




Introduction
Non-communicable diseases remain a major contributor to morbidity and mortality in older ages. Inflammation appears to be a common pathway underlying multiple causes of death in old age [1]. Inflammatory biomarkers predict mortality [2] and age-related disease such as cardiovascular disease [2-4] as well as worsening mobility and frailty in the community [5]. Efforts to identify therapies to target chronic low grade inflammation in older adults and evaluate the impact of reduction of inflammation on important outcomes are needed [6].
Chronic low grade inflammation characterized by an imbalance of inflammatory and anti-inflammatory pathways called "inflamm-aging" is a fundamental mechanism of aging [7,8]. Genes involved in the immune and inflammatory response pathway are associated with longevity [9] and genetic regulation of immunity is associated with human healthspan [10]. Gene expression is considered an important bridge to connect genetic variation, environmental exposures, and lifestyle factors with aging-related diseases and traits. More than one thousand genes are differentially expressed in blood in relation to chronological age, with many involved in innate and adaptive immunity, cytokine and chemokine signaling, and immune function [11].
Biological age is a measure of an individual's predicted age based on multiple biomarkers and may prove more useful in studying the biology of aging as compared to studying chronological age alone [12]. Given the importance of inflammation to aging biology, we developed a biologic age estimate based on inflammatory biomarkers representing pro-inflammatory and anti-inflammatory processes in a sample of older adults. We reported that our inflammatory biologic age measure was significantly associated with age-related morbidity and mortality [13]. The objective of the present study was to assess the association of genome-wide gene expression with inflammatory biologic age in participants from the community-based Framingham Heart Study. We hypothesized that genes associated with inflammatory biologic age would provide mechanistic insights into understanding human aging biology.
Results
Our study sample included 2386 participants (mean age 67A±9 years, 55% women) from the Framingham Offspring Cohort. Descriptive characteristics of the participants are provided in Table 1.
Table 1. Clinical characteristics of the study sample.
| Characteristics | N=2386 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Women, n (%) | 1304 (55%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, year ±SD | 66.8 ± 8.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Inflammatory BA | 66.8 ± 11.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ∆Age | 0.02 ± 7.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Smoker, n (%) | 198 (8.3%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Systolic blood pressure, mm Hg | 129 ± 17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diastolic blood pressure, mm Hg | 74 ± 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hypertension treatment | 1166 (49%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMI kg/m2 | 28.4 ± 5.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total cholesterol mg/dL | 186 ± 38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HDL cholesterol mg/dL | 58 ± 18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lipid treatment, n (%) | 1044 (44%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diabetes mellitus, n (%) | 406 (17%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cardiovascular disease, n (%) | 194 (8.9%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-reactive protein (mg/L) | 1.5 (0.8, 3.2) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Intercellular adhesion molecule 1 (ng/mL) | 277 (234, 342) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Interleukin-6 (pg/mL) | 1.8 (1.2, 2.9) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lipoprotein-Associated Phospholipase A2 (Lp-PLA2) Mass (ng/mL) | 202 (171, 231) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lp-PLA2 Activity (nmol/mL/min) | 137 (115, 160) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Monocyte chemoattractant protein-1 (pg/mL) | 368 (302, 444) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Osteoprotegerin (pmol/L) | 4.7 (3.9, 5.7) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P-Selectin (ng/mL) | 40 (33, 48) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tumor necrosis factor receptor II (pg/mL) | 2383 (1940, 3050) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +Characteristics are represented by mean ± SD or n (%); biomarkers are median and first, third quartile | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ∆Age=Inflammatory biologic age – chronologic age | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Association of inflammatory ∆age with gene expression
We identified 448 genes significantly associated with the difference (∆Age) between biologic age and chronologic age (P<2.8x10-6). Among them, 302 genes were positively associated with inflammatory ∆age, whereas the remaining 146 genes were negatively associated (Supplemental Table 1). Figure 1 shows the volcano plot of all studied genes, and the top 25 associations are shown in Table 2. The most significant gene was FCGR1A (P=3.5x10-26), which encodes a fragment of Immunoglobulin G, known to play an important role in immune processes.
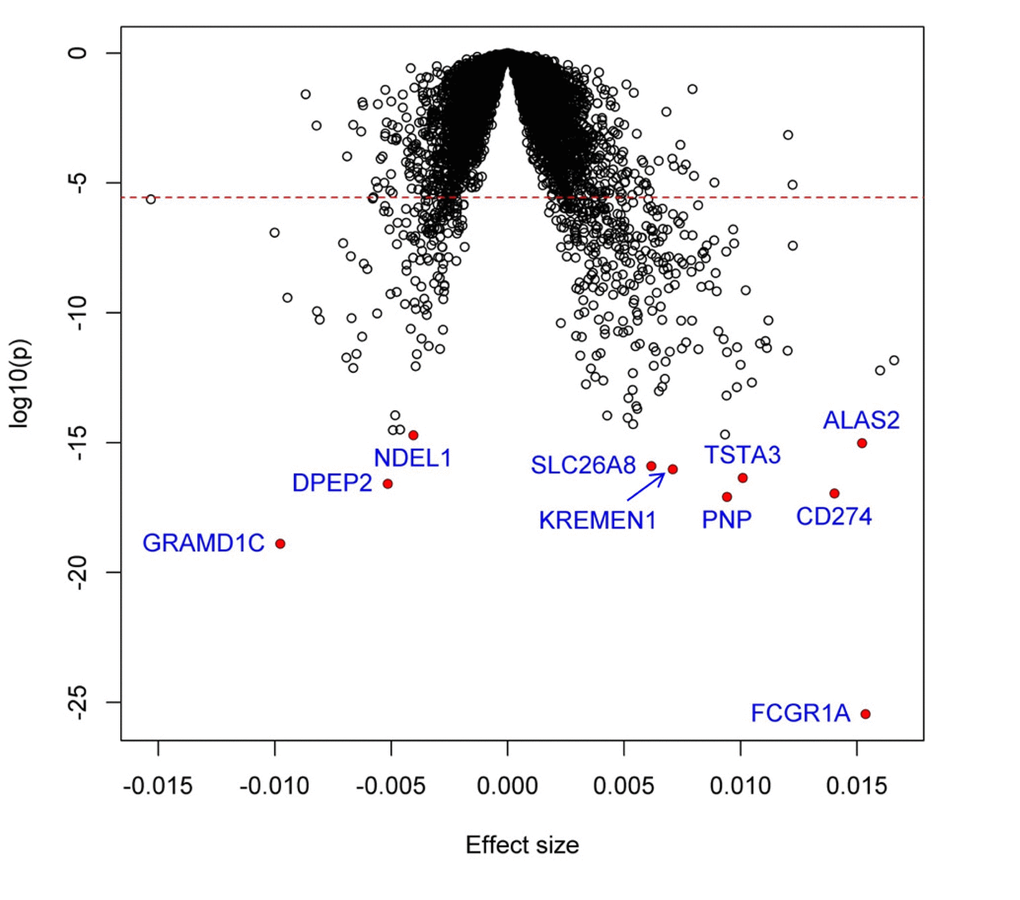
Figure 1. Volcano plot of association with inflammatory ∆age. Each dot represents one gene. The x-axis represents the beta estimation (β) of each gene, whereas the y-axis represents the log10(P). Positive effects represent that the genes were positively associated with inflammatory ∆age, whereas negative effects represent that the genes were negatively associated with inflammatory ∆age. The red dash line indicates P<0.05/17873=2.8x10-6.
Table 2. Top 25 genes associated with inflammatory ∆age.
| Affymetrix Transcript Cluster ID | Gene | Beta* | SE* | P-value | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2357845 | FCGR1A | 0.0154 | 0.0014 | 3.5E-26 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2636626 | GRAMD1C | -0.0098 | 0.0011 | 1.3E-19 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3527514 | PNP | 0.0094 | 0.0011 | 8.1E-18 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3161082 | CD274 | 0.0140 | 0.0016 | 1.1E-17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3696142 | DPEP2 | -0.0051 | 0.0006 | 2.6E-17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3157660 | TSTA3 | 0.0101 | 0.0012 | 4.4E-17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3941793 | KREMEN1 | 0.0071 | 0.0008 | 9.3E-17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2951730 | SLC26A8 | 0.0062 | 0.0007 | 1.2E-16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4009849 | ALAS2 | 0.0152 | 0.0019 | 9.6E-16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3709685 | NDEL1 | -0.0040 | 0.0005 | 1.9E-15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3061456 | SAMD9L | 0.0093 | 0.0012 | 2.1E-15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3576284 | RPS6KA5 | -0.0049 | 0.0006 | 3.0E-15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3690550 | SIAH1 | -0.0046 | 0.0006 | 3.2E-15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2421925 | GBP7 | 0.0054 | 0.0007 | 5.1E-15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2700828 | SIAH2 | 0.0052 | 0.0007 | 9.1E-15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2584258 | KCNH7 | 0.0043 | 0.0005 | 1.1E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3628832 | DAPK2 | -0.0048 | 0.0006 | 1.1E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2828479 | SLC22A4 | 0.0056 | 0.0007 | 2.0E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2369463 | FAM20B | 0.0055 | 0.0007 | 2.5E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2964200 | UBE2J1 | 0.0052 | 0.0007 | 5.2E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3090053 | SLC25A37 | 0.0094 | 0.0012 | 6.5E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2438531 | HDGF | 0.0065 | 0.0009 | 9.6E-14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3651955 | METTL9 | 0.0054 | 0.0007 | 1.1E-13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2421883 | GBP1 | 0.0098 | 0.0013 | 1.4E-13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2909404 | CD2AP | 0.0066 | 0.0009 | 1.4E-13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +The analyses were adjusted for age and sex | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *Beta is in units of one standard deviation change in gene expression per year of inflammatory ∆age; *SE: standard error | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
We created an expression score to represent the overall association between the expression profile and inflammatory ∆age using the weighted sum of all the 448 significant genes. To explore the potential utility of the expression score, we examined its association with mortality with up to ten years follow-up (mean 7.9 years, 270 deaths observed). The expression score was significantly associated with mortality after adjusting for age and sex (P=3.2x10-3, HR=1.62, 95% CI: 1.18-2.23). The association remained significant (P=0.048) after additionally adjusting for other mortality-related covariates, suggesting that not only the inflammatory ∆age itself, but also the expression score, might be a useful biomarker to predict mortality.
Pathway analysis
We then examined the enrichment of inflammatory ∆age associated genes in biological pathways. The top 10 enriched pathways are shown in Table 3. Two pathways were significant after correction for multiple testing, including NOD-like receptor signaling pathway (P=2.0x10-6, false discovery rate=6.0x10-4) and ubiquitin mediated proteolysis (P=1.7x10-4, false discovery rate =0.03). The NOD-like receptor signaling pathway remained significant (P=4.6x10-7, false discovery rate =1.4x10-4) if we included only positively associated genes.
Table 3. Ten most significant canonical pathways enriched with genes associated with inflammatory ∆age.
| KEGG pathway | #Genes in Pathway | Ratio of enrichment | P value | False discovery rate | Inflammatory ∆age related genes in the pathway |
| NOD-like receptor signaling pathway | 170 | 3.62 | 2.0E-06 | 6.0E-04 | GABARAP; GBP4; GBP5; MAPK14; CTSB; NLRP1; GABARAPL1; TAB3; GBP1; BIRC3; IFI16; IL1B; GBP7; MYD88; OAS1; STAT1; CASP5; AIM2 |
| Ubiquitin mediated proteolysis | 137 | 3.25 | 1.7E-04 | 0.03 | UBE2F; UBOX5; NEDD4L; BIRC3; MDM2; UBE2J1; UBE2O; SMURF2; SIAH1; ITCH; CUL4A; UBE2L6; CDC34 |
| Legionellosis | 55 | 4.35 | 1.0E-03 | 0.10 | CLK1; APAF1; IL1B; ARF1; MYD88; TLR5; CASP7 |
| Endocytosis | 260 | 2.24 | 1.5E-03 | 0.11 | ARAP2; AP2M1; AP2S1; ZFYVE27; AP2A1; NEDD4L; IGF1R; IGF2R; ARF1; LDLR; MDM2; PRKCZ; SMURF2; ITCH; PIP5K1B; SNX3; DNAJC6 |
| Central carbon metabolism in cancer | 67 | 3.57 | 3.2E-03 | 0.18 | AKT3; AKT1; FLT3; HK1; SLC1A5; SLC2A1; SLC7A5 |
| Salmonella infection | 86 | 3.18 | 3.5E-03 | 0.18 | MAPK14; KLC3; IL1B; KLC1; MYD88; PKN2; TLR5; WASF1 |
| TNF signaling pathway | 110 | 2.80 | 4.7E-03 | 0.20 | AKT3; MAPK14; AKT1; TAB3; BIRC3; IL1B; ITCH; CASP7; RPS6KA5 |
| Hepatitis C | 133 | 2.57 | 5.4E-03 | 0.21 | AKT3; PSME3; MAPK14; AKT1; DDX58; EIF2AK1; LDLR; OAS1; PPP2R2D; STAT1 |
| Measles | 136 | 2.52 | 6.3E-03 | 0.21 | AKT3; AKT1; DDX58; EIF2AK1; IL1B; JAK3; MYD88; OAS1; IFIH1; STAT1 |
| Pyrimidine metabolism | 105 | 2.61 | 1.1E-02 | 0.32 | CMPK2; DCK; PNP; RRM2B; POLR1D; POLR2B; UPP1; ENTPD5 |
Interaction between genes associated with inflammatory ∆age
We also applied a dense module searching strategy [14] to identify a subnetwork containing genes associated with inflammatory ∆age. As shown in Figure 2, the subnetwork is comprised of 34 nodes and 47 edges, in which each node represents one gene, and each edge represents the interaction between two genes. Two genes, GRB2 and CBL, play a pivotal role in the network. GRB2 itself was not associated with inflammatory ∆age (P=0.55), but it was connected with 10 other genes, of which five (KCNH7, ALAS2, FLT3, CD2AP, MAPK14) were associated with inflammatory ∆age. Similarly, CBL was not associated with inflammatory ∆age (P=0.07), but four (FCGR1A, IGF1R, CD2AP, MAPK14), out of its seven neighbors were associated with inflammatory ∆age.
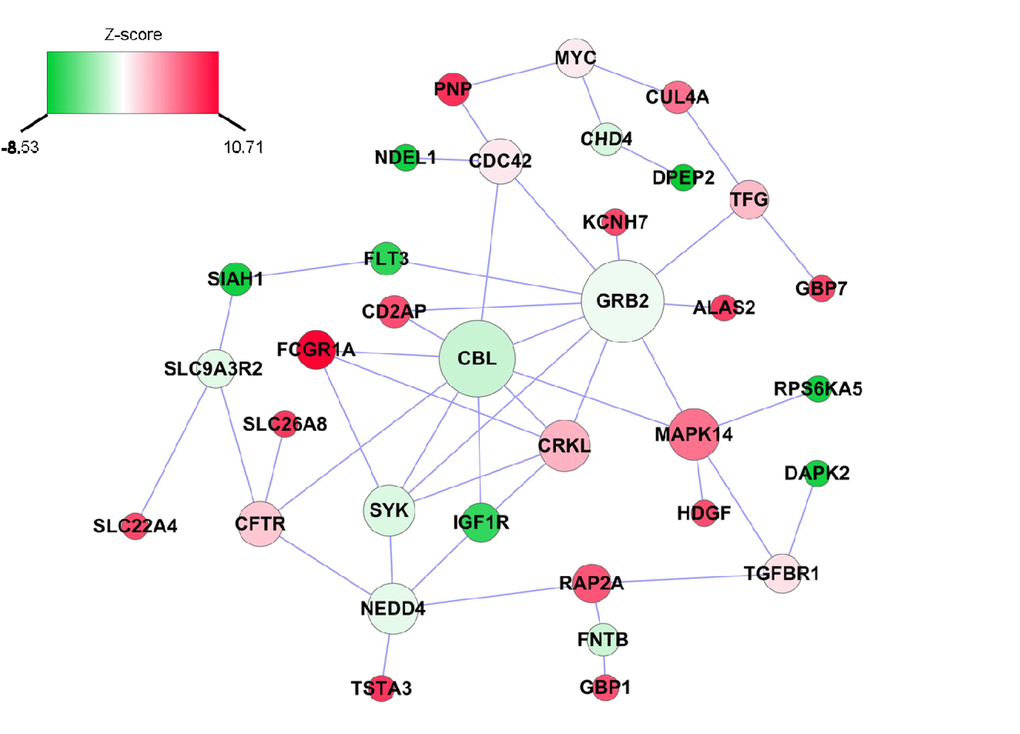
Figure 2. Inflammatory ∆age-related subnetwork derived from protein-protein interaction. Each node represents one gene, whereas each edge represents the interaction between two genes. The nodes were colored to represent their association with inflammatory ∆age by z-score: red represents genes that were positively associated with inflammatory ∆age, whereas green represents genes that were negatively associated with inflammatory ∆age. The node size is proportional to the number of edges that the node connects to.
Comparison with genes associated with chronological age
It has been long recognized that aging is an important factor affecting gene expression. An earlier meta-analysis of whole blood gene expression in 14,983 individuals reported 1,497 genes that were differentially expressed with chronological age [11]. Among them, 56 genes also were associated with inflammatory ∆age, showing a significant enrichment (P=0.0023) by the Fisher's exact test (Supplemental Table 1), suggesting that some common mechanisms might be involved in the regulation of chronological age and inflammatory ∆age.
Enrichment of inflammatory ∆age in methylation genes
Among 448 genes whose expression was associated with inflammatory ∆age, 223 genes contained at least one CpG site in which methylation was associated with inflammatory ∆age (defined as differentially methylated genes [DMGs], Supplemental Table 1). Among the 223 DMGs, 147 were positively associated with inflammatory ∆age, and the remaining 76 genes were negatively associated with inflammatory ∆age. These gene loci showed two complementary assays demonstrating altered genomic regulation in relation to inflammatory ∆age. In order to assess whether this is greater than chance, we generated one million gene sets, containing 448 randomly selected genes. We then checked how many of the 448 randomly selected genes were DMGs. As shown in Figure 3, on average, each randomly matched gene set contained 153 DMGs (min: 109 genes, max: 199 genes), which is much smaller than that of inflammatory ∆age-related genes (empirical p-value <1x10-6), indicating that there was significant overlap between genes whose expression and whose methylation were associated with inflammatory ∆age. In addition, of the 56 genes associated with inflammatory ∆age that were previously reported to be associated with age [11], 35 genes were DMGs (Supplemental Table 1).
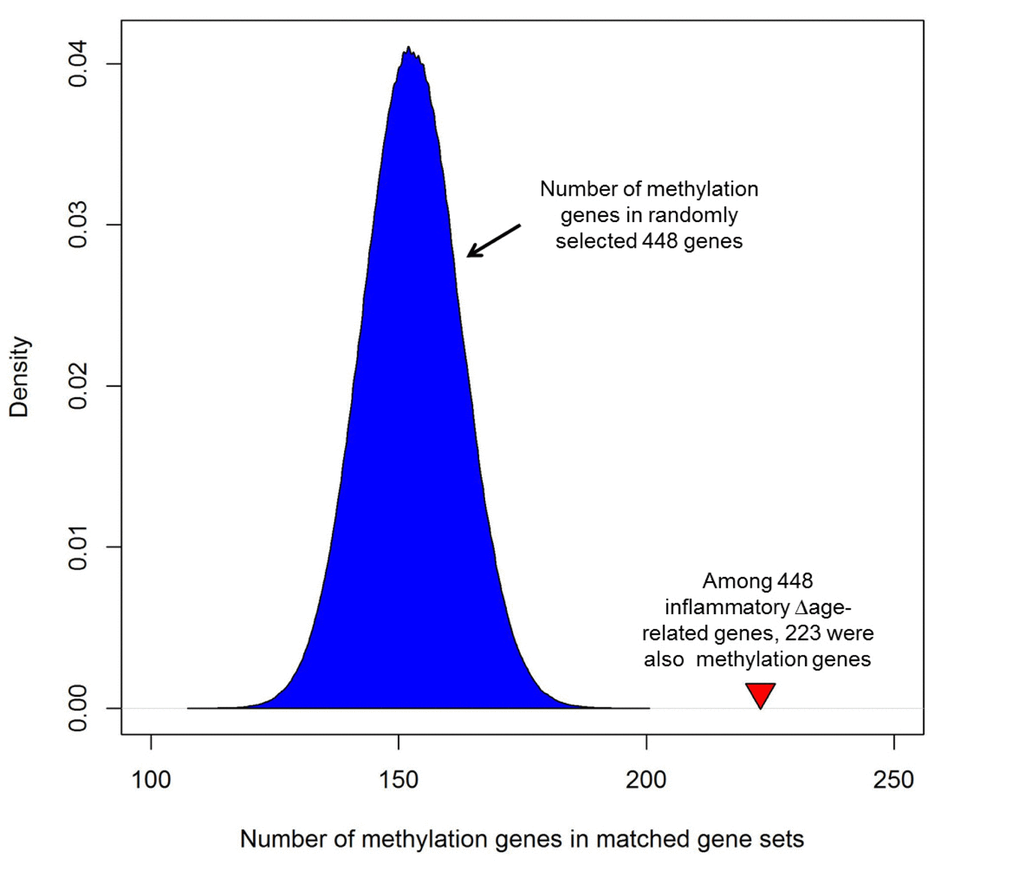
Figure 3. Enrichment of inflammatory ∆age-related genes with corresponding differences in methylation. Among 448 genes whose expression was associated with inflammatory ∆age, 223 genes contained at least one CpG where methylation was associated with inflammatory ∆age (defined as differentially methylated genes [DMGs]). In order to assess its significance, we generated one million gene sets, each one containing 448 randomly selected genes and determined how many of the 448 randomly selected genes were DMGs. As shown, each randomly matched gene set contained a mean of 153 methylation genes (min: 109 genes, max: 199 genes), which is much smaller than that of inflammatory ∆age-related genes (empirical p-value <1x10-6). The red triangle indicates the number of DMGs in inflammatory ∆age-related genes.
We then tested the association of gene expression with inflammatory ∆age by additionally adjusting for DNA methylation. Of 223 DMGs, the associations of 154 (69%) genes with inflammatory ∆age were slightly attenuated after adjusting for the most significant CpG site in the gene, suggesting that DNA methylation might be a potential mechanism regulating the association between gene expression and inflammatory ∆age.
Discussion
We investigated the association of gene expression with inflammatory ∆age in more than 2000 older participants from the community-based Framingham Heart Study Offspring cohort. A total of 448 genes were found to be significantly associated with inflammatory ∆age with two-thirds of the genes demonstrating increased expression with greater inflammatory ∆age. The most significant gene was FCGR1A, encoding the Fc-gamma receptor (FCGR1A) that complexes with Immunoglobulin G, which is known to activate or inhibit various cell functions, and plays a critical role in immune responses. Many inflammatory ∆age-related differentially expressed genes were found to be involved in NOD-like receptor signaling pathway and ubiquitin mediated proteolysis pathway. Among the 448 genes whose expression was associated with inflammatory ∆age, 223 genes contained at least one CpG site associated with inflammatory ∆age suggesting that DNA methylation may be a potential mechanism regulating the association between gene expression and inflammatory ∆age.
As expected, in addition to FCGR1A several of the most significant genes were involved in immune and inflammatory-related functions. For example, mutations in PNP result in nucleoside phosphorylase deficiency that in turn produce defective cell-mediated immunity. Pnp-deficient mice exhibit a progressive T-cell decline with both reduced numbers of thymocytes and splenic T-cells [15]. CD274 encodes an immune inhibitory receptor expressed on T cells, B cells and various tumor cell types. The usefulness of blocking CD274 to enhance anti-cancer immunity is under investigation [16]. Dysregulation of the immune system is one mechanism underlying inflamm-aging that accelerates biologic aging and risk for age-related disease [8].
In contrast, among the top results the SIAH1 and SIAH2 genes code evolutionarily conserved E3 ubiquitin ligases implicated in hypoxia and apoptosis. SIAH1 and SIAH2 also regulate diverse cellular functions such as DNA damage response, mitochondrial dynamics and estrogen receptor signaling [17]. These two genes have been implicated in a number of age-related diseases including Parkinson's disease and several cancers [18,19]. A SNP in SIAH1 was recently reported to reach a suggestive but not genome-wide level of association with exceptional longevity (living past the age that less than 1% of individuals from the 1900 birth cohort survived) [20]. In that study the longevity allele was associated with decreased risk for cardiovascular disease and hypertension. In animal models SIAH2 appears to regulate obesity-related adipose tissue dysfunction and recruitment of immune cells to adipose tissue [21]. Adipose tissue dysfunction is an important physiologic contributor to aging related metabolic derangements, chronic disease, and frailty [22].
The NOD-like receptor signaling pathway is the biologic pathway most significantly enriched with genes associated with inflammatory ∆age. In this pathway was NLRP1 (NLR Family Pyrin Domain Containing 1), a protein coding gene that is part of the NLRP1 inflammasome initiated in response to danger signals [23]. NLRP1 plays a role in generating innate immune responses and apoptosis. Associations with age-related diseases remain under investigation but there may be a role for susceptibility to cancer, atherosclerosis and Alzheimer disease. Other Nod-like family receptor sensors have been associated with aging and amelioration of NLRP3 mediated inflammation improves age-related declines in a number of physiologic systems [24].
DNA methylation has been recognized as an important biomarker associated with aging and age-related diseases [25-27]. Multiple computational models have been developed to predict chronological age using methylation biomarkers [28-30]. We found a significant overlap between genes whose expression was associated with inflammatory ∆age, and genes that contained at least one CpG site associated with inflammatory ∆age. Moreover, in more than two thirds of genes, we observed the association between gene expression and inflammatory ∆age was attenuated after adjusting for the CpG site within the gene. Our results suggest that DNA methylation might be an important mechanism to regulate gene expression and thus inflammatory ∆age. Both genetics and environmental factors influence gene expression and DNA methylation, which together demonstrate a dynamic landscape of chronological changes during the aging process. Future investigation of the interplay between DNA methylation and gene expression might uncover important mechanisms underlying aging, and potentially lead to better strategies to slowdown or even reverse the aging process [31,32].
Several limitations of our study merit comment. Participants were of European descent, thus the generalizability of our findings to other race/ethnicities is unclear. Gene expression was measured from whole blood, which contains a variety of cell types and may have specific cell responses. We thereby accounted for the relative abundance of each cell type in our analyses. Our study was limited to association analyses, we cannot exclude residual confounding, and we cannot infer causality between inflammatory ∆age and gene expression. In addition, the inflammatory ∆age and gene expression was measured during one examination, therefore we cannot comment on longitudinal variation in the relations between gene expression and inflammatory ∆age. Our results need to be replicated in an independent sample.
However, our study also had several strengths. Our study is one of the first efforts to estimate biological age from a set of robust inflammatory biomarkers. We took a hypothesis-free approach to study transcriptome-wide profiling with inflammatory biological age in a large carefully characterized cohort. Our epigenome-wide analysis identified CpGs associated with inflammatory ∆age in the many of the same genes as our transcriptome-wide association study lending support to our findings.
In conclusion, we identified 448 genes that were significantly associated with inflammatory aging in a large community-based cohort. Future functional characterization and direct perturbation of the identified gene regulation network may enable the development of preventative strategies or therapies to arrest or slow biological aging or age-related diseases and declines of physical function.
Methods
Study sample
The Framingham Heart Study is a multi-generational study initiated in 1948 to investigate cardiovascular disease and its risk factors in the community. The current study is limited to the Offspring cohort participants, who are the children of the Framingham Original cohort as well as the offspring spouses [33,34]. They were enrolled in 1971-1975 and they have been examined every 4 to 8 years. To be eligible for this study, participants needed to attend examination 8 (2005 to 2008, n=3021) at which inflammatory biomarker and gene expression data were obtained. Participants were excluded if the following data were missing: inflammatory biomarkers (n= 279), gene expression (n= 353). The final study sample included 2386 participants. All participants provided written informed consent, and the study was approved by the Boston University Medical Center Institutional Review Board.
Derivation of inflammatory biologic age
The inflammatory biologic age estimate comprised nine inflammatory biomarkers measured from fasting morning blood samples. Assay details have been reported and the intra- and inter- assay coefficients of variation were below 10% [35]. The inflammatory biomarkers included c-reactive protein (CRP), intercellular adhesion molecule-1 (ICAM1), interleukin-6 (IL6), lipoprotein-associated phospholipase A2 (LP-PLA2) mass, LP-PLA2 activity, monocyte chemoattractant protein-1 (MCP1), osteoprotegerin, p-selectin, and tumor necrosis factor receptor II (TNFR2). The inflammatory biomarkers function broadly in the inflammation pathway including as acute phase reactants, chemokines, cytokines, selectins, and cell adhesion molecules [36]. These biomarkers were modestly correlated (Pearson correlations 0.06 to 0.27 except C-reactive protein and interleukin-6, r=0.52) (Supplemental Table 2). Seven of the nine biomarkers were significantly correlated with age (Supplemental Table 2a).
We used the Klemera and Doubal method [37] to estimate inflammatory biologic age. The Klemera-Doubal algorithm includes age as one of the biomarkers and demonstrates the best performances in the precision of estimation [37] and predictive ability [12]. We defined I"age as inflammatory biologic age minus chronologic age. Thus, individuals with I"age>0 have greater biologic age than chronologic age, whereas individuals with I"age<0 have biologic age less than chronologic age. The chronologic age and I"age were not correlated (P>0.05) [38].
RNA extraction and gene expression profiling
The details regarding the gene expression profiling have been described previously [39,40]. Total RNA was isolated from whole blood using PAXgene blood tubes (PreAnalytiX, Hombrechtikon, Switzerland) and amplified using the WT-Ovation Pico RNA Amplification System (NuGEN, San Carlos, CA) according to the manufacturers' standard operating procedures. The obtained cDNA was hybridized to the Affymetrix Human Exon 1.0 ST Array (Affymetrix, Inc., Santa Clara, CA). Signal intensities from the image scanner were then quantile-normalized and log2 transformed, followed by summarization using Robust Multi-array Average [41]. The annotation for each transcript was obtained from Affymetrix NetAffx Analysis Center (version 31). Transcript clusters that were not mapped to RefSeq transcripts were excluded. A total of 17,873 distinct transcripts mapping to 17,562 unique genes were included for downstream analysis. In order to adjust for the effects of different cell counts on gene expression, cell counts were imputed using a partial least squares regression method applied to mRNA expression [42].
Statistical analyses
Linear mixed effect models were used to test associations between inflammatory ∆age and gene expression, with gene expression the dependent measure and the inflammatory ∆age the exposure variable. We treated familial relatedness in the Framingham participants as a random variance-covariance factor. The models were additionally adjusted for sex, age, imputed cell counts, and technical covariates. All analyses were performed using the R software package (www.r-project.org), and the linear mixed effect models were implemented in the "lmekin" package. We used Bonferroni adjustment to correct for multiple testing, which was defined as 0.05/17873=2.8x10-6.
We also created an expression score that included genes significantly associated with inflammatory ∆age. Specifically, for any participant j, the score was defined as
,
where i represented the ith significant gene,
Pathway analysis
Pathway analyses were performed to identify potential biologic mechanisms among the significant genes. We used WebGestalt [43], a web-based pathway analysis tool. The enrichment was assessed by the Fisher's exact test for each KEGG pathway, and the multiple testing was accounted for by the false discovery rate approach [44].
Construction of interaction subnetwork associated with inflammatory biologic age
In order to examine the interaction between inflammatory ∆age-related genes, we constructed an interaction network using a dense module searching strategy [14]. The gene interactions were downloaded from the PINA database [45]. Before searching, each gene was assigned a score to represent its association with inflammatory ∆age, equivalent to Z(p-value). Each module starts with one of the top 25 genes associated with inflammatory ∆+age. For each of its neighboring genes, we evaluated if adding it to the module would increase the overall module score [46], which was defined as
,
where k is the number of genes in the module, and
Enrichment of inflammatory ∆age in methylation genes
We also examined if DNA methylation might be involved in the association between gene expression and inflammatory ∆age. For each significant gene (p<2.8x10-6), we examined if the gene contained any CpG site that was associated with inflammatory ∆age. The details of Framingham DNA methylation were described previously [47-49]. Briefly, the genomic DNA was collected from fasting peripheral whole blood, bisulfite-treated and hybridized to the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA) according to the manufacturer's standard protocols [50]. The methylation status was represented by the β value, and the raw data were normalized and corrected for the background noise by "DASEN" R package [51]. After quality control filters, a total of 443,252 CpG sites were then tested for the association with inflammatory ∆age, and the significance cutoff for the CpG site was defined as p<0.05/N, where N was the number of CpG sites within the tested genes.
Abbreviations
DMGs: Differentially methylated genes.
Author Contributions
HL and JMM initiated the study and drafted the manuscript. HL, KLL, QZ, JR, and MGL performed statistical analyses. EJB generated the inflammatory biomarker data. RJ and DL generated and processed the gene expression data. KLL, QZ, JR, EJB, MMM, RJ, DL, and MGL critically reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank participants of Framingham Heart Study to make this study possible.
Conflicts of Interest
The authors declare no commercial conflicts of interest.
Funding
This work is supported by NIH grants R56AG029451 (Murabito), 5R01HL092577 (Benjamin) and 5R01HL128914 (Benjamin). Measurement of inflammatory biomarkers was funded through R01 HL 064753 and R01 HL076784 and further supported by 1R01 HL64753 (Benjamin), R01AG028321 (Benjamin). Framingham gene expression profiling was funded through the Division of Intramural Research (Levy), National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD. Additional support was from the NIH NHLBI K99 HL136875 (Mendelson). This project was partly supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through BU-CTSI Grant Number 1UL1TR001430. The Framingham Heart Study is supported by the National Heart, Lung and Blood Institute's Framingham Heart Study (Contracts No. N01-HC-25195 and HHSN-268201500001I). The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
References
- 1. Newman AB, Sachs MC, Arnold AM, Fried LP, Kronmal R, Cushman M, Psaty BM, Harris TB, Robbins JA, Burke GL, Kuller LH, Lumley T. Total and cause-specific mortality in the cardiovascular health study. J Gerontol A Biol Sci Med Sci. 2009; 64:1251–61. https://doi.org/10.1093/gerona/glp127 [PubMed]
- 2. Schnabel RB, Yin X, Larson MG, Yamamoto JF, Fontes JD, Kathiresan S, Rong J, Levy D, Keaney JF
Jr , Wang TJ, Murabito JM, Vasan RS, Benjamin EJ. Multiple inflammatory biomarkers in relation to cardiovascular events and mortality in the community. Arterioscler Thromb Vasc Biol. 2013; 33:1728–33. https://doi.org/10.1161/ATVBAHA.112.301174 [PubMed] - 3. Cesari M, Penninx BW, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, Rubin SM, Ding J, Simonsick EM, Harris TB, Pahor M. Inflammatory markers and onset of cardiovascular events: results from the Health ABC study. Circulation. 2003; 108:2317–22. https://doi.org/10.1161/01.CIR.0000097109.90783.FC [PubMed]
- 4. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJ, Cornel JH, Pais P, et al, and CANTOS Trial Group. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017; 377:1119–31. https://doi.org/10.1056/NEJMoa1707914 [PubMed]
- 5. Bandeen-Roche K, Walston JD, Huang Y, Semba RD, Ferrucci L. Measuring systemic inflammatory regulation in older adults: evidence and utility. Rejuvenation Res. 2009; 12:403–10. https://doi.org/10.1089/rej.2009.0883 [PubMed]
- 6. Manini TM, Anton SD, Beavers DP, Cauley JA, Espeland MA, Fielding RA, Kritchevsky SB, Leeuwenburgh C, Lewis KH, Liu C, McDermott MM, Miller ME, Tracy RP, et al, and ENRGISE Pilot study investigators. ENabling Reduction of Low-grade Inflammation in SEniors Pilot Study: Concept, Rationale, and Design. J Am Geriatr Soc. 2017; 65:1961–68. https://doi.org/10.1111/jgs.14965 [PubMed]
- 7. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007; 128:92–105. https://doi.org/10.1016/j.mad.2006.11.016 [PubMed]
- 8. Fougere B, Boulanger E, Nourhashemi F, Guyonnet S, Cesari M. Chronic Inflammation: Accelerator of Biological Aging. J Gerontol A Biol Sci Med Sci. 2017; 72:1218–25. https://doi.org/10.1093/gerona/glw240 [PubMed]
- 9. Franceschi C, Olivieri F, Marchegiani F, Cardelli M, Cavallone L, Capri M, Salvioli S, Valensin S, De Benedictis G, Di Iorio A, Caruso C, Paolisso G, Monti D. Genes involved in immune response/inflammation, IGF1/insulin pathway and response to oxidative stress play a major role in the genetics of human longevity: the lesson of centenarians. Mech Ageing Dev. 2005; 126:351–61. https://doi.org/10.1016/j.mad.2004.08.028 [PubMed]
- 10. Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell. 2012; 11:727–31. https://doi.org/10.1111/j.1474-9726.2012.00871.x [PubMed]
- 11. Peters MJ, Joehanes R, Pilling LC, Schurmann C, Conneely KN, Powell J, Reinmaa E, Sutphin GL, Zhernakova A, Schramm K, Wilson YA, Kobes S, Tukiainen T, et al, and NABEC/UKBEC Consortium. The transcriptional landscape of age in human peripheral blood. Nat Commun. 2015; 6:8570. https://doi.org/10.1038/ncomms9570 [PubMed]
- 12. Levine ME. Modeling the rate of senescence: can estimated biological age predict mortality more accurately than chronological age? J Gerontol A Biol Sci Med Sci. 2013; 68:667–74. https://doi.org/10.1093/gerona/gls233 [PubMed]
- 13. Murabito JM, Zhao Q, Larson MG, Rong J, Lin H, Benjamin EB, Levy D, Lunetta KL. Measures of Biologic Age in a community sample predict mortality and age-related disease. The Journals of Gerontology: Series A. 2017, https://doi.org/110.1093/gerona/glx1144.
- 14. Jia P, Zheng S, Long J, Zheng W, Zhao Z. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 2011; 27:95–102. https://doi.org/10.1093/bioinformatics/btq615 [PubMed]
- 15. Snyder FF, Jenuth JP, Mably ER, Mangat RK. Point mutations at the purine nucleoside phosphorylase locus impair thymocyte differentiation in the mouse. Proc Natl Acad Sci USA. 1997; 94:2522–27. https://doi.org/10.1073/pnas.94.6.2522 [PubMed]
- 16. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515:563–67. https://doi.org/10.1038/nature14011 [PubMed]
- 17. Gopalsamy A, Hagen T, Swaminathan K. Investigating the molecular basis of Siah1 and Siah2 E3 ubiquitin ligase substrate specificity. PLoS One. 2014; 9:e106547. https://doi.org/10.1371/journal.pone.0106547 [PubMed]
- 18. van der Willik KD, Timmermans MM, van Deurzen CH, Look MP, Reijm EA, van Zundert WJ, Foekens R, Trapman-Jansen AM, den Bakker MA, Westenend PJ, Martens JW, Berns EM, Jansen MP. SIAH2 protein expression in breast cancer is inversely related with ER status and outcome to tamoxifen therapy. Am J Cancer Res. 2016; 6:270–84. [PubMed]
- 19. Franck T, Krueger R, Woitalla D, MA1/4ller T, Engelender S, Riess O. Mutation analysis of the seven in absentia homolog 1 (SIAH1) gene in Parkinson's disease. J Neural Transm (Vienna). 2006; 113:1903–08. https://doi.org/10.1007/s00702-006-0480-z [PubMed]
- 20. Sebastiani P, Gurinovich A, Bae H, Andersen S, Malovini A, Atzmon G, Villa F, Kraja AT, Ben-Avraham D, Barzilai N, Puca A, Perls TT. Four Genome-Wide Association Studies Identify New Extreme Longevity Variants. J Gerontol A Biol Sci Med Sci. 2017; 72:1453–64. https://doi.org/10.1093/gerona/glx027 [PubMed]
- 21. Kilroy G, Carter LE, Newman S, Burk DH, Manuel J, Möller A, Bowtell DD, Mynatt RL, Ghosh S, Floyd ZE. The ubiquitin ligase Siah2 regulates obesity-induced adipose tissue inflammation. Obesity (Silver Spring). 2015; 23:2223–32. https://doi.org/10.1002/oby.21220 [PubMed]
- 22. Stout MB, Justice JN, Nicklas BJ, Kirkland JL. Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology (Bethesda). 2017; 32:9–19. https://doi.org/10.1152/physiol.00012.2016 [PubMed]
- 23. Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140:821–32. https://doi.org/10.1016/j.cell.2010.01.040 [PubMed]
- 24. Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev. 2015; 265:63–74. https://doi.org/10.1111/imr.12295 [PubMed]
- 25. Johnson AA, Akman K, Calimport SR, Wuttke D, Stolzing A, de Magalhães JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012; 15:483–94. https://doi.org/10.1089/rej.2012.1324 [PubMed]
- 26. Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol. 2015; 13:7. https://doi.org/10.1186/s12915-015-0118-4 [PubMed]
- 27. Richardson B. Impact of aging on DNA methylation. Ageing Res Rev. 2003; 2:245–61. https://doi.org/10.1016/S1568-1637(03)00010-2 [PubMed]
- 28. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. https://doi.org/10.1186/gb-2013-14-10-r115 [PubMed]
- 29. Vidaki A, Ballard D, Aliferi A, Miller TH, Barron LP, Syndercombe Court D. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci Int Genet. 2017; 28:225–36. https://doi.org/10.1016/j.fsigen.2017.02.009 [PubMed]
- 30. Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS One. 2011; 6:e14821. https://doi.org/10.1371/journal.pone.0014821 [PubMed]
- 31. Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014; 124:24–29. https://doi.org/10.1172/JCI69735 [PubMed]
- 32. Martin GM. Epigenetic drift in aging identical twins. Proc Natl Acad Sci USA. 2005; 102:10413–14. https://doi.org/10.1073/pnas.0504743102 [PubMed]
- 33. Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study. Design and preliminary data. Prev Med. 1975; 4:518–25. https://doi.org/10.1016/0091-7435(75)90037-7 [PubMed]
- 34. Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979; 110:281–90. https://doi.org/10.1093/oxfordjournals.aje.a112813 [PubMed]
- 35. Fontes JD, Yamamoto JF, Larson MG, Wang N, Dallmeier D, Rienstra M, Schnabel RB, Vasan RS, Keaney JF
Jr , Benjamin EJ. Clinical correlates of change in inflammatory biomarkers: TheA Framingham Heart Study. Atherosclerosis. 2013; 228:217–23. https://doi.org/10.1016/j.atherosclerosis.2013.01.019 [PubMed] - 36. Schnabel RB, Lunetta KL, Larson MG, Dupuis J, Lipinska I, Rong J, Chen MH, Zhao Z, Yamamoto JF, Meigs JB, Nicaud V, Perret C, Zeller T, et al. The relation of genetic and environmental factors to systemic inflammatory biomarker concentrations. Circ Cardiovasc Genet. 2009; 2:229–37. https://doi.org/10.1161/CIRCGENETICS.108.804245 [PubMed]
- 37. Klemera P, Doubal S. A new approach to the concept and computation of biological age. Mech Ageing Dev. 2006; 127:240–48. https://doi.org/10.1016/j.mad.2005.10.004 [PubMed]
- 38. Murabito JM, Zhao Q, Larson MG, Rong J, Lin H, Benjamin EJ, Levy D, Lunetta KL. Measures of Biologic Age in a Community Sample Predict Mortality and Age-Related Disease: The Framingham Offspring Study. J Gerontol A Biol Sci Med Sci. 2017. https://doi.org/10.1093/gerona/glx144 [PubMed]
- 39. Joehanes R, Ying S, Huan T, Johnson AD, Raghavachari N, Wang R, Liu P, Woodhouse KA, Sen SK, Tanriverdi K, Courchesne P, Freedman JE, O'Donnell CJ, et al. Gene expression signatures of coronary heart disease. Arterioscler Thromb Vasc Biol. 2013; 33:1418–26. https://doi.org/10.1161/ATVBAHA.112.301169 [PubMed]
- 40. Lin H, Yin X, Lunetta KL, Dupuis J, McManus DD, Lubitz SA, Magnani JW, Joehanes R, Munson PJ, Larson MG, Levy D, Ellinor PT, Benjamin EJ. Whole blood gene expression and atrial fibrillation: the Framingham Heart Study. PLoS One. 2014; 9:e96794. https://doi.org/10.1371/journal.pone.0096794 [PubMed]
- 41. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–64. https://doi.org/10.1093/biostatistics/4.2.249 [PubMed]
- 42. Huan T, Rong J, Liu C, Zhang X, Tanriverdi K, Joehanes R, Chen BH, Murabito JM, Yao C, Courchesne P, Munson PJ, O'Donnell CJ, Cox N, et al. Genome-wide identification of microRNA expression quantitative trait loci. Nat Commun. 2015; 6:6601. https://doi.org/10.1038/ncomms7601 [PubMed]
- 43. Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013; 41:W77-83. https://doi.org/10.1093/nar/gkt439 [PubMed]
- 44. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995; 57:289–300.
- 45. Cowley MJ, Pinese M, Kassahn KS, Waddell N, Pearson JV, Grimmond SM, Biankin AV, Hautaniemi S, Wu J. PINA v2.0: mining interactome modules. Nucleic Acids Res. 2012; 40:D862–65. https://doi.org/10.1093/nar/gkr967 [PubMed]
- 46. Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002 (Suppl 1); 18:S233–40. https://doi.org/10.1093/bioinformatics/18.suppl_1.S233 [PubMed]
- 47. Lin H, Yin X, Xie Z, Lunetta KL, Lubitz SA, Larson MG, Ko D, Magnani JW, Mendelson MM, Liu C, McManus DD, Levy D, Ellinor PT, Benjamin EJ. Methylome-wide Association Study of Atrial Fibrillation in Framingham Heart Study. Sci Rep. 2017; 7:40377. https://doi.org/10.1038/srep40377 [PubMed]
- 48. Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, Just AC, Duan Q, Boer CG, Tanaka T, Elks CE, Aslibekyan S, Brody JA, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2016; 192. https://doi.org/10.1038/mp.2016.192 [PubMed]
- 49. Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, Colicino E, Waite LL, Joehanes R, Guan W, Brody JA, Elks C, Marioni R, et al, and WHI-EMPC Investigators, and CHARGE epigenetics of Coronary Heart Disease. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol. 2016; 17:255. https://doi.org/10.1186/s13059-016-1119-5 [PubMed]
- 50. Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan JB, Shen R. High density DNA methylation array with single CpG site resolution. Genomics. 2011; 98:288–95. https://doi.org/10.1016/j.ygeno.2011.07.007 [PubMed]
- 51. Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013; 14:293. https://doi.org/10.1186/1471-2164-14-293 [PubMed]