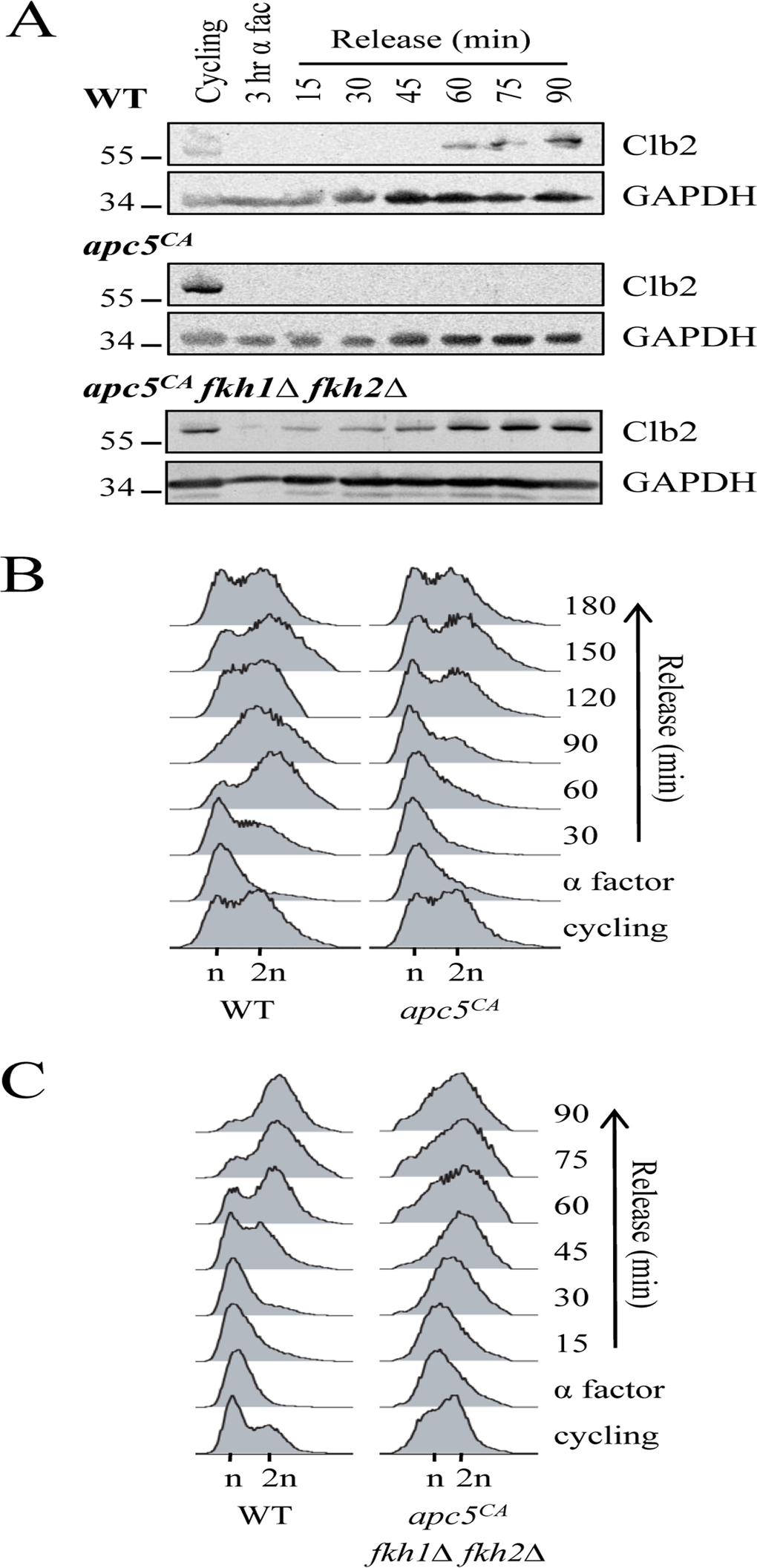
Figure 9. Removal of FKH1 and FKH2 re-establishes Clb2 synthesis and rapid cell cycle re-entry in apc5CA cells (A) WT, apc5CA and apc5CA fkh1Δ fkh2Δ cells were arrested in G1 and then synchronously released back into the cell cycle. Samples were removed every 15 minutes for Western analyses using antibodies against Clb2 and GAPDH. (B) WT and apc5CA cells used for the experiment in (A) were allowed to grow for 3 hours with samples removed every 30 minutes for analysis by flow cytometry. (C) Flow cytometry was used on the WT and apc5CA fkh1Δ fkh2Δ samples used in (A).
Stabilization of Fkh1 alters chronological lifespan and genomic stability
Additional important functions of the yeast Forkhead proteins includes lifespan and genomic stability, and here we assess whether Fkh1 protein stability is biologically important in these processes. Fkh1-TAP and Fkh1-TAP fkh2Δ cells exhibited near superimposable lifespans, when measured for chronological lifespan (CLS) (Fig. 10A). In contrast, fkh1K-R fkh2Δ cells have a remarkably shorter lifespan, with 99% of the cell population dead by four days, compared to 99% dead by day nine in the FKH1 WT strains. The presence of WT FKH2 in fkh1K-R cells partially abrogated the shortening of the K-R lifespan, supporting its compensatory effect for Fkh1 function.
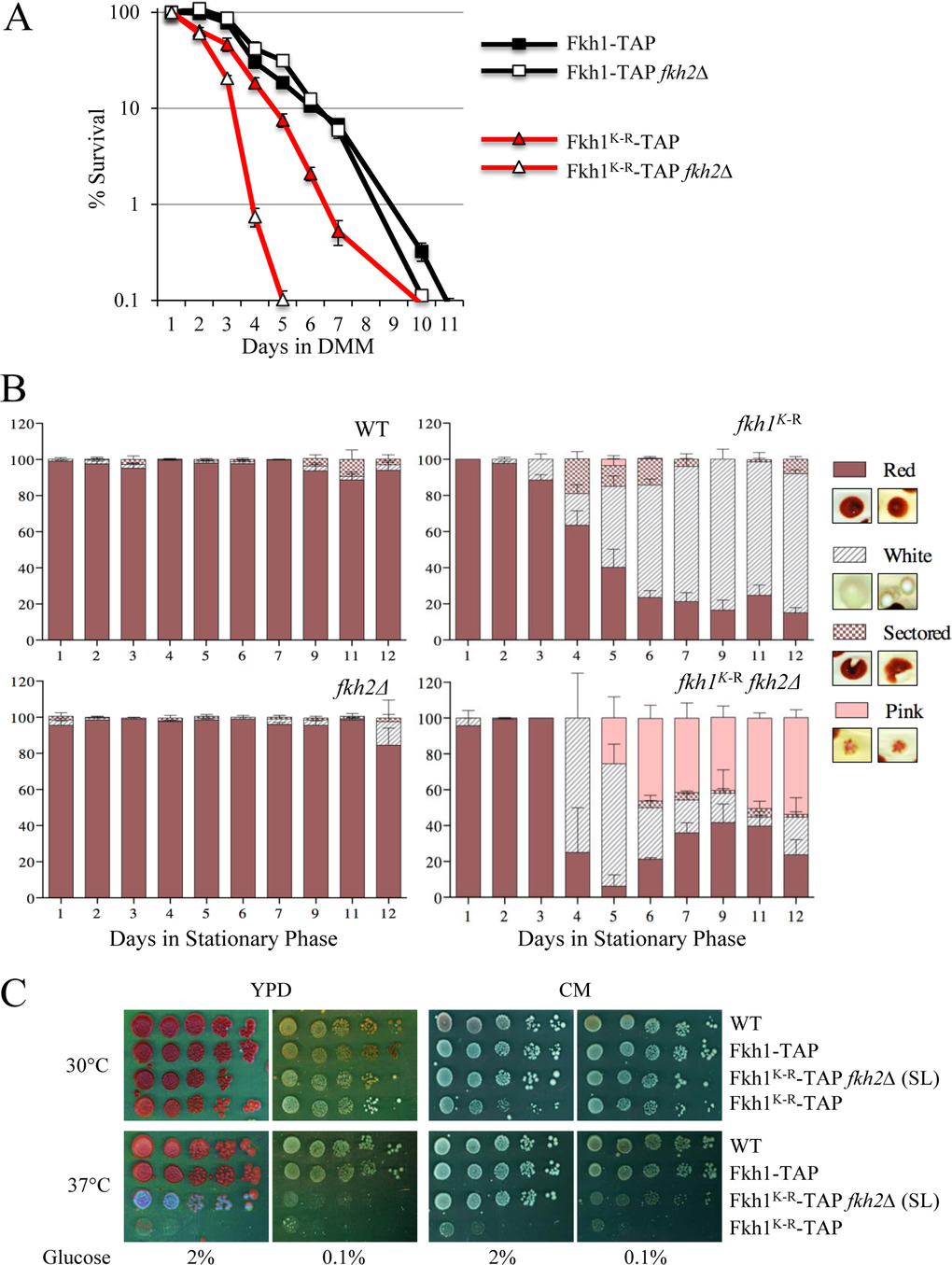
Figure 10. The Fkh1K-R mutation decreases chronological lifespan, increases genomic instability, and confers temperature sensitive growth (A) Chronological lifespan (CLS) was performed on cells expressing the FKH1K-R allele, in the presence or absence of FKH2. Cells were grown in Minimal Media (MM) and maintained in the Depleted Minimal Media (DMM) throughout the experiment. The experiments were performed in duplicate, at least twice. Standard error of the mean is shown. (B) FKH1K-R ade2 cells, in the presence or absence of FKH2, and in stationary phase, reverted from red to white/sectored. Each day following stationary phase, cells were plated as part of the CLS experiment. The plates were left for a week to fully develop color. The number of cells that were white, sectored, or a mottled pink were counted and plotted as a percentage of the total. (C) Strains expressing FKH1K-R, +/− FKH2, were grown overnight in 2% YPD media, then spot diluted onto plates containing rich YPD or complete medium (CM). Plates were supplemented with either 2 or 0.1% glucose. The plates were incubated for 3 to 6 days at 30 and 37°C.
Color sectoring of yeast colonies is a marker of genomic instability, and we serendipitously observed that individual cells plated during the CLS experiment were sectoring, depending on genetic background. Colonies derived from the WT and fkh2Δ strains did not sector (Fig. 10B). Samples of sectored colonies are shown in the inset for Fig. 10B. However, the red fkh1K-R FKH2 mutant colonies generated significant numbers of sectored red:white, and white colonies. This occurs when the red phenotype of ade2 mutants revert to white, due to spontaneous genomic mutation [47]. Mutations leading to red:white sectoring occur after plating, whereas the white colonies are due to mutations occurring prior to plating. The longer the cells sat in stationary phase, the more red:white colony sectoring occurred (Fig. 10B). Strikingly, it was most prevalent in the short-lived fkh1K-R fkh2Δ cells. Pink speckled colonies began to appear suggesting that mutations causing color change were ongoing and persistent despite being in stationary phase. Thus, the lifespan defect in Fkh1K-R cells appears to be linked with severe genomic instability.
Lastly, we questioned if stress response was impaired in the Fkh1K-R mutant, along with its shortened lifespan and elevated genomic instability. We directly assessed this by spot diluting cells onto rich media (YPD) or complete media (CM) in the presence of temperature and carbon (2% vs 0.1% glucose) stress. Growth on 0.1% glucose requires metabolic adaptation to utilize alternative carbon sources; complete media (CM) is a much more nutritionally restricted growth medium than YPD; and higher temperatures contribute further to adverse growth conditions (Fig. 10C). In general we observed that fkh1K-R FKH2 cells were ts regardless of plate conditions. The shorter-lived fkh1K-R fkh2Δ were more resilient, and were only ts when carbon stress was introduced (0.1% glucose). This investigation therefore confirms that Fkh1 degradation also plays an important biological role in carbon and temperature stress responses, which may be important for controlling genomic stability, as well as in the previous lifespan requirement. In conclusion, we have shown that Fkh1 is targeted for degradation by the APCCdc20 complex in mitosis, and have identified a highly conserved lysine that when mutated results in reduced CLS, genomic instability and ts growth.
Discussion
In this report we establish that Fkh1 is an APCCdc20 substrate targeted for degradation during mitosis. This is an important observation as it demonstrates that yeast Fox proteins are structurally and functionally conserved with higher eukaryotic systems. Considering the ease of yeast genetic manipulations, yeast will provide valuable insight into how Fox proteins are regulated at the molecular level. It is known that ubiquitination of Fox proteins plays a vital role in their regulation. It is primarily believed that nuclear Fox proteins are first phosphorylated, then shuttled from the nucleus to the cytoplasm, where they are targeted for degradation [48, 49]. Nuclear FOXO3A, for example, can be phosphorylated followed by Ub-dependent degradation by either AKT and the E3 SCF [50], or Erk and the E3 Mdm2 [51]. Likewise, FOXM1 protein is sequentially targeted by PLK1, then APCCdh1 activity during G1 [52-54]. However, knowledge of the fine details of this regulation, and the result of defective regulation, are lacking. Here, we show that mutation of a highly conserved lysine residue that stabilizes Fkh1 also results in a shortened yeast lifespan, increased genomic instability, increased sensitivity to stress, and delayed progression through G1. Expression of the stabilizing fkh1K-R allele does not, however, alter Apc5/Fkh1 interactions, nor disrupt recruitment of Fkh1 to CLB2 or FKH2 promoters. These observations provide a framework for further investigations towards a thorough understanding of Fox protein function and regulation.
Fkh1 is an APC substrate
Genetic interactions between FKH and APC mutants have been previously established, as deletion of both FKH1 and FKH2 exacerbates APC mutant cell cycle, lifespan and stress response phenotypes [5]. Nonetheless, while deletion of both FKH1 and FKH2 impair apc5CA defects, we show that mutation of only FKH1 suppressed these defects (Fig. 1). Mutations that suppress APC defects have been proposed to define potential APC substrates, as removal of these proteins blocks their ability to accumulate and impair cell cycle function [32]. In addition, many proteins that activate the APC, later become APC substrates [24, 25]. Fkh1, therefore, constituted a likely APC substrate. Our previous observation that the Fkhs act upstream of the APC, as activators [5], provided further evidence to suggest that Fkh1 had the potential to be an APC substrate. Our results presented here provide compelling evidence to support this hypothesis, as Fkh1 1) accumu-lates and is stabilized in APC and proteasome mutants, and with proteasome inhibitors (Fig. 2), 2) is reduced when APC subunits are overexpressed (Fig. 3), 3) is unstable during mitosis (Fig. 4), 4) is specifically stable in cdc20-1, but not cdh1Δ mutants (Fig. 5), and 5) is stabilized when a highly conserved lysine is mutated to arginine (Fig. 6). The stabilized Fkh1K-R protein resulted in reduced yeast CLS, increased genomic instability and increased sensitivity to stress (Fig. 10). Previous work from the Aparicio laboratory showed that Fkh1 and Fkh2 are required for early replication origin clustering, acting by facilitating long-range origin interactions [55]. More recently this lab showed that Fkh protein levels limit early origin firing, and that Fkh overexpression resulted in an increase in origin firing in early S phase. This increase was established in late G1 and slowed the progression of the replication fork [56]. Considering these observations, the inappropriate stabilization of Fkh1 during mitosis and G1 could lead to early firing of replication origins, thus further destabilizing cell cycle progression. Taken together, our results clearly show that the inability to target Fkh1 for degradation following mitotic onset has dire consequences to the cell.
Although Fox proteins are known in higher eukaryotic systems to be targeted for degradation, it was not at all clear in yeast how Fox proteins are regulated. Our detection of a physical interaction between Fkh1 with Apc5 lasting throughout the cell cycle supports the idea that Fkh1 is an APC substrate (Fig. 8). It is interesting that Apc5 and Fkh1 remain physically associated throughout the cell cycle, but that Fkh1 is only targeted for degradation during mitosis. One possible explanation for this association may be to allow the flexibility to rapidly respond to stress regardless of the cell cycle position. Both the APC and Fkh1 respond to stress [5, 57], thus it is possible a tight association of the two throughout the cell cycle allows for a rapid response to stress through the coordinated action of these two stress response pathways.
Accumulation of Fkh1 during mitosis delays cell cycle re-entry
We have observed that Clb2 accumulates in asynchronous apc5CA cells, yet does not accumulate in G1 arrested apc5CA or apc10Δ cells (Figs. 2A, 4F, 9A, S4A). Knowing that Fkh1 can repress CLB2 transcription in G1, we considered whether the low levels of Clb2 protein during G1 in APC mutants is a consequence of simultaneously elevated Fkh1 levels during G1. Our findings presented in Fig. 9 demonstrate that Clb2 protein synthesis and cell cycle progression through G1 was indeed delayed in apc5CA cells, supporting this concept. Interestingly, deletion of FKH1 and FKH2 in these cells not only reversed this effect, but accelerated Clb2 protein synthesis and cell cycle re-entry. Therefore, cell cycle progression through mitosis and G1 could rely on the downregulation of Fkh1.
In summary, our work presented here illuminates how Fkh1 function is regulated in a cell cycle-dependent manner. We identified a highly conserved Fkh1 lysine residue (K373) that regulates cell cycle-dependent protein stability, cellular lifespan, and genomic stability. This provides valued insight into how this family of conserved stress responsive transcription factors is regulated and what occurs when this regulation fails. Much remains to be learned regarding how the Fkhs interact in yeast with each other and with their binding partners. Our results therefore pave the way for future studies in higher eukaryotic systems.
Methods
Yeast strains and plasmids
The S. cerevisiae strains used in this study are shown in Table 1. All strains were isogenic derivatives of S288c strains except where noted. BY4741 (YTH1029) and the rpn10Δ::kanMX6 and cdh1Δ::kanMX6 derivatives, were obtained from the ResGen Library (provided by W. Xiao, U of Saskatchewan). The cdc20-1 strain and its associated WT (W303) were a kind gift from A. Amon (David Koch Institute for Integrative Cancer Research at MIT). PCR based methods were used to integrate the TAP::HIS3 cassette into the various cells used. Cells harboring deletions marked by the same phenotypic marker were generated by crossing. The plasmid encoding GAL-driven FKH1-HA (hemaglutinin epitope tag) was obtained from Open Biosytems and kindly provided by W. Xiao. The copper-driven (50 μM CuSO4 for 2 hours) GST-APC5, GST-APC10, and the empty vector control (pYEX4T-1) were obtained from the Exclone Library.
Table 1. Saccharomyces cerevisiae strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| YTH457 | MATa ade2 his3 leu2 ura3 apc5CA-PA::His5 | Harkness et al. 2002 |
| YTH1029 | MATa his3 leu2 met15 ura3 | W. Xiao |
| YTH1235 | MATa ade2 his3 leu2 lys2 ura3 | Harkness et al. 2002 |
| YTH1693 | MATa his3 leu2 met15 ura3 apc10Δ::kanMX6 | Harkness et al. 2005 |
| YTH2427 | MATa ade2 his3 leu2 lys2 met15 ura3 fkh1Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2431 | MATa ade2 his3 leu2 lys2 met15 ura3 apc5CA-PA::His5 fkh1Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2444 | MATa ade2 his3 leu2 lys2 met15 ura3 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2449 | MATa ade2 his3 leu2 lys2 met15 ura3 apc5CA-PA::His5 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2578 | MATa ade2 his3 leu2 lys2 met15 ura3 fkh1Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2579 | MATα ade2 his3 leu2 lys2 met15 ura3 fkh1Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2581 | MAT(?) ade2 his3 lys2 met15 ura3 apc5CA-PA::His5 fkh1Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH2582 | MATa ade2 his3 leu2 lys2 met15 ura3 apc5CA-PA::His5 fkh1Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH3124 | MAT(?) ade2 his3 leu2 lys2 met15 ura3 apc10Δ::kanMX6 fkh1Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH3346 | MAT(?) ade2 his3 leu2 lys2 met15 ura3 apc10Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH3408 | MAT(?) ade2 his3 leu2 lys2 met15 ura3 apc10Δ::kanMX6 fkh1Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH3409 | MAT(?) ade2 his3 leu2 lys2 met15 ura3 apc10Δ::kanMX6 fkh1Δ::kanMX6 fkh2Δ::kanMX6 | Postnikoff et al. 2012 |
| YTH3638 | MATa his3 leu2 met15 ura3 rpn10Δ::kanMX6 | W. Xiao |
| YTH3785 | MATa his3Δ1 leu2 met15Δ ura3 cdh1Δ::kanMX6 | W. Xiao |
| YTH3862 | MATa ade2 ura3 leu trp1 his3 can1 GAL psi+ | A. Amon |
| YTH3863 | MATα ade2 ura3 leu2 trp1 his3 can1 cdc20-1 | A. Amon |
| YTH3926 | as YTH1235, but FKH1-TAP::HIS3 | Postnikoff et al. 2012 |
| YTH3930 | as YTH457, but FKH1-TAP::HIS3 | Postnikoff et al. 2012 |
| YTH3933 | as YTH1693, but FKH1-TAP::HIS3 | Postnikoff et al. 2012 |
| YTH4004 | as YTH457, but bar1Δ::kanMX6 | Menzel et al. 2014 |
| YTH4112 | as YTH3785, but FKH1-TAP::HIS3 | This study |
| YTH4117 | as YTH1029, but FKH1-TAP::HIS3 | This study |
| YTH4267 | MATa ade2 his3 leu2 ura3 | Menzel et al. 2014 |
| YTH4373 | as YTH3638, but FKH1-TAP::HIS3 | This Study |
| YTH4379 | as YTH3638, but APC5-TAP::HIS3 | Islam et al. 2011 |
| YTH4617 | MatA ade2 leu2 trp1 ura3 FKH1-TAP::HIS3 | This Study |
| YTH4675 | as YTH1235, but fkh1K373RTAP::HIS3 | This Study |
| YTH4753 | MatA ade2 lys2 met15 ura3 FKH1-TAP::HIS3 fkh2Δ::kanMX6 | This Study |
| YTH4755 | MatA ade2 lys2 met15 ura3 fkh1K373RTAP::HIS3 fkh2Δ::kanMX6 | This Study |
| YTH4798 | as YTH4617, but cdc20-1 Mat A | This Study |
Media and methods
Yeast growth media used in this study, yeast/peptone/dextrose (YPD) and drop out (DO) supplemented with required amino acids, was described previously [5, 28]. Yeast Minimal Media (MM) used in chronological lifespan (CLS) experiments contained 0.17% yeast nitrogen base/0.5% ammonium sulfate/0.003% essential amino acids, and 2% glucose unless otherwise indicated, with the pH adjusted using sodium hydroxide. Yeast Complete Media (CM) was also used in CLS experiments (0.17% yeast nitrogen base/0.5% ammonium sulfate/2% glucose/0.13% dropout powder including all amino acids, and 1 sodium hydroxide tablet). Methods including spot dilutions, Westerns, reverse transcriptase PCR, E. coli and yeast transformations, flow cytometry, DNA mini-preps, oxidative stress plate assays, and replicative and chronological lifespan assays were all performed according to our previous publications [5, 28, 29, 58]. For MG132 experiments, cultures were started in YPD to ensure log phase growth, then transferred to synthetic media (0.17% yeast nitrogen base without ammonium sulfate) supplemented with 0.1% proline, essential amino acids, and 2% glucose, with addition of 0.003% SDS the next day for an additional 3 hours at 30°C. At this point 75 μM MG132 (Enzo Life Sciences) or the drug vehicle (dimethyl sulfoxide; DMSO) were added, and cells were incubated at 30°C for 3 hours before harvesting and storage at −80°C. Genomic instability was determined by scoring the number of red, white, red/white sectored and pink speckled colonies following stationary phase growth. Four separate plates were counted for each strain on each day. Antibodies against Clb2 (Santa Cruz Biotechnology), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Sigma), α-tubulin (Sigma), and Tandom Affinity Purification (TAP; Pierce) where purchased from the indicated suppliers. The GST J90 GST antibody was described previously [33].
TAP Co-immunoprecipitation (Co-IP)
Lysates were generated using a standard beadbeat protocol [26] in RIPA buffer lacking SDS. TAP tagged proteins were IPed using IgG Sepharose 6 Fast Flow beads (GE Healthcare), essentially as described [33], and eluted lysates were analyzed by western blot to identify co-purified proteins. To generate lysates used in Co-IP, frozen cell pellets were resuspended in 2X cell volume of ice-cold RIPA (Radio Immuno Precipitation Assay Buffer) lysis buffer containing non-ionic detergents, a 1:500 dilution of protease inhibitor cocktail (BioVision), 10 μM n-ethylmaleimide, 1.2 mM dithiothreitol (DTT), and 5 mM EDTA. Iced 0.5 mm glass beads were added to the suspension and the frozen cells were mechanically disrupted using a Mini-Beadbeater (Biospec products). Lysates were clarified and total protein concentration was determined using the Bradford method (BioRad). Protein lysates were added to washed IgG Sepharose 6 Fast Flow beads (GE Healthcare) and rotated end-over-end at 4°C for 90 minutes. Following incubation, beads were washed 2X with lysis buffer, and proteins were eluted off beads by boiling in 2X Laemmli sample buffer and then SDS-PAGE was used to separate proteins for western analyses.
Cell cycle arrest
Cell cycle arrest was performed according to our standard procedures [6, 21]. Hydroxyurea (Sigma) was used at 0.3 M for approximately 3 hours for S phase arrest. For mitotic arrest, Nocodazole (Calbiochem) was used for approximately 3 hours, with 5 μg/ml added initially and then an additional 2.5 μg/ml added after 90 minutes. For G1 arrest of BAR1 strains, cells were first washed 2X with fresh YPD, and then resuspended in YPD at pH 3.5 containing 2 μg/ml of α-factor (Zymo Research) and cultured for approximately 3 hours, with an additional 2 μg/ml added after 90 min. Cultures were considered arrested once >90% of the cells were in the desired phase of the cell cycle according to bud morphology. To release cells from arrest, cultures were washed twice with fresh media. To stop protein synthesis Cycloheximide (CHX; Calbiochem) was used at 10 μg/ml. Arrests were confirmed by flow cytometry (Saskatoon Cancer Center/Pharmacy & Nutrition), as performed previously [5, 26].
Quantitative PCR (Q-PCR)
Cells were cultured in YPD at 30°C, and samples were collected in mid log phase (ie.OD600=0.46-0.90). Equivalent cell amounts were harvested for each sample, for a total of 6×108 total cells per sample. Total RNA was extracted using the hot phenol method with DNA digestion completed using DNAse I (Fermentas) in the presence of RNAse inhibitors (Fermentas). The quality of RNA was assessed spectrophotometrically by measuring the OD260/280 ratio. Only samples that measured a ratio between 1.8-2.0 were used. The RNA integrity was assessed by visual inspection of the 28S/18S subunits on a formaldehyde agarose gel. First-strand synthesis was performed on 1 μg of total RNA using RevertAid Reverse Transcriptase (Fermentas) following manufacturers’ recommendations. Gene-specific primers targeted against yeast ACT1 and FKH1 (Table 2) were designed based on full-length sequences available from the Saccharomyces Genome Database (SGD). The expression of FKH1 was normalized with reference to the ACT1 endogenous control gene. Standard PCR reactions were completed in the mid-linear range for each message using our previously published protocol [29].
Table 2. Primer pairs
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| FKH1 | 5′-TCTCTTTCAGCAGTGGTG | 5′-CTTGTACTCTTCCGGTAG |
| ACT1 | 5′-CGCCTCTGTGACAAGACAGC | 5′-GTCGTAGTAGAAATTTGGCC |
| FKH2 Promoter | 5′-CACTTCAAAGCCAATCTTCCC | 5′-TTCTTGCGCATTGATAAAGT |
| CLB2 Promoter | 5′-GACAGATTTTATTCCAAATGCGG | 5′-CGCTTTTCAGAAGTATCAATTCG |
| FKH1K373R | 5′-GTGCCCAAAAGAGCCGGCCAACAAGGTAGAGGG | 5′-TTTCCAATTCATCCCTCTACCTTGTTGGCCGGCTC |
| FKH1DB | 5′-TGGCAGAACTCCGTTGCGCATAATGCGTCC | 5′-GGCTTTATTTAGGGACGCATTATGCGCAACGG |
| rDNA Internal | 5′-TAAGCGGAGGAAAAGAAACC | 5′-CTCTTCGAAGGCACTTTACA |
Chromatin Immunoprecipitation (ChIP)
CHIP was performed with modifications to those previously described [59]. Following cross-linking of the cell lysate (1% formaldehyde at room temperature for 15 min), and quenching of the cross-linker (5 min incubation in 125 mM glycine) cells were washed 2× with iced TBS (20 mM Tris HCL pH 7.6, 150 mM NaCl) and frozen at −80°C. Frozen cells were resuspended in lysis buffer (50 mM Hepes-KOH pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na deoxycholate, 1:500 dilution of protease inhibitor cocktail (BioVision)) and lysed by mechanical disruption using iced 0.5 mm glass beads with two 30-sec pulses in a Mini-BeadBeater (Biospec Products). Lysates were sonicated 5 × 10 sec at output 2 (Branson Sonifier 250). Sonicated lysates were clarified and total protein was quantified using the Bradford method (BioRad). Equivalent amounts of total protein were incubated with washed IgG Sepharose 6 Fast Flow beads (GE Healthcare) and rotated end-over-end at 4°C for 90 minutes. The IPs were washed 2× with lysis buffer, 1x with lysis buffer with 500 mM NaCl, 1x with CHIP wash buffer (10 mM Tris-HCl pH 8.0, 0.25 M LiCl, 0.5% NP-40, 0.5% Na deoxycholate, and 1 mM EDTA), and 1x with TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Samples were eluted from the beads by incubating for 15 min at 65°C with 50 mM Tris-HCl pH 8.0, 10 mM EDTA, and 1% SDS. The beads were then washed with TE/0.67% SDS and the wash was pooled with the eluted sample. Cross-links were reversed by overnight incubation at 65°C, followed by protein digestion with Proteinase K (Sigma), and RNA digestion with RNAse A (Sigma). DNA was phenol-extracted, ethanol-precipitated, and resuspended in H2O. Crude input samples were made up to 250 μL with TE/1% SDS and the cross-links were reversed and the DNA purified as indicated above.
Equivalent amounts of input DNA for each sample were used in PCR. Two sets of primers were used in each reaction. Primer sets were designed to amplify the promoter region of CLB2 or FKH2, or part of the RDN1 coding sequence, which served as a loading control. PCR involved an initial denaturation of 30 sec at 98°C, then 36-40 cycles at: 98°C for 15 sec, 55°C for 20 sec, and 72°C for 20 sec, with a final extension at 72°C for 5 min. Control PCR reactions were carried out with a range of cycles to ensure that we were in the linear portion of the assay.
Statistics
The Standard Error of the Mean was calculated in Microsoft Excel and was used for error bars. Significance was calculated using the Student T-test for comparing 2 Independent Means (http://www.socscistatistics.com).
Supplementary Materials
Author Contributions
MEM and SDLP each contributed to experimental design, completion and interpretation. TGA contributed to figure production, writing, and editing of the manuscript. TAAH funded and designed the project, and wrote the manuscript.
Acknowledgments
We appreciate the kind gifts of strains and plasmids from Drs. A. Amon and W. Xiao. Dr. B.F. Eames is thanked for thoughtful review of the manuscript.
Funding
Funds for this work were provided by the Canadian Institutes for Health Research (CIHR) to TAAH.
Conflicts of Interest
The authors have no conflict of interests to declare.
References
- 1. Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans.Science.2010;328:321-326..
- 2. Kennedy BK, Austriaco NR Jr, Guarente L. Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span.J. Cell Biol.1994;127:1985-1993..
- 3. Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and chronological aging in Saccharomyces cerevisiae.Cell Metab.2012;16:18-31..
- 4. Harkness TA, Shea KA, Legrand C, Brahmania M, Davies GF. A functional analysis reveals dependence on the anaphase-promoting complex for prolonged life span in yeast.Genetics.2004;168:759-774..
- 5. Postnikoff SD, Malo ME, Wong B, Harkness TA. The yeast forkhead transcription factors fkh1 and fkh2 regulate lifespan and stress response together with the anaphase-promoting complex.PLoS Genet.2012;8:e1002583.
- 6. Menzel J, Malo ME, Chan C, Prusinkiewicz M, Arnason TG, Harkness TA. The anaphase promoting complex regulates yeast lifespan and rDNA stability by targeting Fob1 for degradation.Genetics.2014;196:693-709..
- 7. Chandler-Brown D, Choi H, Park S, Ocampo BR, Chen S, Le A, Sutphin GL, Shamieh LS, Smith ED, Kaeberlein M. Sorbitol treatment extends lifespan and induces the osmotic stress response in Caenorhabditis elegans.Front. Genet.2015;6:316.
- 8. Huang X, Leggas M, Dickson RC. Drug synergy drives conserved pathways to increase fission yeast lifespan.PLoS One.2015;10:e0121877.
- 9. Cui HJ, Liu XG, McCormick M, Wasko BM, Zhao W, He X, Yuan Y, Fang BX, Sun XR, Kennedy BK, Suh Y, Zhou ZJ, Kaeberlein M, Feng WL. PMT1 deficiency enhances basal UPR activity and extends replicative lifespan of Saccharomyces cerevisiae.Age.2015;37:9788.
- 10. Shapira M, Segal E, Botstein D. Disruption of yeast forkhead-associated cell cycle transcription by oxidative stress.Mol. Biol. Cell.2004;15:5659-5669..
- 11. Postnikoff SD and Harkness TA. Mechanistic insights into aging, cell-cycle progression, and stress response.Front Physiol.2012;3:183.
- 12. Charitou P and Burgering BM. Forkhead box (O) in control of reactive oxygen species and genomic stability to ensure healthy lifespan.Antioxid. Redox. Signal.2013;19:1400-1419..
- 13. Coomans de Brachène A and Demoulin JB. FOXO transcription factors in cancer development and therapy.Cell. Mol. Life Sci.2016;73:1159-1172..
- 14. Martins R, Lithgow GJ, Link W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity.Aging Cell.2016;15:196-207..
- 15. Huang H and Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation.Biochim. Biophys. Acta.2011;1813:1961-1964..
- 16. Zhao Y, Wang Y, Zhu WG. Applications of post-translational modifications of FoxO family proteins in biological functions.J. Mol. Cell. Biol.2011;3:276-282..
- 17. Chiacchiera F and Simone C. The AMPK-FoxO3A axis as a target for cancer treatment.Cell Cycle.2010;9:1091-1096..
- 18. Murakami H, Aiba H, Nakanishi M, Murakami-Tonami Y. Regulation of yeast forkhead transcription factors and FoxM1 by cyclin-dependent and polo-like kinases.Cell Cycle.2010;9:3233-3242..
- 19. Linke C, Klipp E, Lehrach H, Barberis M, Krobitsch S. Fkh1 and Fkh2 associate with Sir2 to control CLB2 transcription under normal and oxidative stress conditions.Front Physiol.2013;4:173.
- 20. Wang JJ, Qiu L, Cai Q, Ying SH, Feng MG. Transcriptional control of fungal cell cycle and cellular events by Fkh2, a forkhead transcription factor in an insect pathogen.Sci. Rep.2015;5:10108.
- 21. Jiao R, Postnikoff S, Harkness TA, Arnason TG. The SNF1 Kinase Ubiquitin-associated Domain Restrains Its Activation, Activity, and the Yeast Life Span.J. Biol. Chem.2015;290:15393-15404..
- 22. Koranda M, Schleiffer A, Endler L, Ammerer G. Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters.Nature.2000;406:94-98..
- 23. Sherriff JA, Kent NA, Mellor J. The Isw2 chromatin-remodeling ATPase cooperates with the Fkh2 transcription factor to repress transcription of the B-type cyclin gene CLB2.Mol. Cell. Biol.2007;27:2848-2860..
- 24. Craney A and Rape M. Dynamic regulation of ubiquitin-dependent cell cycle control.Curr. Opin. Cell Biol.2013;25:704-710..
- 25. Zhang J, Wan L, Dai X, Sun Y, Wei W. Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis.Biochim. Biophys. Acta.2014;1845:277-293..
- 26. Harkness TA, Davies GF, Ramaswamy V, Arnason TG. The ubiquitin-dependent targeting pathway in Saccharomyces cerevisiae plays a critical role in multiple chromatin assembly regulatory steps.Genetics.2002;162:615-632..
- 27. Harkness TA, Arnason TG, Legrand C, Pisclevich MG, Davies GF, Turner EL. Contribution of CAF-I to anaphase-promoting-complex-mediated mitotic chromatin assembly in Saccharomyces cerevisiae.Eukaryot. Cell.2005;4:673-684..
- 28. Turner EL, Malo ME, Pisclevich MG, Dash MD, Davies GF, Arnason TG, Harkness TA. The Saccharomyces cerevisiae anaphase-promoting complex interacts with multiple histone-modifying enzymes to regulate cell cycle progression.Eukaryot. Cell.2010;9:1418-1431..
- 29. Islam A, Turner EL, Menzel J, Malo ME, Harkness TA. Antagonistic Gcn5-Hda1 interactions revealed by mutations to the Anaphase Promoting Complex in yeast.Cell Div.2011;6:13.
- 30. Jia L, Kim S, Yu H. Tracking spindle checkpoint signals from kinetochores to APC/C.Trends Biochem. Sci.2013;38:302-311..
- 31. Chang L and Barford D. Insights into the anaphase-promoting complex: a molecular machine that regulates mitosis.Curr. Opin. Struct. Biol.2014;29:1-9..
- 32. Thornton BR and Toczyski DP. Securin and B-cyclin/CDK are the only essential targets of the APC.Nat. Cell Biol.2003;5:1090-1094..
- 33. Arnason TG, Pisclevich MG, Dash MD, Davies GF, Harkness TA. Novel interaction between Apc5p and Rsp5p in an intracellular signaling pathway in Saccharomyces cerevisiae.Eukaryot. Cell.2005;4:134-146..
- 34. Yu H, Peters JM, King RW, Page AM, Hieter P, Kirschner MW. Identification of a cullin homology region in a subunit of the anaphase-promoting complex.Science.1998;279:1219-1222..
- 35. Zachariae W, Shevchenko A, Andrews PD, Ciosk R, Galova M, Stark MJ, Mann M, Nasmyth K. Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins.Science.1998;279:1216-1219..
- 36. Belew AT and Dinman JD. Cell cycle control (and more) by programmed −1 ribosomal frameshifting: implications for disease and therapeutics.Cell Cycle.2015;14:172-178..
- 37. Hollenhorst PC, Bose ME, Mielke MR, Müller U, Fox CA. Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae.Genetics.2000;154:1533-1548..
- 38. Morillon A, O'Sullivan J, Azad A, Proudfoot N, Mellor J. Regulation of elongating RNA polymerase II by forkhead transcription factors in yeast.Science.2003;300:492-495..
- 39. Goh PY, Lim HH, Surana U. Cdc20 protein contains a destruction-box but, unlike Clb2, its proteolysisis not acutely dependent on the activity of anaphase-promoting complex.Eur. J. Biochem.2000;267:434-449..
- 40. Isasa M, Katz EJ, Kim W, Yugo V, González S, Kirkpatrick DS, Thomson TM, Finley D, Gygi SP, Crosas B. Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome.Mol. Cell.2010;38:733-745..
- 41. Pramila T, Wu W, Miles S, Noble WS, Breeden LL. The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle.Genes Dev.2006;20:2266-2278..
- 42. Teixeira LK and Reed SI. Ubiquitin ligases and cell cycle control.Annu. Rev. Biochem.2013;82:387-414..
- 43. Pesin JA and Orr-Weaver TL. Regulation of APC/C activators in mitosis and meiosis.Annu. Rev. Cell Dev Biol.2008;24:475-499..
- 44. Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression.Nat. Cell Biol.2004;6:138-145..
- 45. Simpson-Lavy KJ and Breis M. Clb2 and the APC/C(Cdh1) regulate Swe1 stability.Cell Cycle.2010;9:3046-3053..
- 46. Ostrow AZ, Nellimoottil T, Knott SR, Fox CA, Tavaré S, Aparicio OM. Fkh1 and Fkh2 bind multiple chromosomal elements in the S. cerevisiae genome with distinct specificities and cell cycle dynamics.PLoS One.2014;9:e87647.
- 47. Hieter P, Mann C, Snyder M, Davis RW. Mitotic stability of yeast chromosomes: a colony color assay that measures nondisjunction and chromosome loss.Cell.1985;40:381-392..
- 48. Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation.Proc. Natl. Acad. Sci. USA.2003;100:11285-11290..
- 49. Plas DR and Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome.J. Biol. Chem.2003;278:12361-12366..
- 50. Huang H, Regan KM, Wang F, Wang D, Smith DJ, van Deursen JM, Tindall DJ. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation.Proc. Natl. Acad. Sci. USA.2005;102:1649-1654..
- 51. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, Huang H, Kuo HP, Lee DF, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation.Nat. Cell Biol.2008;10:138-148..
- 52. Laoukili J, Alvarez-Fernandez M, Stahl M, Medema RH. FoxM1 is degraded at mitotic exit in a Cdh1-dependent manner.Cell Cycle.2008;7:2720-2726..
- 53. Park HJ, Costa RH, Lau LF, Tyner AL, Raychaudhuri P. Anaphase-promoting complex/cyclosome-CDH1-mediated proteolysis of the forkhead box M1 transcription factor is critical for regulated entry into S phase.Mol. Cell. Biol.2008;28:5162-5171..
- 54. Zhang J, Yuan C, Wu J, Elsayed Z, Fu Z. Polo-like kinase 1-mediated phosphorylation of Forkhead box protein M1b antagonizes its SUMOylation and facilitates its mitotic function.J. Biol. Chem.2015;290:3708-3719..
- 55. Knott SR, Peace JM, Ostrow AZ, Gan Y, Rex AE, Viggiani CJ, Tavaré S, Aparicio OM. Forkhead transcription factors establish origin timing and long-range clustering in S.cerevisiae. Cell.2012;148:99-111..
- 56. Peace JM, Villwock SK, Zeytounian JL, Gan Y, Aparicio OM. Quantitative BrdU immunoprecipitation method demonstrates that Fkh1 and Fkh2 are rate-limiting activators of replication origins that reprogram replication timing in G1 phase.Genome Res.2016;26:365-375..
- 57. Rodríguez-Colman MJ, Sorolla MA, Vall-Llaura N, Tamarit J, Ros J, Cabiscol E. The FOX transcription factor Hcm1 regulates oxidative metabolism in response to early nutrient limitation in yeast. Role of Snf1 and Tor1/Sch9 kinases.Biochim. Biophys. Acta.2013;1833:2004-2015..
- 58. Postnikoff SD and Harkness TA. Replicative and chronological life-span assays.Methods Mol. Biol.2014;1163:223-227..
- 59. Pramila T, Miles S, GuhaThakurta D, Jemiolo D, Breeden LL. Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle.Genes Dev.2002;16:3034-3045..