



Introduction
The nematode Caenorhabditis elegans has been invaluable to biological research of mechanisms that slow aging processes and may prevent age-related diseases. C. elegans is well suited for molecular studies of aging because of its easy genetic tractability and lifespan of less than 3 weeks. Aging has been linked to a variety of different signaling pathways, most notably the reproductive system. Removing the germline of C. elegans extends lifespan by up to 60% [1]. The role of germline signaling in the regulation of health and longevity has been shown to be evolutionarily conserved in several species, including C. elegans, D. melanogaster, and mice [1-4]. Also, in humans it has been suggested that the reproductive state and lifespan correlate [5,6]. The lifespan extension associated with ablation of the germline in C. elegans is caused specifically by loss of proliferating germline stem cells and requires the preservation of the somatic gonad [1,7]. These findings suggest that longevity is not simply a result of sterility but is regulated by counterbalancing signals produced by the germline and somatic gonad. In C. elegans, the effects of germline absence on longevity have been studied either by laser ablation of germline precursor cells or by genetic mutants that show defects in germ cell proliferation. The most studied gene in this context is glp-1, which encodes a homolog of Notch that is expressed in germline stem cells and whose normal role is to promote proliferation of germ cells [8]. Mutation of glp-1 reduces the number of germline stem cells and thus promotes longevity [7].
The FOXO-family transcription factor DAF-16 is needed for germline removal to extend lifespan [1]. DAF-16 is best known for its ability to promote longevity in response to reduced insulin/insulin-like growth factor 1 (IGF-1) signaling [reviewed in 9]. However, the mechanisms by which insulin/IGF-1 signaling and germline loss activate DAF-16 seem to be distinct. For example, KRI-1/KRIT ankyrin repeat protein and the TCER-1/TCERG1 transcription elongation factor are required for germline absence to induce DAF-16 nuclear accumulation and promote longevity but are not involved in insulin/IGF-1 signaling [10,11]. Moreover, loss of germ cells further increases the lifespan of already long-lived insulin pathway mutants [1]. Ablation of the germline leads to DAF-16 activation and accumulation primarily in nuclei of intestinal cells. The intestine seems to play a key role in this pathway, as expression of DAF-16 exclusively in this tissue is sufficient to extend lifespan in germline-less animals [12]. Steroid hormone signaling also plays an important role for gonadal longevity. In germline-deficient animals, the nuclear hormone receptor DAF-12 and DAF-9, a cytochrome P450 synthesizing DAF-12 ligands, stimulate nuclear accumulation of DAF-16 and promote longevity [10,13].
Genetic experiments revealed that longevity signaling from the reproductive system involves several other transcription factors in addition to DAF-16. Recently, the transcription factor SKN-1, orthologous to mammalian Nrf (NF-E2 related factor) proteins has been implicated in long life from germline-less animals [14-16]. SKN-1 mediates a wide range of oxidative stress defense, detoxification, has important metabolic functions, and promotes longevity in various species [reviewed in 17].
The cell cycle is a well-coordinated set of events culminating in cell growth and division. Evolutionarily conserved regulators of this process include cyclins, cyclin-dependent kinases (CDKs) and CDK inhibitors (CKI). CDKs partner with regulatory subunits, the cyclins, which control kinase activity and substrate specificity. CDK/cyclin complexes thus ensure sequential progression through the cell cycle in an ordered fashion [reviewed in 18]. A key regulator for progression of the cell cycle from the G1 to the S phase is the CDK2/cyclin E complex. Once activated, this complex phosphorylates and therefore inhibits the retinoblastoma protein Rb, hence releasing the E2F transcription factor which activates gene expression for cell cycle progression [19-21].
In addition to their main functions in cell cycle control, recent research has indicated that mammalian CDKs, cyclins, and CKIs play diverse roles in a variety of cellular processes such as transcription, DNA-damage repair, epigenetic regulation, metabolism, proteolytic degradation, and stem cell self-renewal [reviewed in 22]. Interestingly, CDKs and cyclins can accomplish these functions at least in part without complex formation. Notably, studies in C. elegans revealed important functions of cell cycle regulators during development beyond their traditional role in cell cycle. CDK-1 and cyclin B contribute to transition from oocyte to embryo, asymmetric cell division, and cell fate specification by regulating the localization and timely elimination of cell fate determinants [23-25]. CDK-2 and cyclin E have been shown to control the balance between mitotic proliferation and meiotic differentiation in the C. elegans germline by reducing abundance of the GLD-1 translational repressor [26,27]. CDC25 phosphatases are key positive cell cycle regulators through their ability to remove inhibitory phosphate from CDK/cyclin complexes [28]. Intriguingly, knockdown of cdc-25 in adult C. elegans has been shown to promote stress tolerance and longevity [29]. However, the molecular mechanisms how cdc-25 influences aging remain to be elucidated. Together, these studies suggest that CDKs, cyclins, and regulatory proteins can also influence cellular and developmental processes in addition to the cell cycle.
The genetic evidence that CDC-25 is important for stress response and aging raises the question of whether further cell cycle regulators or the entire cell cycle machinery may have broader roles in these processes. Here, we have investigated how longevity is affected by genetic inhibition of cdk-2 and cyclin E (encoded by cye-1) in C. elegans. We find that heat tolerance, oxidative stress resistance, and longevity are increased when cdk-2 and cye-1 are inhibited. We localize this effect to the germline and identify that germline signaling plays a central role in its influence on aging. SKN-1 and DAF-16 activate protective gene expression and extend lifespan when cdk-2/cye-1 is inactivated. Genetic interference with further cell cycle genes also increases lifespan and triggers a similar SKN-1-dependent response, suggesting the notion that cell cycle factors might influence aging through their regulatory functions in germline proliferation.
Results
Inhibition of cell cycle regulates adult lifespan in C. elegans
To assess the function of cell cycle factors in regulation of longevity we first knocked down the cyclin-dependent kinase cdk-2 by RNA interference (RNAi). We used RNAi feeding starting from L4/early adulthood to inhibit cdk-2 and obtain results that were not confounded by its developmental functions. We found that post-developmental inactivation of cdk-2 had a strong effect on lifespan with an increase of 28% compared to wild type control (24.1 d in cdk-2(RNAi) vs 18.8 d control(RNAi)) (Figure 1A and Table 1). Thus, inhibition of cdk-2 function during adulthood is sufficient to confer longevity. The activity of cyclin-dependent kinases (CDKs) and their substrate specificity is regulated by cyclins. CDK-2 acts in concert with the cyclin E homolog CYE-1 to govern mitotic cell cycle [reviewed in 30]. Therefore we next tested knockdown of cye-1. cye-1(RNAi) resulted in lifespan extension similar to cdk-2(RNAi) (24.9 d in cye-1(RNAi) vs 19.5 d control(RNAi)) (Figure 1B and Table 1).
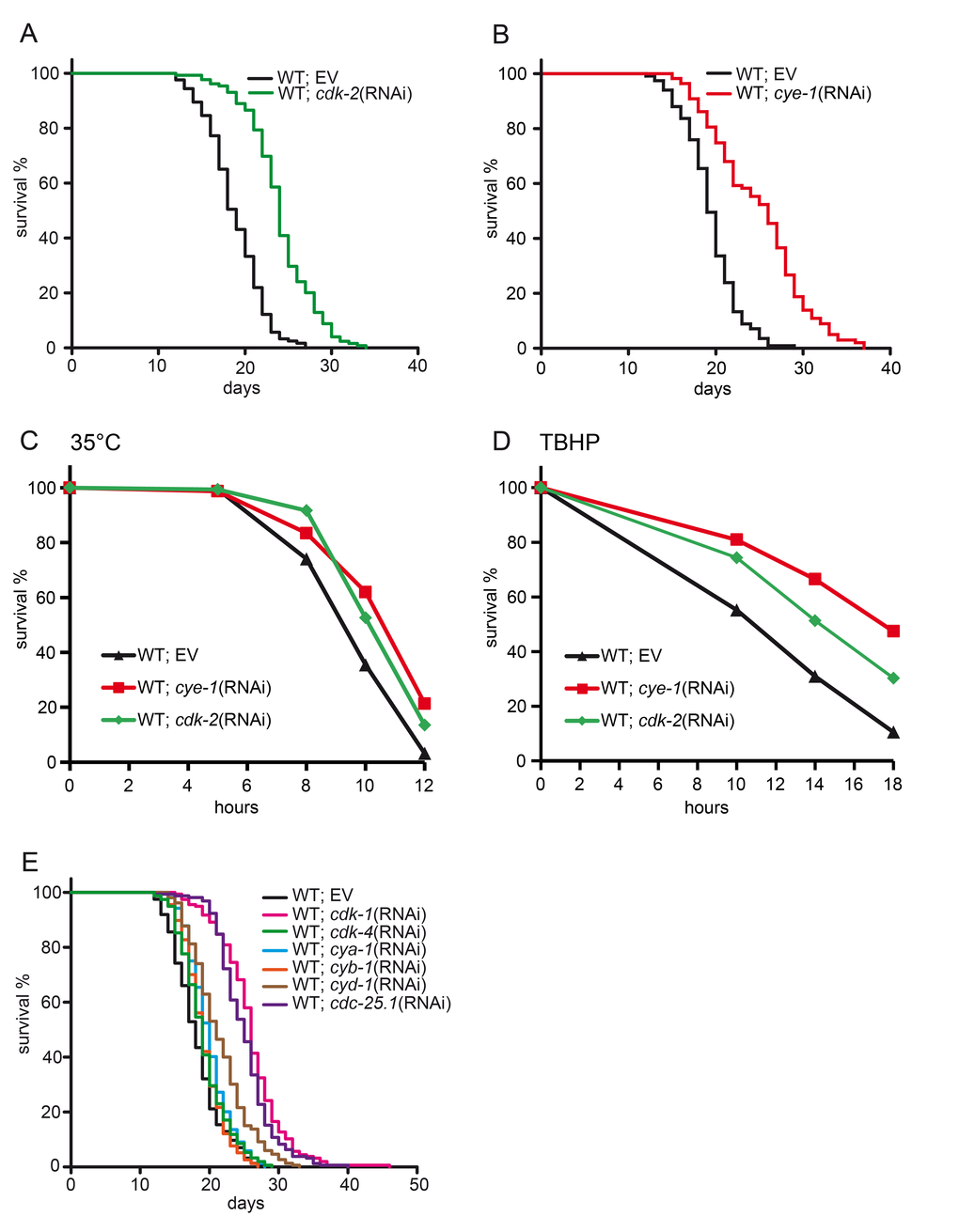
Figure 1. Inhibition of cell cycle extends lifespan and enhances stress tolerance. (A) Lifespan analysis in wild type animals fed with cdk-2(RNAi) or control. Inhibition of cdk-2 increases the mean lifespan by 28%. (B) Lifespan analysis in wild type animals fed with cye-1(RNAi) or control. (C) Increased resistance to heat (35°C) deriving from cdk-2 and cye-1(RNAi). See Table S3 for statistics. (D) Inhibition of cye-1 and cdk-2 by RNAi increases resistance to oxidative stress from TBHP. Statistical analyses are presented in Table S4. (E) Inhibition of key components of the cell cycle machinery extends lifespan. EV refers to empty RNAi vector control. All survival plots show combined data from at least two experiments. See also Table 1 for corresponding data and statistics, and Table S1 for individual experiments.
Table 1. Lifespan analyses. Pooled lifespan data shown in Figures 1-3
| Strain | RNAi | Mean lifespan ± SEM (days) | Median lifespan (days) | 75th Percentile | 25th Percentile | p value vs. N2 (control) | p value vs. mutant | % mean lifespan change vs. N2 (control) | % mean Lifespan change vs. mutant | N | No. of Exp | Figure | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 18,8±0,3 | 19 | 21 | 17 | 123/129 | 2 | 1A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cdk-2 | 24,1±0,3 | 24 | 26 | 22 | 2,0E-27 | 28% | 125/141 | 2 | 1A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 19,5±0,3 | 19 | 21 | 18 | 114/120 | 2 | 1B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 24,9±0,5 | 26 | 29 | 20 | 5,7E-19 | 28% | 103/120 | 2 | 1B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 18,2±0,3 | 18 | 20 | 15 | 158/161 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cdk-1 | 26±0,4 | 26 | 28 | 23 | 9,6E-46 | 43% | 157/160 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cdk-4 | 19,2±0,3 | 19 | 21 | 17 | 0,035 | 6% | 153/160 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cya-1 | 19,9±0,3 | 20 | 22 | 18 | 2,9E-04 | 10% | 155/160 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cyb-1 | 19,2±0,2 | 19 | 21 | 17 | 0,087 | 6% | 157/160 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cyd-1 | 21,5±0,3 | 21 | 24 | 18 | 6,7E-12 | 18% | 153/160 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cdc-25.1 | 25,1±0,3 | 25 | 27 | 22 | 1,2E-41 | 38% | 158/160 | 2 | 1E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 19±0,2 | 19 | 21 | 17 | 267/275 | 4 | 2A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 26,1±0,3 | 26 | 29 | 24 | 6,8E-84 | 38% | 247/270 | 4 | 2A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| glp-1(e2141) | control | 23,9±0,3 | 24 | 27 | 21 | 7,0E-54 | 26% | 315/413 | 4 | 2A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| glp-1(e2141) | cye-1 | 24,5±0,3 | 24 | 28 | 21 | 1,8E-58 | 0,075 vs glp-1(ctr) | 29% | 3% | 305/401 | 4 | 2A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 18,9±0,3 | 19 | 21 | 16 | 122/124 | 2 | 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 24,2±0,4 | 24 | 26 | 22 | 8,8E-20 | 28% | 105/113 | 2 | 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mes-1(bn7) (fertile) | control | 18,2±0,4 | 18 | 20 | 15 | 0,31 | -4% | 116/122 | 2 | 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mes-1(bn7) (fertile) | cye-1 | 22,1±0,5 | 22 | 25 | 19 | 6,9E-08 | 1,9E-09 vs mes-1F(ctr) | 17% | 21% | 84/97 | 2 | 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mes-1(bn7) (sterile) | control | 24,2±0,7 | 23 | 28 | 19 | 1,4E-12 | 28% | 88/115 | 2 | 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mes-1(bn7) (sterile) | cye-1 | 23,7±0,7 | 23 | 27 | 18 | 1,5E-10 | 0,84 vs mes-1S(ctr) | 25% | -2% | 90/111 | 2 | 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 19,9±0,3 | 20 | 23 | 17 | 110/111 | 2 | 2C+2D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 24±0,3 | 24 | 26 | 22 | 3,8E-14 | 21% | 102/118 | 2 | 2C+2D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-12 (rh61rh411) | control | 17,6±0,3 | 18 | 20 | 15 | 4,5E-07 | -11% | 147/147 | 2 | 2C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-12 (rh61rh411) | cye-1 | 19,3±0,3 | 19 | 22 | 17 | 0,38 | 1,9E-05 vs daf-12(ctr); 9,9E-017 vs N2(cye-1) | -3% | 10% | 133/149 | 2 | 2C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-9(rh50) | control | 18,5±0,4 | 18 | 22 | 16 | 0,026 | -7% | 128/130 | 2 | 2D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-9(rh50) | cye-1 | 19,7±0,4 | 20 | 23 | 16 | 0,51 | 9,7E-03 vs daf-9(ctr); 9,8E-021 vs N2(cye-1) | -1% | 6% | 123/123 | 2 | 2D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 19,4±0,3 | 19 | 21 | 16 | 129/134 | 2 | 3A+3D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 25,3±0,4 | 25 | 28 | 23 | 9,2E-28 | 30% | 127/136 | 2 | 3A+3D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-16(mgDf47) | control | 17,2±0,3 | 17 | 20 | 14 | 4,3E-05 | -11% | 161/169 | 2 | 3A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-16(mgDf47) | cye-1 | 19,2±0,4 | 19 | 23 | 15 | 0,29 | 8,3E-06 vs daf-16(ctr) | -1% | 11% | 168/188 | 2 | 3A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| skn-1(zu67) | control | 16,1±0,3 | 16 | 18 | 14 | 2,8E-10 | -17% | 132/135 | 2 | 3D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| skn-1(zu67) | cye-1 | 15,7±0,3 | 16 | 18 | 13 | 7,0E-12 | 0,53 vs skn-1(ctr) | -19% | -3% | 124/129 | 2 | 3D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 19,4±0,3 | 19 | 21 | 17 | 115/123 | 2 | 3B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 25,5±0,3 | 25 | 28 | 23 | 6,3E-29 | 31% | 113/139 | 2 | 3B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| kri-1(ok1251) | control | 19±0,2 | 19 | 21 | 17 | 0,42 | -2% | 149/154 | 2 | 3B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| kri-1(ok1251) | cye-1 | 21±0,4 | 20 | 24 | 18 | 7,7E-04 | 7,1E-06 vs kri-1(ctr) | 8% | 10% | 113/131 | 2 | 3B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | control | 19,3±0,3 | 19 | 22 | 17 | 106/139 | 2 | 3C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | cye-1 | 25±0,4 | 24 | 28 | 22 | 4,4E-19 | 29% | 75/130 | 2 | 3C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-2(e1370) | control | 52,3±0,8 | 55 | 60 | 47 | 4,1E-83 | 171% | 179/190 | 2 | 3C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| daf-2(e1370) | cye-1 | 58,2±0,8 | 59 | 66 | 54 | 7,4E-90 | 9,1E-010 vs daf-2(ctr) | 202% | 11% | 178/199 | 2 | 3C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| These combined results were derived from individual experiments that are described in Supplementary Table S1. Experiments are grouped and graphed in the indicated figures. Wild type N2 animals were used for RNAi treatment. SEM: standard error of the mean. 75th and 25th percentiles refer to the day at which 75% or 25% of the analyzed population is dead. N represents number of observed deaths / total number of worms in experiment. P values were calculated by log-rank test. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To determine whether the timing of cell cycle gene knockdown might influence lifespan, we initiated cye-1 RNAi feeding either during development (starting from L1 throughout life), in L4, or in adulthood (day 3) when the worms produced eggs. Interestingly, we found that knockdown of cye-1 during development as well as in adulthood resulted in longevity (Supplementary Figure 1A and Supplementary Table S2) suggesting that cell cycle events in the larvae may also modulate the aging process. Furthermore, inhibition of progeny production by supplementation of fluorodeoxyuridine (FUDR) did not affect the longevity of cye-1(RNAi). We obtained comparable results with or without inclusion of FUDR (Supplementary Figure 1B and Supplementary Table S2).
Many C. elegans mutations and manipulations that increase lifespan also confer resistance towards diverse forms of stress [31]. We analyzed heat and oxidative stress response of cye-1 and cdk-2 deficient worms. Knockdown of cye-1 and cdk-2 by RNAi enhanced tolerance to thermal stress (Figure 1C) and oxidative stress (Figure 1D) compared to wild type control.
C. elegans displays age-related degenerative changes including reduction of pharyngeal pumping and body movement during its lifespan [32]. There is a positive correlation between the decline of neuromuscular behavior and survival probability. We therefore assessed whether inhibition of cell cycle genes could also attenuate the decline of locomotory functions. We observed that cye-1(RNAi) and cdk-2(RNAi) delayed the age-related decline of pharyngeal pumping (Supplementary Figure 1C) and body movements (Supplementary Figure 1D). We also quantified the number of progeny after cye-1 and cdk-2 knockdown by RNAi. Consistent with essential roles of cye-1 and cdk-2 for cell cycle progression and embryonic development, cye-1(RNAi) and cdk-2(RNAi) significantly reduced the number of progeny compared to wild type (Supplementary Figure 1E), although the effect was only partially penetrant. We further assessed the morphology of the gonad after 2 generations of RNAi feeding against cye-1 and cdk-2, and observed that a portion of the animals were sterile or even arrested during larval development (Supplementary Figure 1E and 1F).
Given the profound impact that inhibition of cye-1 and cdk-2 had on longevity, we wondered if other components of the cell cycle also control aging. We therefore tested activators of the cell cycle including the cyclin-dependent kinases cdk-1 and cdk-4, their cyclin partners cya-1, cyb-1 and cyd-1, and the CDK-activating phosphatase cdc-25.1. We observed that RNAi-knockdown of these core components of the cell cycle machinery during adulthood significantly extended lifespan (Figure 1E and Table 1). We focused our analyses on cdk-2 and the corresponding cyclin cye-1 because they are well described key regulators of the cell cycle and robustly extended lifespan.
Cell cycle genes regulate longevity through the germline
To determine if an intact germline is necessary for lifespan regulation by cell cycle genes, we examined the effect of cye-1 and cdk-2 knockdown in glp-1 mutant worms. glp-1 encodes a Notch family receptor that is essential for mitotic proliferation of germline stem cells [33,34]. We used the glp-1 temperature-sensitive mutant allele e2141. When grown at the non-permissive temperature of 25°C, these mutants lack the germline and are long lived [7,34]. We observed that knockdown of cye-1 and cdk-2 by RNAi did not further extend the longevity of glp-1(e2141) mutant worms (Figures 2A and S2A, Table 1 and S2) suggesting that cell cycle genes require the germline to regulate longevity. We found that this effect could be reproduced by another germline-defective mutant, mes-1(bn7). The mes-1 gene controls initial development of the embryonic germline [35]. At 20°C, about 50% of mes-1(bn7) mutants lack the germline, and these sterile animals live up to 50% longer than their fertile siblings [7]. Knockdown of cye-1(RNAi) was able to extend the lifespan of fertile mes-1 worms (21%), but not sterile mes-1 animals (Figure 2B and Table 1). We further assessed the function of cell cycle regulators specifically in the germline using rrf-1 mutants. RRF-1 is mainly required for RNAi in somatic tissues. Hence, in animals lacking rrf-1 activity RNAi is efficient in the germline but it functions poorly in somatic cells [36]. We found the cye-1(RNAi) strongly extended lifespan in rrf-1(pk1417) mutants (18% compared to rrf-1 mutants fed with control(RNAi)). The effect was comparable to wild type (Supplementary Figure 2B and Supplementary Table S2). Together, these findings indicate that cell cycle genes function in the germline to influence lifespan.
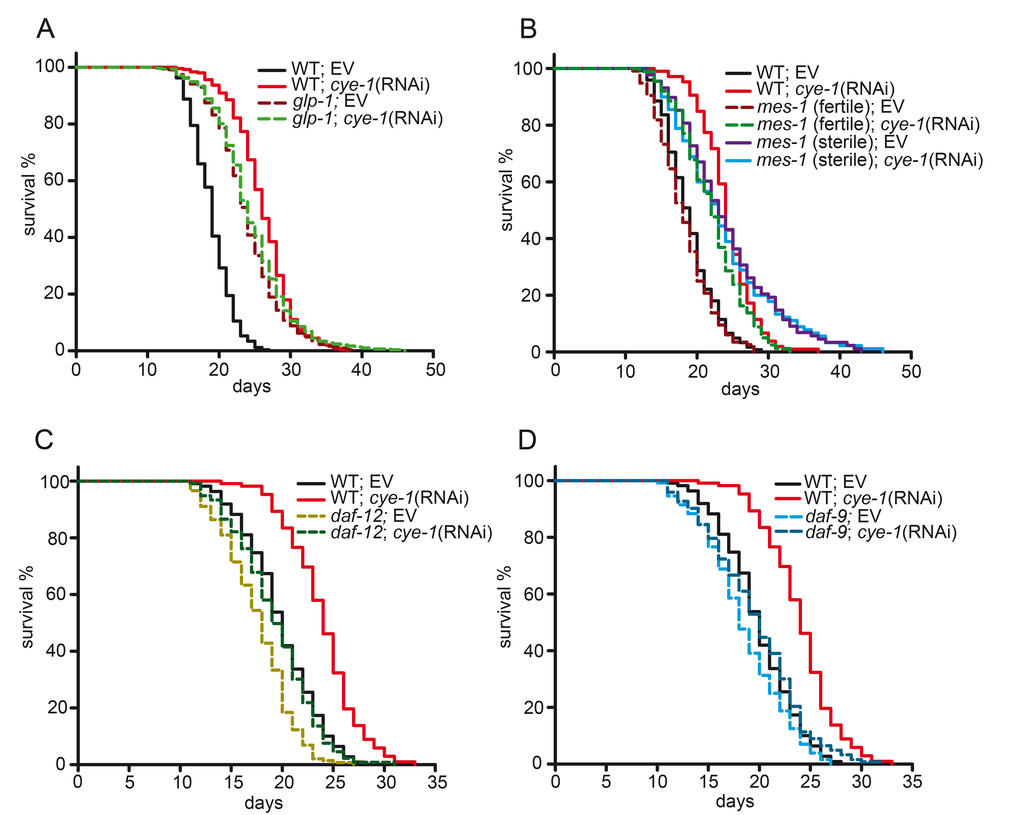
Figure 2. Cell cycle genes function in the germline longevity pathway. (A) cye-1(RNAi) does not alter longevity of germline-defective glp-1(e2141) mutants. glp-1 mutants were raised at 25°C until L4/early adulthood to eliminate germ cells and then shifted to 20°C for the rest of the assay. (B) Lifespans of germline-defective mes-1 mutants. mes-1(bn7) induced sterility is approximately 50% penetrant at 20°C [7]. The sterile and fertile mes-1 animals were identified by their appearance using a dissecting microscope. cye-1(RNAi) extends lifespan in fertile mes-1 animals but not in sterile mes-1 mutants. (C) The steroid hormone signaling pathway is required for cye-1(RNAi) longevity. The nuclear hormone receptor daf-12(rh61rh411) mutants greatly suppress the longevity of cye-1(RNAi) treated worms. (D) Inactivation of the steroid hormone biosynthesis enzyme daf-9 abolishes lifespan extension caused by cye-1(RNAi). Survival plots show combined data from at least two experiments. Quantitative data and statistical analyses of lifespan assays are shown in Table 1, individual experiments are presented in Table S1. EV refers to empty control vector.
The longevity of animals lacking the germline depends on steroid hormone signaling pathways. Mutations in the nuclear hormone receptor daf-12 prevent the lifespan-extending effect when germline stem cells are removed [1,10,37]. To test whether cye-1 genetically interacts with daf-12 to regulate lifespan, we examined the effect of cye-1 knockdown in the daf-12(rh61rh411) null mutant. We found that daf-12(rh61rh411);cye-1(RNAi) animals have significantly shortened lifespans compared with cye-1(RNAi) (Figure 2C and Table 1). Enzymes that produce DAF-12 ligands, such as the cytochrome P450 DAF-9 also contribute to steroid hormone signaling and mutation of daf-9 abrogates the longevity of germline-less animals [13,37]. The loss of function mutant daf-9(rh50) displays a slightly reduced lifespan, presumably because DAF-12 is not fully activated [37]. We observed that the lifespan extension of cye-1(RNAi) was strongly suppressed in the daf-9 mutant background (Figure 2D and Table 1). Together, these observations suggest that cye-1 exerts its effect on lifespan through the steroid hormone signaling pathway including DAF-9 and the nuclear hormone receptor DAF-12.
Longevity associated with inhibition of cell cycle requires DAF-16
It is well established that the activity of the transcription factor DAF-16/FOXO is required for germline removal to extend lifespan, as daf-16 mutants completely abolish longevity of germline-less animals [1]. Upon germline elimination DAF-16 accumulates in the nucleus of intestinal cells and stimulates expression of target genes [12,38,39]. We first analyzed the role of cell cycle genes in the DAF-16-dependent germline longevity pathway. We found that the daf-16(mgDf47) null mutation [40] significantly suppressed the longevity of cye-1(RNAi) (Figure 3A and Table 1) and cdk-2(RNAi) treated worms (Supplementary Figure 2C and Supplementary Table S2). Interestingly, the daf-16(mgDf47) mutation did not fully block the pro-longevity effect of cell cycle inhibition. RNAi against cye-1 and cdk-2 produced a modest but consistent increase in the mean lifespan of the daf-16 mutant (11% and 10%, respectively), but to a much lesser extent than in wild type (30% and 28%) (Table 1 and S2). We tested the genetic interaction with a different daf-16 null allele: again knockdown of cye-1 slightly extended the mean lifespan of daf-16(mu86) mutants (14% compared to 23% in wild type) (Supplementary Figure 2D and Supplementary Table S2). These findings indicate that the longevity benefits of cell cycle inhibition largely depend upon DAF-16 but also raise the possibility that a different mechanism might be involved.
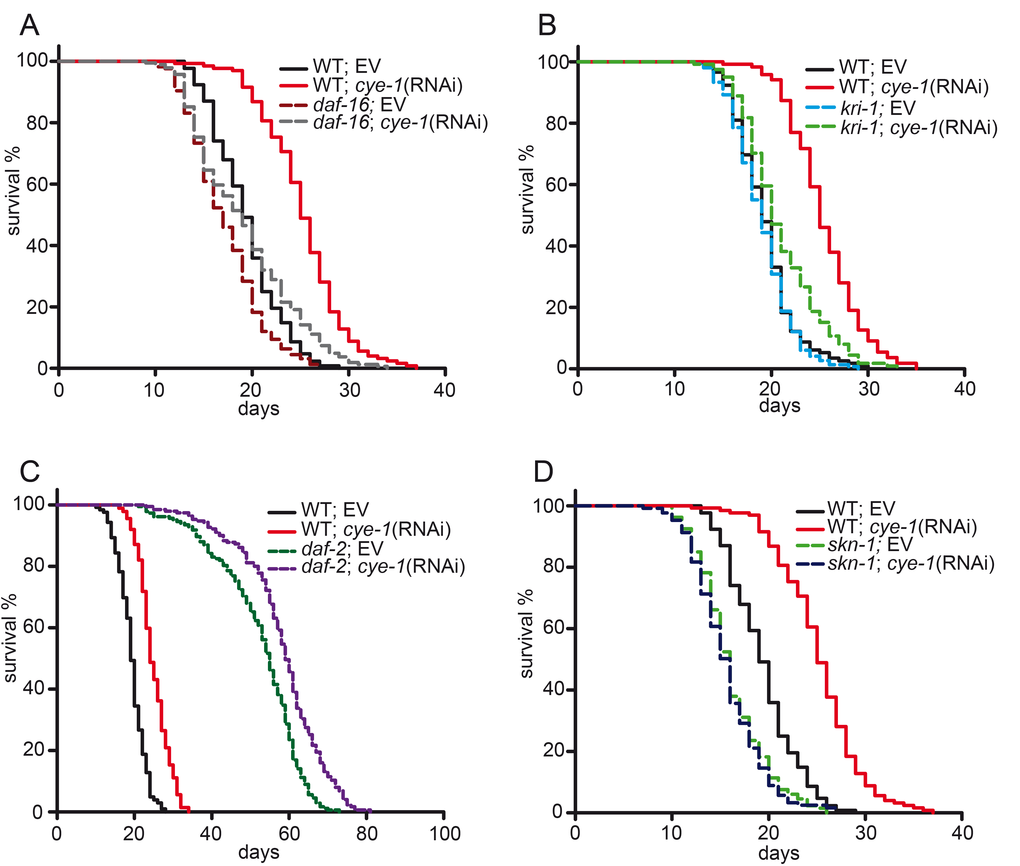
Figure 3. DAF-16 and SKN-1 are required for longevity from reduced cell cycle activity. (A) Lifespan extension in response to cell cycle inhibition is mediated by DAF-16. daf-16(mgDf47) null mutants and wild type worms were fed with cye-1 and control(RNAi). The longevity associated with cye-1(RNAi) was greatly decreased but not eliminated by daf-16 mutation. (B) Longevity of cye-1(RNAi) was greatly reduced by kri-1(ok1251) mutants. (C) Knockdown of cye-1 by RNAi further increases longevity of daf-2(e1370) mutants. (D) Longevity extension by cye-1(RNAi) was eliminated by skn-1(zu67) mutation. Survival plots show combined data from at least two experiments and are summarized in Table 1, individual experiments are presented in Table S1. EV refers to empty control vector.
The ankyrin-repeat protein KRI-1/KRIT1 is needed for loss of the germline to target DAF-16 to the nucleus and extend lifespan [10]. Next, we investigated if kri-1 is also required for the longevity of cell cycle deficient worms. We observed that the longevity associated with cye-1(RNAi) and cdk-2(RNAi) was strongly suppressed by lack of kri-1, shortening mean lifespan by up to 18% (Figure 3B and S2E, Tables 1 and S2).
The reproductive pathway functions in a synergistic manner with the daf-2/insulin-like signaling pathway to regulate DAF-16, as loss of the germline greatly extends the long lifespan of daf-2 mutants [1]. Accordingly, we observed that knockdown of cye-1(RNAi) in daf-2(e1370) mutants further increased their longevity (Figure 3C and Table 1) indicating that cell cycle genes act in the DAF-16-dependent germline longevity pathway but not in the insulin signaling pathway.
SKN-1/Nrf is required for longevity from reduced cell cycle activity
It was intriguing that daf-16 null mutants did not completely suppress longevity associated with inactivation of cye-1 and cdk-2. Our findings suggested that inhibition of the cell cycle might affect longevity through a pathway in parallel to DAF-16. The transcription factor SKN-1/Nrf has conserved functions in stress defense, protein homeostasis, and metabolism and promotes longevity [41,42]. Recently, SKN-1 has been shown to promote longevity in the absence of germline stem cells [15,16]. We wondered whether SKN-1 might act in parallel to DAF-16 for longevity under conditions in which cell cycle processes are inactive. We therefore tested the effects of cye-1 and cdk-2 RNAi knockdown in the skn-1(zu67) mutant and wild type, and found that the pro-longevity effect of cye-1 and cdk-2(RNAi) was essentially prevented by mutation of skn-1 (Figure 3D and Supplementary Figure 2F, Table 1 and Supplementary Table S2). These observations indicate that SKN-1 is fully required for the effects of cell cycle factors on longevity.
Cell cycle inhibition induced a transcriptional stress response that involves SKN-1 and DAF-16
Our results raise the question if the activity of SKN-1 is actually controlled by inhibition of the cell cycle. SKN-1 accumulates in intestinal nuclei and upregulates expression of detoxification genes in response to certain stresses or when mechanisms that limit SKN-1 activation are inhibited [41-44]. We further investigated how RNAi knockdown of cell cycle factors activates SKN-1 dependent transcriptional response and analyzed the expression of the SKN-1 target gene glutathione S-transferase, gst-4 using a transcriptional GFP reporter (Pgst-4::GFP) [45]. In the intestine the Pgst-4::GFP reporter was expressed at very low levels under normal conditions (Figure 4A). Transcription from the transgenic gst-4 promoter was robustly induced by RNAi knockdown of cye-1 and cdk-2 (Figure 4A). This gst-4 induction was greatly abolished by mutation of skn-1 (Figure 4B). Similarly, inhibition of the cyclin cya-1, cyb-1, and cyd-1, the cyclin-dependent kinases cdk-1 and cdk-4, and the CDK activator cdc-25.1 resulted in robust gst-4 activation through skn-1-dependent mechanisms (Supplementary Figure 3 A-C).
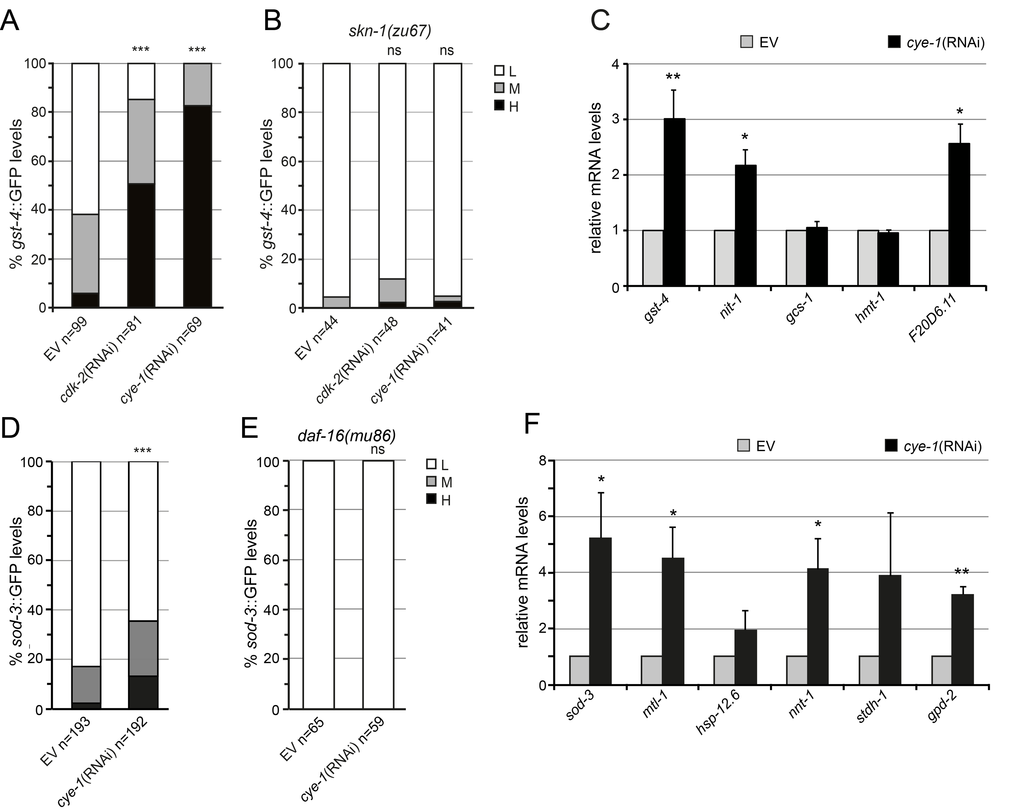
Figure 4. Cell cycle inhibits SKN-1 and DAF-16-driven stress response. (A) Activation of the Pgst-4::GFP reporter in L4/young adult animals that were exposed to cye-1(RNAi), cdk-2(RNAi), or control beginning from L1. (B) Analysis of Pgst-4::GFP expression in skn-1(zu67) mutants. In (A) and (B) induction of the Pgst-4::GFP reporter in the intestine was quantified as low (L), medium (M) and high (H) (see Supplementary Figure 3C). Pooled data from 2 experiments. p-values were calculated by the Chi-square test. ***p < 0.001. ns, not significant. n, number of worms analyzed. (C) Induction of endogenous SKN-1 target gene expression in response to cye-1(RNAi) analyzed by qPCR. RNAi was performed from L4 to day four of adulthood. Data are presented as fold change compared to wild-type on control(RNAi) averaged from at least three independent experiments, error bars represent SEM. p-values were derived from a student’s t-test. *p< 0.05. **p<0.01. (D) cye-1(RNAi) induces expression of Psod-3::GFP (see also Supplementary Figure 4). (E) Analysis of Psod-3::GFP expression in daf-16(mu86) mutants. In (D) and (E) Psod-3::GFP quantification with high (H), medium (M), and low (L) scoring. Pooled data from at least 2 experiments. p-values were calculated by the Chi-square-test. ***p < 0.001. ns, not significant. n, number of worms analyzed. (F) DAF-16 target gene expression assayed by qPCR. Worms were exposed to cye-1(RNAi) or empty vector control during adulthood. The nnt-1, stdh-1 and gpd-2 genes have been shown previously to be upregulated by germline removal. Data are mean ± SEM. p-values were derived from a student’s t-test. *p< 0.05. **p<0.01.
We next analyzed effects on endogenous SKN-1–regulated gene expression in adult worms after inhibition of cye-1 through RNAi. Here, we focused on cye-1 representative for different cell cycle factors. We used quantitative PCR to analyze the mRNA levels of a set of well characterized SKN-1 targets involved in various stress processes like glutathione S-transferase gst-4, γ-glutamyl cysteine synthetase gcs-1, oxidoreductase F20D6.11, nitrilase nit-1, and ABC transporter hmt-1 [42,43]. mRNA production of gst-4 and nit-1 has also been shown to depend on SKN-1 under normal (unstressed) conditions, while gcs-1, hmt-1, and F20D6.11 are upregulated by SKN-1 in response to stress [42]. We observed that RNAi knockdown of cye-1 increased the expression of several endogenous SKN-1 target genes in adult worms (Figure 4C). We conclude that inhibition of cell cycle factors promotes the transcriptional activity of SKN-1 for detoxification and stress defense.
Given the importance of DAF-16 for longevity associated with cell cycle inhibition we investigated whether cye-1 influences the expression of DAF-16 target genes. First, we analyzed the expression of the conserved DAF-16/FOXO target superoxide dismutase (sod-3). In adult worms, a sod-3::GFP transgenic reporter was expressed at low levels under normal conditions, but was elevated by cye-1(RNAi) (Figure 4D). The up-regulation of sod-3 was strongly attenuated in daf-16 mutants (Figure 4E). Next, we used qPCR to analyze how knockdown of cye-1 affected the transcriptional profile of well-identified DAF-16 target genes [46,47]. Several stress resistance genes like sod-3, mtl-1 encoding a metallothionein protein, and hsp-12.6 encoding a small heat shock protein were upregulated under cye-1(RNAi) conditions (Figure 4F). We also analyzed the expression of genes known to be upregulated by germline removal, including gpd-2, stdh-1 and nnt-1 [11]. We found that the expression of these genes was strongly induced by cye-1(RNAi) knockdown (Figure 4F). Together, our data indicate that when cell cycle factors are inhibited, SKN-1 and DAF-16 induce a protective transcriptional response.
Discussion
The last few decades have unraveled a variety of different mechanisms and signaling pathways that actively influence aging in C. elegans. Focusing on cyclin E cye-1 and cyclin-dependent kinase 2 cdk-2, we here characterize a yet unknown role of cell cycle genes in the regulation of longevity, health and stress resistance (Figure 1). Additionally, inactivation of several other core components of the cell cycle machinery similarly extended lifespan (Figure 1E) supporting the idea that cell cycle regulators play a role for longevity in C. elegans.
Several lines of evidence suggest that these longevity benefits are mediated through specific regulatory pathways involving the germline. Proliferating cells are the likeliest target in this mechanism and the germline is the only tissue in adult C. elegans with a continuously proliferating pool of cells, whereas somatic tissues are entirely post-mitotic [48]. Consistent with this idea, genetic inhibition of cdk-2 and cye-1 did not further extend the lifespan of glp-1 mutants in which the number of germline stem cell is markedly reduced (Figure 2A and Supplementary Figure 2A). Similarly, inactivation of cye-1 did not increase longevity in mes-1 mutants that fail to develop a germline and are sterile, by contrast to results obtained in fertile mes-1 mutants (Figure 2B). Moreover, cye-1(RNAi) increased lifespan in rrf-1 mutants in which RNAi-mediated gene knockdown functions most efficiently in the germline (Supplementary Figure 2B). Importantly, the longevity effect was still apparent when RNAi-mediated cye-1 knockdown was initiated at different stages, during larval development or later adulthood (day 3) (Supplementary Figure 1A). Of note, lifespan can be extended in C. elegans when germline proliferation is inhibited by glp-1 inactivation during adulthood [7]. In flies, similar findings have been described [3]. Together, our data show that the benefits associated with genetic inhibition of cell cycle regulators involve germline mechanisms.
It is well established that germline-mediated longevity requires the activity of the FOXO transcription factor DAF-16 and a lipophilic hormone/steroid signaling pathway comprising DAF-9 and DAF-12 [1,7,13,37]. We found that these canonical germline pathway components also play a critical role in the increases in lifespan that derive from cell cycle inhibition (Figure 2C and D, Figure 3A, Supplementary Figure 2C and D). Interestingly, the dramatic lifespan extension associated with knockdown of cye-1 and cdk-2 was not fully eliminated in two daf-16 null mutants indicating that DAF-16 is central for the influence of cell cycle regulators on longevity, but additional factors contribute to those effects. Inhibition of cye-1 by RNAi led to induction of DAF-16 target genes (gpd-2, stdh-1 and nnt-1) (Figure 4D-F) whose expression is known to be changed following germline-loss [11]. Previous work has shown that KRI-1 is needed for loss of germline to trigger DAF-16 activity and extend lifespan [10]. We observed that the increases in lifespan that derive from cell cycle inhibition largely depend upon the activity of kri-1 (Figure 3B, Supplementary Figure 2E) also consistent with DAF-16 being important.
It was particularly striking that SKN-1/Nrf2 was substantially required for lifespan extension associated with cell cycle inhibition. Mutation of skn-1 completely blunted the beneficial effect of cye-1/cdk-2 RNAi on longevity (Figure 3D and Supplementary Figure 2F) and therefore must be critical for the influence of cell cycle factors on aging. Genetic inactivation of cell cycle regulators led to activation of the direct SKN-1 target gene gst-4 apparently in the intestine through skn-1-dependent mechanisms (Figure 4A, B and Supplementary Figure 3A-C). Moreover, RNAi knockdown of cye-1 induced endogenous SKN-1 target gene expression (Figure 4C) supporting that the SKN-1 transcriptional response is central for cell cycle factor-mediated longevity and protection from stress. Only two recent studies indicated that SKN-1/Nrf2 is a target of germline-signaling for longevity [15,16]. When germline stem cells are ablated (by mutation of glp-1), lipid-based signaling activates SKN-1 in the intestine which induces a broad transcriptional program involved in detoxification, proteasome maintenance, extracellular matrix, and lipid metabolism thereby increasing stress tolerance and longevity [15]. Diverse genetic regulatory pathways and interventions, including insulin/IGF-1, TOR, dietary restriction, mitochondrial ROS production as well as germline signaling impact on SKN-1/Nrf2 to control aging [reviewed in 17]. Our data show that cell cycle factors influence SKN-1 through germline-based signaling and further support the idea that germ cells involve SKN-1/Nrf proteins to promote longevity.
It was intriguing that we observed particularly strong effects on lifespan extension after inhibition of cye-1 and cdk-2. The CYE-1/CDK-2 complex is a master regulator of the cell division cycle facilitating G1/S transition. A basic model would be that cell cycle inhibition leads to reduction of germ cell numbers and therefore the same lifespan regulating mechanisms are initiated as in germless animals. The mitotic cell cycle of C. elegans germ cells features a rapid progression with a highly abbreviated or absent G1 phase [26,49]. S and G2 may thus be the major phases for regulation of cell cycle dynamics. The rapid kinetic is thought to be exerted by high CYE-1 activity during the cell cycle which may bypass G1 and drive entry into S phase. Additionally, CYE-1 is found in high levels throughout the proliferative zone [26,50,51]. Thus, high CYE-1 levels and an atypical cell cycle structure may be necessary to promote the pool of undifferentiated germ cells. Interestingly, other animals including flies, frogs, and zebrafish display similar characteristics in early embryonic cell divisions [reviewed in 52] suggesting evolutionarily conserved mechanisms. We assume that the potency of our tested cell cycle factors to delay aging correlates with the impact on germline proliferation. Consistent with this, disruption of cye-1 and cdk-2 resulted in a strongly reduced number of germ cells and sterility [27] (Supplementary Figure 1).
In addition to forward germ cell proliferation by regulating cell cycle structure per se, cye-1/cdk-2 also contribute to the regulation of the proliferative fate versus meiotic entry decision [26,27]. CYE-1/CDK-2 have been shown to directly phosphorylate and downregulate GLD-1 [27] which normally facilitates the switch of germ cells from mitotic into meiotic cell cycle and differentiation [53]. Moreover, GLD-1 also represses cye-1 mRNA translation allowing a negative feedback loop [54]. Thus, cye-1/cdk-2 may function through gld-1 to influence the mitosis/meiosis transition. Interestingly, the ability of cye-1/cdk-2 to influence the proliferative fate appears to be a specific function of these factors and not simply a consequence of disruption of cell cycle, as knockdown of other cell cycle factors does not induce premature meiotic entry [26]. Given that gld-1 and likewise glp-1 have been implicated in aging processes [7,55], and both interact with cye-1 and cdk-2, this encouraged our experimental focus on these two cell cycle factors. Possibly, these two and other cell cycle regulators might have further specific functions or yet unknown substrates in the germline directly linked to longevity and health. Notably, it was previously described that cell cycle factors, i.e. checkpoint proteins might influence lifespan by targeting postmitotic cells [29]. We show here that reduced germline proliferation is a substantial determinant for prolongation of lifespan by inhibition of cell cycle genes. Although conclusively clarifying the role of every cell cycle regulator for germline-mediated longevity will be a necessary task of future studies.
Interestingly, knockdown of not all cell cycle genes that we tested had the same impact on aging (Figure 1E). Why may this be the case? Individual cell cycle genes have differential effects in the germline [56]. In other words, cell cycle factors appear to be of dissimilar importance for maintenance of germline proliferation per se. As stated above, activity in the germline and prolongation of lifespan seem to be positively correlated. Beside cye-1 and cdk-2, knockdown of the cell cycle regulators cdk-1 and cdc-25.1 induced the strongest lifespan extension (Figure 1E). These two genes have been shown to be critical for germline proliferation and their depletion robustly reduces the number of germ cells [26,57-59]. cdc-25.1 executes unique functions in the germline, while its activity is likely redundant in some somatic tissues [60]. CDK-1, in turn, is a target of CDC-25.1 and is required for mitotic and meiotic cell cycle progression as well as for oocyte maturation in the germline [59,61,62].
By contrast, cdk-4(RNAi) had very little impact on lifespan (Figure 1E), and likewise cdk-4 only plays a subliminal role in germline proliferation in the literature [26,59,63]. CDK-4 acts together with CYD-1 in a canonical complex to regulate G1/S phase progression in postembryonic cells [63]. Interestingly, cyd-1 mutants display developmental defects which are not found in cdk-4 mutants, possibly because CYD-1 has further binding partners or CDK-4 is more stable [reviewed in 61]. Congruously with this, inhibition of cyd-1 showed a mild but solid increase of lifespan compared to the rather weak cdk-4(RNAi) in our experiments. The effect was also weaker for cyclin B cyb-1. B-type cyclins together with Cdk1 control progression through mitosis. Three typical B-type cyclins (cyb-1, cyb-2.1, and cyb-2.2) and a distinct cyclin B3 (cyb-3) are expressed in C. elegans. It has been reported that these mitotic cyclins have partially redundant functions and only inactivation of all four cyclins fully resembles the defects in cdk-1 inhibition [64]. Presumably, inactivation of just cyb-1 by RNAi might therefore result in a weak cdk-1 inhibition and longevity phenotype. Although, we cannot exclude that reduced efficiency of dsRNA-dependent inactivation of mRNA could also account for mild extension in longevity observed for these genes.
Together, our data show that inhibition of core cell cycle factors provokes stress tolerance and longevity in C. elegans. These effects critically involve proliferation of the germline stem cells and depend on the action of the conserved transcription factor DAF-16/FOXO, the nuclear hormone receptor DAF-12, and SKN-1/Nrf2. It will now be of interest to determine how interference with cell cycle triggers these specific germline-associated mechanisms. We propose that reduction of germ cell numbers is pivotal for cell cycle-associated longevity, but specific functions of distinct cell cycle factors in the germline as described for cye-1/cdk-2 might also play a crucial role. By revealing that longevity benefits of germline inhibition can be conferred by modulating the cell cycle, our data also suggest that this could be instrumental for developing new methods to study germline-dependent aging so that benefits are not outweighed by detriments.
Materials and Methods
C. elegans strains
C. elegans strains were grown on NGM agar plates with E. coli OP50 lawns [65]. Mutant strains are described in Supplementary Table S5.
RNA interference
RNAi plasmids were picked from the Ahringer [66] or ORF-RNAi [67] libraries and confirmed by sequencing (see Supplementary Table S6). For RNAi feeding experiments cultures were grown overnight in LB-medium with 12.5µg/ml tetracycline and 50µg/ml ampicillin. The following day, cultures were diluted 1:10 in 50µg/ml ampicillin LB-medium and grown to an OD595 of 0.8-1.1. Bacteria were spun down and resuspended in 1/10 of the original volume containing 50µg/ml ampicillin and induced with 0.7mM IPTG. This culture was seeded onto NGM agar plates containing 50µg/ml ampicillin, 12.5µg/ml tetracycline and 1mM IPTG. Empty vector plasmid pPD129.36 (L4440) was used as control.
Lifespan assays
Lifespan assays were performed unless otherwise indicated at 20°C. Animals were age-synchronized by timed egg laying and allowed to develop on control empty vector RNAi plates at 20°C. At L4 larval stage, worms were transferred on NGM RNAi plates containing 50 µM FUDR to prevent hatching of larvae. Animals were examined every day until the end of the assay. Worms that did not respond to gentle prodding with movement were scored as dead. Worms that crawled off the plates, showed extruded organs, or exploded due to internally hatching larvae were censored. All lifespans were plotted with L4 as time-point 0. Data were analyzed using a log-rank test in SigmaPlot 11.0.
To induce sterility in glp-1(e2141) mutants, adult animals were allowed to lay eggs at 15°C and the progeny were shifted to 25°C at L1 until L4/early adulthood. They were then placed on plates containing FUDR and RNAi bacteria for lifespan assays at 20°C. To prevent dauer formation of daf-2(e1370), daf-12(rh61rh411), and daf-9(rh50) mutants, these strains were allowed to develop to early adulthood at 15°C before starting the lifespan at 20°C.
Stress resistance assays
For the analysis of resistance against oxidative stress and heat, wild type worms were fed with RNAi for 4 days starting at L4 stage at 20°C. Worms were then either placed on plates containing 7.5 mM tert-butyl hydroperoxide (TBHP) or at 35°C preheated seeded RNAi plates. The criteria for analysis and censoring were essentially the same as described for lifespan assays.
Healthspan assays
Worms were raised as described for lifespan assays. To quantify pharyngeal pumping rates, worms were examined under a dissecting microscope every other day starting at the first day of adulthood. The number of contractions of the terminal bulb was counted for 30 sec.
For thrashing assays, worms were transferred into phosphate-buffered saline and observed for thrashing movements after 5 to 10 minutes of recovery. Quantification of thrashing movements was performed on alternate days beginning on the first day of adulthood. A thrash was defined as a full change in the lateral bending direction of the whole worm corpus. Pairwise statistical comparison was performed using a Mann-Whitney U-Test.
Brood size
Hermaphrodites were allowed to lay eggs on empty vector control. Their progeny was raised at 20°C until L4. Individual larvae were placed on fresh RNAi plates containing either empty vector control, cye-1(RNAi), or cdk-2(RNAi) and transferred on new plates every day until egg-laying ceased. The total number of progeny was counted. Pairwise statistical comparison was performed using a Mann-Whitney U-Test.
Phenotypic analysis
Worms were fed with empty vector control, cye-1(RNAi), or cdk-2(RNAi) for two generations at 20°C. At day 2 of adulthood worms were mounted on 2% agar pads, immobilized and images were taken with a Zeiss Axioplan-2 microscope.
Microscopy and scoring of transgenic GFP reporters
Microscopy was performed using a Zeiss Axioplan-2 microscope equipped with an AxioCam camera and AxioVision-Software Rel.4.8. Worms were placed on 2% agarose pads on slides and stunned with 2 mM levamisole. GFP was detected using an EGFP-filter set (480/20 nm excitation, 510/20 nm emission). Images were processed with AxioVision Rel 4.8 and Adobe Photoshop CS6.
Intestinal Pgst-4::GFP expression was assessed as described [43]. Worms were raised on RNAi until L4/young adult stage. Scoring was as follows: “high”, strong GFP levels throughout most of the intestine; “medium”, intense GFP levels anteriorly and/or posteriorly; “low”, only barely visible or no GFP expression observed.
For Psod-3::GFP expression L4 larvae were placed on RNAi and four days later GFP intensity was scored in the adult worms. Fluorescence intensity was categorized in “high”, worms displayed robust GFP intensity throughout their bodies or showed strong nuclear signal; “medium”, easily detectable nuclear signal at weak levels; “low”, no nuclear GFP signal observed. p values were determined from a Chi-square test.
RNA isolation and quantitative PCR
For RNA preparation approximately 500 animals were grown at 20°C on empty vector or cye-1(RNAi) plates for 4 days starting from L4 stage. Worms were washed in M9 buffer to remove bacteria. RNA was extracted with TriReagent (Sigma), treated with DNAse (Quiagen), and purified using a RNA purification column (ZYMO Research). RNA concentration and quality was assessed using a NanoPhotometer P-Class. cDNA was synthesized with reverse transcriptase (SuperScript III First-Strand Synthesis System, Invitrogen) and oligo-dT primer. qPCR runs were performed in technical triplicates on a Roche Light Cycler 480 using the Takyon No Rox SYBR MasterMix blue dTTP (Eurogentec).
Samples were analyzed by the 2-ΔΔCt method with normalization to the geometric mean of the reference genes cdc-42 and Y45F10D.4. At least three biological replicates were examined for each sample. Primer sequences are listed in Supplementary Table S7.
Supplementary Materials
Acknowledgements
We thank Collin Ewald for helpful discussions. We thank Roland Nitschke from the Life Imaging Center (LIC) in the Center for Systems Biology, Albert-Ludwigs-University Freiburg for support in image recording and analysis, and Alexandra Schwierzock for excellent assistance. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Funding
This study was supported by grants from the DFG (KFO 201) and the European Social Fund and the Ministry of Science, Research, and Arts Baden-Württemberg to ENH (Margarete von Wrangell Programm).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1. Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 1999; 399:362–66. https://doi.org/10.1038/20694 [PubMed]
- 2. Mason JB, Cargill SL, Anderson GB, Carey JR. Transplantation of young ovaries to old mice increased life span in transplant recipients. J Gerontol A Biol Sci Med Sci. 2009; 64:1207–11. https://doi.org/10.1093/gerona/glp134 [PubMed]
- 3. Flatt T, Min KJ, D’Alterio C, Villa-Cuesta E, Cumbers J, Lehmann R, Jones DL, Tatar M. Drosophila germ-line modulation of insulin signaling and lifespan. Proc Natl Acad Sci USA. 2008; 105:6368–73. https://doi.org/10.1073/pnas.0709128105 [PubMed]
- 4. Cargill SL, Carey JR, Müller HG, Anderson G. Age of ovary determines remaining life expectancy in old ovariectomized mice. Aging Cell. 2003; 2:185–90. https://doi.org/10.1046/j.1474-9728.2003.00049.x [PubMed]
- 5. Westendorp RG, Kirkwood TB. Human longevity at the cost of reproductive success. Nature. 1998; 396:743–46. https://doi.org/10.1038/25519 [PubMed]
- 6. Min KJ, Lee CK, Park HN. The lifespan of Korean eunuchs. Curr Biol. 2012; 22:R792–93. https://doi.org/10.1016/j.cub.2012.06.036 [PubMed]
- 7. Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C. Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science. 2002; 295:502–05. https://doi.org/10.1126/science.1065768 [PubMed]
- 8. Crittenden SL, Troemel ER, Evans TC, Kimble J. GLP-1 is localized to the mitotic region of the C. elegans germ line. Development. 1994; 120:2901–11. [PubMed]
- 9. Kenyon CJ. The genetics of ageing. Nature. 2010; 464:504–12. https://doi.org/10.1038/nature08980 [PubMed]
- 10. Berman JR, Kenyon C. Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell. 2006; 124:1055–68. https://doi.org/10.1016/j.cell.2006.01.039 [PubMed]
- 11. Ghazi A, Henis-Korenblit S, Kenyon C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 2009; 5:e1000639. https://doi.org/10.1371/journal.pgen.1000639 [PubMed]
- 12. Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003; 115:489–502. https://doi.org/10.1016/S0092-8674(03)00889-4 [PubMed]
- 13. Gerisch B, Weitzel C, Kober-Eisermann C, Rottiers V, Antebi A. A hormonal signaling pathway influencing C. elegans metabolism, reproductive development, and life span. Dev Cell. 2001; 1:841–51. https://doi.org/10.1016/S1534-5807(01)00085-5 [PubMed]
- 14. Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012; 489:263–68. https://doi.org/10.1038/nature11315 [PubMed]
- 15. Steinbaugh MJ, Narasimhan SD, Robida-Stubbs S, Moronetti Mazzeo LE, Dreyfuss JM, Hourihan JM, Raghavan P, Operaña TN, Esmaillie R, Blackwell TK. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. eLife. 2015; 4:4. https://doi.org/10.7554/eLife.07836 [PubMed]
- 16. Wei Y, Kenyon C. Roles for ROS and hydrogen sulfide in the longevity response to germline loss in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2016; 113:E2832–41. https://doi.org/10.1073/pnas.1524727113 [PubMed]
- 17. Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY and Isik M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic Biol Med. 2015; 88:290-301.
- 18. Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000; 100:71–78. https://doi.org/10.1016/S0092-8674(00)81684-0 [PubMed]
- 19. Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994; 79:551–55. https://doi.org/10.1016/0092-8674(94)90540-1 [PubMed]
- 20. Levine EM. Cell cycling through development. Development. 2004; 131:2241–46. https://doi.org/10.1242/dev.01180 [PubMed]
- 21. Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005; 24:2776–86. https://doi.org/10.1038/sj.onc.1208613 [PubMed]
- 22. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013; 140:3079–93. https://doi.org/10.1242/dev.091744 [PubMed]
- 23. Shirayama M, Soto MC, Ishidate T, Kim S, Nakamura K, Bei Y, van den Heuvel S, Mello CC. The Conserved Kinases CDK-1, GSK-3, KIN-19, and MBK-2 Promote OMA-1 Destruction to Regulate the Oocyte-to-Embryo Transition in C. elegans. Curr Biol. 2006; 16:47–55. https://doi.org/10.1016/j.cub.2005.11.070 [PubMed]
- 24. Kim S, Ishidate T, Sharma R, Soto MC, Conte D
Jr , Mello CC, Shirayama M. Wnt and CDK-1 regulate cortical release of WRM-1/β-catenin to control cell division orientation in early Caenorhabditis elegans embryos. Proc Natl Acad Sci USA. 2013; 110:E918–27. https://doi.org/10.1073/pnas.1300769110 [PubMed] - 25. Rabilotta A, Desrosiers M, Labbé JC. CDK-1 and two B-type cyclins promote PAR-6 stabilization during polarization of the early C. elegans embryo. PLoS One. 2015; 10:e0117656. https://doi.org/10.1371/journal.pone.0117656 [PubMed]
- 26. Fox PM, Vought VE, Hanazawa M, Lee MH, Maine EM, Schedl T. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development. 2011; 138:2223–34. https://doi.org/10.1242/dev.059535 [PubMed]
- 27. Jeong J, Verheyden JM, Kimble J. Cyclin E and Cdk2 control GLD-1, the mitosis/meiosis decision, and germline stem cells in Caenorhabditis elegans. PLoS Genet. 2011; 7:e1001348. https://doi.org/10.1371/journal.pgen.1001348 [PubMed]
- 28. Moreno S, Nurse P, Russell P. Regulation of mitosis by cyclic accumulation of p80cdc25 mitotic inducer in fission yeast. Nature. 1990; 344:549–52. https://doi.org/10.1038/344549a0 [PubMed]
- 29. Olsen A, Vantipalli MC, Lithgow GJ. Checkpoint proteins control survival of the postmitotic cells in Caenorhabditis elegans. Science. 2006; 312:1381–85. https://doi.org/10.1126/science.1124981 [PubMed]
- 30. Kipreos ET. C. elegans cell cycles: invariance and stem cell divisions. Nat Rev Mol Cell Biol. 2005; 6:766–76. https://doi.org/10.1038/nrm1738 [PubMed]
- 31. Shore DE, Carr CE, Ruvkun G. Induction of cytoprotective pathways is central to the extension of lifespan conferred by multiple longevity pathways. PLoS Genet. 2012; 8:e1002792. https://doi.org/10.1371/journal.pgen.1002792 [PubMed]
- 32. Collins JJ, Huang C, Hughes S and Kornfeld K. The measurement and analysis of age-related changes in Caenorhabditis elegans. WormBook. 2008:1-21.
- 33. Austin J, Kimble J. glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell. 1987; 51:589–99. https://doi.org/10.1016/0092-8674(87)90128-0 [PubMed]
- 34. Priess JR, Schnabel H, Schnabel R. The glp-1 locus and cellular interactions in early C. elegans embryos. Cell. 1987; 51:601–11. https://doi.org/10.1016/0092-8674(87)90129-2 [PubMed]
- 35. Strome S, Martin P, Schierenberg E, Paulsen J. Transformation of the germ line into muscle in mes-1 mutant embryos of C. elegans. Development. 1995; 121:2961–72. [PubMed]
- 36. Sijen T, Fleenor J, Simmer F, Thijssen KL, Parrish S, Timmons L, Plasterk RH, Fire A. On the role of RNA amplification in dsRNA-triggered gene silencing. Cell. 2001; 107:465–76. https://doi.org/10.1016/S0092-8674(01)00576-1 [PubMed]
- 37. Gerisch B, Rottiers V, Li D, Motola DL, Cummins CL, Lehrach H, Mangelsdorf DJ, Antebi A. A bile acid-like steroid modulates Caenorhabditis elegans lifespan through nuclear receptor signaling. Proc Natl Acad Sci USA. 2007; 104:5014–19. https://doi.org/10.1073/pnas.0700847104 [PubMed]
- 38. Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001; 28:139–45. https://doi.org/10.1038/88850 [PubMed]
- 39. McCormick M, Chen K, Ramaswamy P, Kenyon C. New genes that extend Caenorhabditis elegans’ lifespan in response to reproductive signals. Aging Cell. 2012; 11:192–202. https://doi.org/10.1111/j.1474-9726.2011.00768.x [PubMed]
- 40. Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997; 389:994–99. https://doi.org/10.1038/40194 [PubMed]
- 41. Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008; 132:1025–38. https://doi.org/10.1016/j.cell.2008.01.030 [PubMed]
- 42. Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009; 8:524–41. https://doi.org/10.1111/j.1474-9726.2009.00501.x [PubMed]
- 43. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15:713–24. https://doi.org/10.1016/j.cmet.2012.04.007 [PubMed]
- 44. An JH, Vranas K, Lucke M, Inoue H, Hisamoto N, Matsumoto K, Blackwell TK. Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proc Natl Acad Sci USA. 2005; 102:16275–80. https://doi.org/10.1073/pnas.0508105102 [PubMed]
- 45. Link CD, Johnson CJ. Reporter transgenes for study of oxidant stress in Caenorhabditis elegans. Methods Enzymol. 2002; 353:497–505. https://doi.org/10.1016/S0076-6879(02)53072-X [PubMed]
- 46. Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003; 300:644–47. https://doi.org/10.1126/science.1083614 [PubMed]
- 47. Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003; 424:277–83. https://doi.org/10.1038/nature01789 [PubMed]
- 48. Hirsh D, Oppenheim D, Klass M. Development of the reproductive system of Caenorhabditis elegans. Dev Biol. 1976; 49:200–19. https://doi.org/10.1016/0012-1606(76)90267-0 [PubMed]
- 49. Crittenden SL, Leonhard KA, Byrd DT, Kimble J. Cellular analyses of the mitotic region in the Caenorhabditis elegans adult germ line. Mol Biol Cell. 2006; 17:3051–61. https://doi.org/10.1091/mbc.E06-03-0170 [PubMed]
- 50. Brodigan TM, Liu J, Park M, Kipreos ET, Krause M. Cyclin E expression during development in Caenorhabditis elegans. Dev Biol. 2003; 254:102–15. https://doi.org/10.1016/S0012-1606(02)00032-5 [PubMed]
- 51. Chiang M, Cinquin A, Paz A, Meeds E, Price CA, Welling M, Cinquin O. Control of Caenorhabditis elegans germ-line stem-cell cycling speed meets requirements of design to minimize mutation accumulation. BMC Biol. 2015; 13:51. https://doi.org/10.1186/s12915-015-0148-y [PubMed]
- 52. Ruijtenberg S, van den Heuvel S. Coordinating cell proliferation and differentiation: antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle. 2016; 15:196–212. https://doi.org/10.1080/15384101.2015.1120925 [PubMed]
- 53. Crittenden SL, Bernstein DS, Bachorik JL, Thompson BE, Gallegos M, Petcherski AG, Moulder G, Barstead R, Wickens M, Kimble J. A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature. 2002; 417:660–63. https://doi.org/10.1038/nature754 [PubMed]
- 54. Biedermann B, Wright J, Senften M, Kalchhauser I, Sarathy G, Lee MH, Ciosk R. Translational repression of cyclin E prevents precocious mitosis and embryonic gene activation during C. elegans meiosis. Dev Cell. 2009; 17:355–64. https://doi.org/10.1016/j.devcel.2009.08.003 [PubMed]
- 55. Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006; 313:971–75. https://doi.org/10.1126/science.1121908 [PubMed]
- 56. Kimble J, Crittenden SL. Germline proliferation and its control. WormBook. 2005; 1–14. [PubMed]
- 57. Boxem M, Srinivasan DG, van den Heuvel S. The Caenorhabditis elegans gene ncc-1 encodes a cdc2-related kinase required for M phase in meiotic and mitotic cell divisions, but not for S phase. Development. 1999; 126:2227–39. [PubMed]
- 58. Ashcroft N, Golden A. CDC-25.1 regulates germline proliferation in Caenorhabditis elegans. Genesis. 2002; 33:1–7. https://doi.org/10.1002/gene.10083 [PubMed]
- 59. Yoon S, Kawasaki I, Shim YH. CDC-25.1 controls the rate of germline mitotic cell cycle by counteracting WEE-1.3 and by positively regulating CDK-1 in Caenorhabditis elegans. Cell Cycle. 2012; 11:1354–63. https://doi.org/10.4161/cc.19755 [PubMed]
- 60. Kim J, Lee AR, Kawasaki I, Strome S, Shim YH. A mutation of cdc-25.1 causes defects in germ cells but not in somatic tissues in C. elegans. Mol Cells. 2009; 28:43–48. https://doi.org/10.1007/s10059-009-0098-8 [PubMed]
- 61. Boxem M. Cyclin-dependent kinases in C. elegans. Cell Div. 2006; 1:6. https://doi.org/10.1186/1747-1028-1-6 [PubMed]
- 62. Burrows AE, Sceurman BK, Kosinski ME, Richie CT, Sadler PL, Schumacher JM, Golden A. The C. elegans Myt1 ortholog is required for the proper timing of oocyte maturation. Development. 2006; 133:697–709. https://doi.org/10.1242/dev.02241 [PubMed]
- 63. Park M, Krause MW. Regulation of postembryonic G(1) cell cycle progression in Caenorhabditis elegans by a cyclin D/CDK-like complex. Development. 1999; 126:4849–60. [PubMed]
- 64. van der Voet M, Lorson MA, Srinivasan DG, Bennett KL, van den Heuvel S. C. elegans mitotic cyclins have distinct as well as overlapping functions in chromosome segregation. Cell Cycle. 2009; 8:4091–102. https://doi.org/10.4161/cc.8.24.10171 [PubMed]
- 65. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974; 77:71–94. [PubMed]
- 66. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001; 2:H0002. [PubMed]
- 67. Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, Vidal M. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 2004; 14:2162–68. https://doi.org/10.1101/gr.2505604 [PubMed]