



Introduction
Human lifespan has long been observed as a complex trait with approximately 25% genetic contributions [1]. To date, only very few genes have been shown consistently associated with it [2–5]. Recent studies reported that copy number variation (CNV) may directly contribute to human lifespan [6–10]. CNV is a general term for all the chromosomal rearrangements, such as deletions, duplications [11]. CNVs can change gene structures, thus affecting gene expression and phenotypes. In human, CNVs have been implicated in numerous diseases, such as autism and diabetes [12,13]. CNVs also contribute significantly to the genome instability of cancer cells [14–16]. For example, recently, Pelttari [14] reported a unique duplication encompassing most of the RAD51 homolog C gene in breast and ovarian cancers. Habibi [15] showed that copy number changes in the gene loci, UDP Glucuronosyltransferase Family 2 members, B28 and B17 were associated with prostate cancer. In addition, Zhou and his colleagues linked 93 CNVs to hepatocellular carcinoma [16].
A few studies have investigated the association of CNV with human lifespan using genome-wide approaches [6–10]. Kuningas [7] first reported this association in 11442 human samples representing two cohorts with the ages ranging from 62.0 to 75.3 and 34.0 to 69.8. They found large deletions in 11p15.5 (p = 2.8×10-6, Hazard Ratio (HR) = 1.59) and 14q21.3 (p = 1.5×10-3, HR = 1.57) among the oldest people. Another study uncovered a deletion in the CNTNAP4 gene in a female group of 80 years of age (Odds Ratio (OR) = 0.41, p = 0.007), but not the male group (OR = 0.97, p = 1) [9]. Recently, a study in Caucasians (n = 388; cases: 81-90 year-olds; controls: 65-75 year-olds) revealed an insertion allele of the CNTNAP2 gene in esv11910 CNV of males (OR = 0.29, 95%; CI: 0.14–0.59; p = 4×10-4), but not females (OR = 0.82, 95%; CI: 0.42–1.57, p = 0.625) [10].
In this study, we investigated the association of CNVs and longevity in Han Chinese by genotyping 4007 individuals obtained from the Chinese Longitudinal Healthy Longevity Survey (CLHLS) database. We have identified a few CNVs, and most of them were new. Some of them encode the known genes and pathways that have been associated with longevity only in this study. The deletions of the cancer-causing and aging-related genes encoded in these CNVRs may extend healthy lifespan for long-lived individuals. Future research on these CNVRs will shed light on the regulatory mechanism controlling human longevity.
Results
The numbers of CNVs increase in the genomes of long-lived individuals
To identify the CNVs associated with longevity in Chinese population, we recruited 1950 long-lived individuals called “cases”, and 2057 middle-aged Chinese as “controls. These individuals were further separated into four cohorts: 1000 long-lived and 1215 middle-aged who lived in the north of China, and the rest of 950 long-lived and 842 middle-aged who resided in the south. The two Northern cohorts were also called “discovery cohorts”, and the two Southern cohorts were referred as “replicate cohorts”.
Using the Principal Component Analysis (PCA) method, no extra sub-cluster was found between case and control comparison (Supplementary Figure 1). Our data obtained from the Northern cohorts revealed a significantly increased number of CNVs among long-lived individuals compared to the controls (6.94 ± 0.35 vs 5.96 ± 0.27, p = 0.027). The similar results were found in the Southern replicate cohorts (5.96 ± 0.27 vs 4.60 ± 0.17; p = 0.001). Total identified CNVs were summarized in detail in Table 1.
Table 1. Summary of CNVs.
| N | Mean age | Female% | Mean CNV length(kb) | Total CNV length(kb) | Mean Count CNV(N) | Mean DEL length(kb) | Total DEL length(kb) | Mean Count del(N) | Mean DUP length(kb) | Total DUP length(kb) | Mean Count dup(N) | ||
| Combined samples | Case | 1950 | 101.51(90-117) | 74.30 | 83.78±1.49 | 508.44±19.78 | 6.46±0.22 | 47.11±1.19 | 224.23±14.04 | 4.15±0.20 | 104.43±2.61 | 284.20±14.01 | 2.30±0.10 |
| Control | 2057 | 48.22(30-65) | 60.77 | 85.57±1.86 | 450.53±16.32 | 5.40±0.18 | 49.92±1.33 | 201.81±11.88 | 3.25±0.16 | 93.94±2.67 | 248.72±11.56 | 2.14±0.07 | |
| P | 0.457 | 0.024 | 0.001 | 0.118 | 0.221 | 0.001 | 0.005 | 0.051 | 0.193 |
In all participating subjects including both cases and the controls, we identified 10046 deletions and 6932 duplications in total. We found that the numbers of CNVs increased significantly in older ages (Spearman rho = 0.386, p = 0.002; Figure 1), particularly, the deletions increased much more than the duplications (Spearman rho = 0.356, p = 0.004). On average, the centenarians (101.51 ± 0.07 years of age) contained 4.15 ± 0.20 deletions and 2.30 ± 0.10 duplications, showing a significant increase (p = 0.001) compared to the middle-aged (48.22 ± 0.16 years of age), who had 3.25 ± 0.16 deletions and 2.14 ± 0.07 duplications. The Spearman correlation was also calculated to show the significance between ages and total added lengths of CNVs (Spearman rho = 0.31, p = 0.017); long-lived people had 508.54 ± 19.8 kb of total CNVs, which was significantly longer than 453.69 ± 17.25 kb in middle-aged controls (p = 0.024). In summary, we have shown that the CNV numbers, especially the deletion numbers, increased significantly in the genomes of long-lived people.
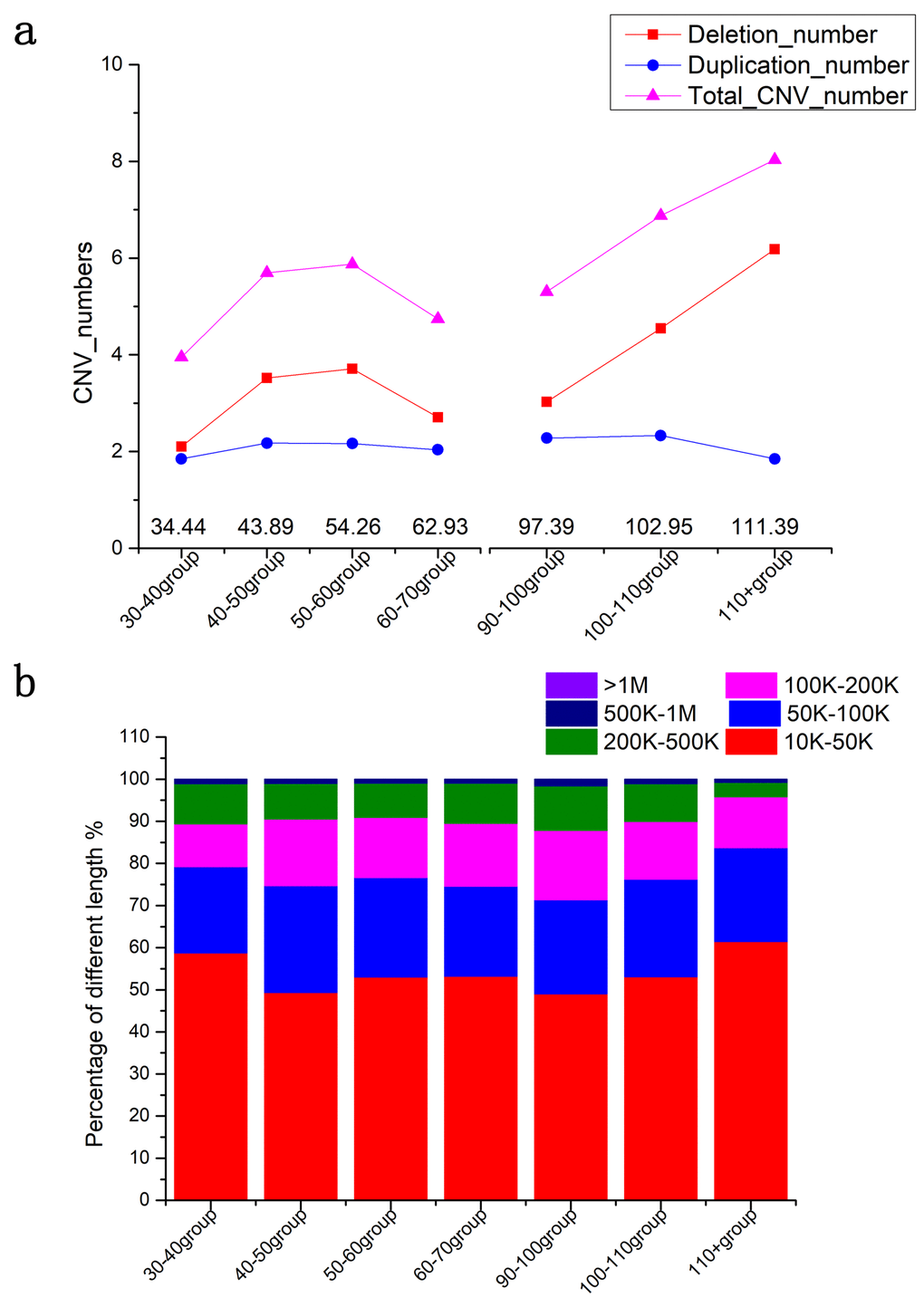
Figure 1. CNV burden in different age groups. (a) The numbers of CNVs in different age groups. (b) The added lengths of CNVs in different age groups. Triangles represent the numbers of the CNVs. The areas in each histogram represent the percentage of different lengths. * p<0.05 by t-test.
Identification of CNVRs associated with longevity
We identified the CNVRs associated with longevity, according to a previously published method [17]. These regions included 62 deletions and 5 duplications identified from the Northern cohorts, 21 deletions and 6 duplications in the Southern cohorts, and 99 deletions and 27 duplications in the combined samples (Figure 2). Among them, we identified eleven CNVs (2 duplications and 9 deletions) with case frequency > 1%, p value < 3.97×10-4 and length > 10k in long-lived individuals from the north as well as the north and south combined samples (Table 2, Supplementary Table 1). These CNVRs have been previously deposited in the database of genomic variants (DGV, http://dgv.tcag.ca/dgv/app/home) through other studies unrelated to longevity (Supplementary Figure 2). In order to confirm the accuracy of genotyping and CNV detecting methods, top six CNVRs (with p-value less than 10-5) were chose to do the quantitative PCR. (Supplementary Figure 3) and all these six CNVRs were validated accurate.
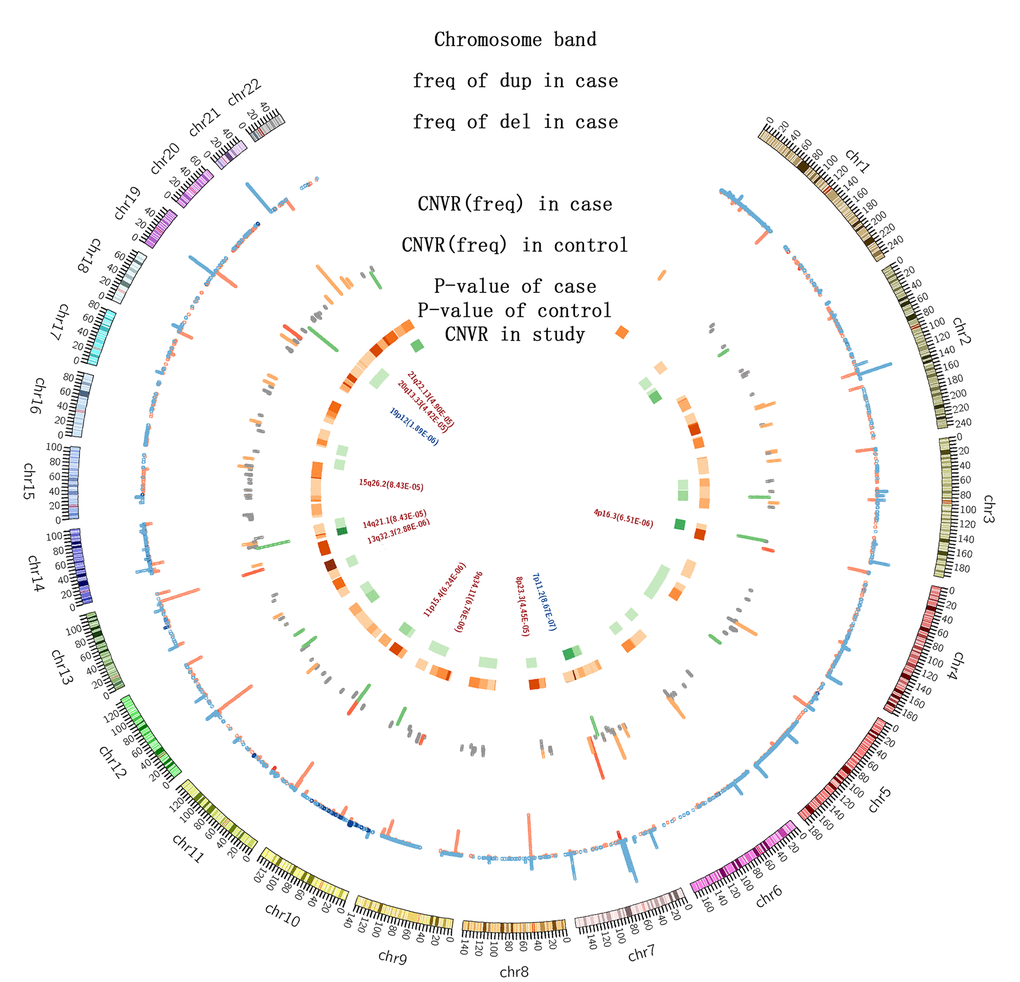
Figure 2. The distribution of CNVs among 4007 individuals. The first circle indicates the positions of chromosomal bands; the second is a histogram representing the frequencies of CNVs in long-lived individuals (red: deletions; blue: duplications; height: CNV frequencies); the third is also a histogram showing the CNV frequencies in the long-lived (orange) and middle-aged (orange) individuals. Frequency: orange and green, >1%; grey, <1%; red, the validated CNVRs by qPCR. The heatmap shows the p-values of CNVRs: orange for the long-lived, green for the middle-aged, and the color gradience towards dark indicates decreasing p-values. The text presents the names of the CNVRs identified in this study.
Table 2. The longevity CNVRs and their encoded genes.
| CNVR(hg19) | Cytoband | Type | p value | Odds Ratio | Frequency in long-lived% | Frequency in middle-aged% | Gene encoded | CNVR Length(kb) |
| chr7:57787663-57839005 | 7p11.2 | Dup | 8.67×10-7 | 1.59 | 15.90 | 5.74 | ZNF716 | 51.34 |
| chr19:22145147-22231026 | 19p12 | Dup | 1.89×10-6 | 4.11 | 2.36 | 0.63 | ZNF208,ZNF257 | 85.88 |
| chr13:100620882-100630973 | 13q32.3 | Del | 2.88×10-6 | 3.88 | 2.41 | 0.63 | ZIC5,ZIC2 | 10.09 |
| chr11:2939050-2949861 | 11p15.4 | Del | 6.24×10-6 | 4.81 | 1.85 | 0.39 | PHLDA2,SLC22A18 | 10.81 |
| chr4:1610872-1621223 | 4p16.3 | Del | 6.51×10-6 | 12.21 | 1.18 | 0.10 | FAM53A | 10.35 |
| chr9:130479233-130504070 chr20:60900481-60927421 | 9q34.11 20q13.33 | Del Del | 6.76×10-6 4.42×10-5 | 21.3 10.65 | 1.03 1.08 | 0.05 0.10 | PTRH1,SH2D3C,TOR2A,TTC16 LAMA5 | 24.84 26.93 |
| chr8:1789006-1799964 | 8p23.3 | Del | 4.45×10-5 | 10.65 | 1.03 | 0.15 | ARHGEF10 | 10.96 |
| chr21:38442208-38453183 | 21q22.13 | Del | 4.90×10-5 | 5.16 | 1.54 | 0.29 | PIGP,TTC3 | 10.98 |
| chr14:38058065-38081005 | 14q21.1 | Del | 8.43×10-5 | 4.57 | 1.54 | 0.34 | TTC6,FOXA1 | 22.94 |
| chr15:96898166-96909819 | 15q26.2 | Del | 8.43×10-5 | 4.57 | 1.54 | 0.34 | NR2F2 | 11.65 |
Functional analysis of eleven CNVRs
To determine if our identified CNVRs can affect biological function, we analyzed these loci for encoded genes. We found several genes within or near 11 CNVRs including Zinc Finger Protein 716 (ZNF716), Zinc Finger Protein 208 (ZNF208), Zinc Finger Protein 257 (ZNF257), Zic Family Member 5 (ZIC5), Zic Family Member 2 (ZIC2), Pleckstrin Homology Like Domain Family A Member 2 (PHLDA2), Solute Carrier Family 22 Member 18 (SLC22A18), Family With Sequence Similarity 53 Member A (FAM53A), Peptidyl-TRNA Hydrolase 1 Homolog (PTRH1), SH2 Domain Containing 3C (SH2D3C), Torsin Family 2 Member A (TOR2A), Tetratricopeptide Repeat Domain 16 (TTC16), Laminin Subunit Alpha 5 (LAMA5), Rho Guanine Nucleotide Exchange Factor 10 (ARHGEF10), Phosphatidylinositol Glycan Anchor Biosynthesis Class P (PIGP), Tetratricopeptide Repeat Domain 3 (TTC3), Tetratricopeptide Repeat Domain 6 (TTC6), Forkhead Box A1 (FOXA1) and Nuclear Receptor Subfamily 2 Group F Member 2 (NR2F2) (Table 2).
Some of these genes have previously been linked to longevity. For example, it has been shown that the rs4925386 in LAMA5 gene in 20q13.33 belongs to laminin alpha family. The age-stratified analyses showed that the rs4925386-T allele was positively associated with longevity (p = 0.001) [18]. The CNVR in 9q34.1 encodes genes SH2D3C and TOR2A. SH2D3C is a signaling adapter protein required for T cell activation, and a decrease in SH2D3C might cause B cell dysfunction and impaired immune function, which could affect lifespan [19]. In addition, the CNVR in 14q21.1 encodes the gene, FOXA1; upregulation of FOXA1 has been shown to decrease diet-restriction-induced longevity in C. elegans [20]. Additionally, NR2F2, a gene about 14kb away from 15q26.2 (p = 8.43×10-5, OR = 4.57) showed higher expression in older samples and was related to vascular development during oxidative stress-induced cellular senescence; vascular-related disease could be used as a marker for aging people [21].
To further understand whether these CNVRs affect specific pathways regulating aging process, The Database for Annotation, Visualization and Integrated Discovery (DAVID), Gene Ontology (GO), and Functional Enrichment analysis tool (FunRich) [22–24] were used for the enriched pathway analyses (Table 3). Six pathways were found to be enriched using FunRich including FOXA1 and FOXA transcription factor networks, which were most directly related to the regulation of longevity.
Table 3. Longevity genes and pathways enriched in the genomes.
| Database Category | Term | Genes | p-value |
| GOTERM_BP_FAT | epithelium development | ZIC2,ZIC5,FOXA1,LAMA5,NR2F2 | 1.2×10-4 |
| GOTERM_BP_FAT | tube morphogenesis | ZIC2,ZIC5,FOXA1,LAMA5 | 4.1×10-4 |
| GOTERM_BP_FAT | tube development | ZIC2,ZIC5,FOXA1,LAMA5 | 2.0×10-3 |
| GOTERM_BP_FAT | morphogenesis of embryonic epithelium | ZIC2,ZIC5,LAMA5 | 2.1×10-3 |
| GOTERM_BP_FAT | epithelial tube morphogenesis | ZIC2,ZIC5,LAMA5 | 2.8×10-3 |
| Biological pathway | FOXA1 transcription factor network | FOXA1,NR2F2 | 1.30×10-3 |
| Biological pathway | Mesenchymal-to-epithelial transition | PHLDA2,SLC22A18,FOXA1 | 2.16×10-3 |
| Biological pathway | FOXA transcription factor networks | FOXA1,NR2F2 | 4.36×10-3 |
| Biological pathway | Alpha6 beta4 integrin-ligand interactions | LAMA5 | 1.39×10-2 |
| Biological pathway | Synthesis of glycosylphosphatidylinositol (GPI) | SLC22A18 | 2.14×10-2 |
| Biological pathway | Post-translational modification: synthesis of GPI-anchored proteins | PIGP | 3.26×10-2 |
To determine if the difference in CNVR could be found between the nonagenarians (90-99) and centenarians (>100), we conducted the CNVR analyses in these two groups separately We detected four new CNVRs that were unique for the nonagenarians, and six new CNVRs only in the centenarians (Supplementary Table 2).
The variants in the CNVRs and eQTL
To determine if SNPs could alter gene expression in CNVRs, we investigated our 11 CNVRs using the Haploreg database [25]. Four of them were found to contain multiple SNPs, defining ≥10 eQTLs in tissue database that might affect gene function (Supplementary Table 4). These CNVRs are the deletions in 21q22.13, 20q13.33, and 11p15.4, and a duplication in 19p12. The eQTLs in the 21q22.13 deletion region could alter the expressions of the PIGP and TTC3 genes primarily in brain tissue. According to the annotation database Genecards (http://www.genecards.org/), PIGP resides in the protein complex that catalyzes the transfer of N-acetylglucosamine from UDP-GlcNAc to phosphatidylinositol, the first step of the glycosylphosphatidylinositol (GPI) biosynthesis. TTC3 is an ubiquitin-protein ligase that mediates the ubiquitination and subsequent degradation of phosphorylated AKT proteins. Several SNPs in the 20q13.33 deletion might affect the LAMA5 and CABLES2 gene expression. LAMA5 has been shown previously to affect cellular adhesion, migration, and organization. Two SNPs, rs1661052 and rs450244, in the 11p15.4 deletion region, showed strong cis-eQTL characteristics and might affect the SLC22A18 expression and its function in transporting organic cations. Finally, the SNPs located in the 19p12 duplication region can affect the expression of the ZNF gene family member such as ZNF208 and ZNF 257.
Discussion
In a genome-wide association study, we investigated the genomes obtained from 1950 long-lived and 2057 middle-aged Han Chinese people and identified 11 CNVRs that were associated with longevity. Four of them had partially overlapping regions with the CNVRs uncovered from the long-lived Danish or U.S population, while the rest seven were first reported in this study. Our statistical analyses indicated that the four overlapped CNVRs in the 7p11.2, 20q13.33, 19p12, and 8p23.3 bands were the strongest candidates with p values, 8.68×10-7, 4.42×10-5, 1.89×10-6, and 4.45×10-5, respectively.
It has been well-accepted that an increasing number or total length of deletions or duplications indicate genomic instability. Forsberg et al. has shown that genome instability increases with longevity [27]. In this study, we observed a significant increase in CNV numbers between long-lived and middle aged individuals (Figure 1). The added length of total CNVs also reflected this increase. Nygaard et al. has shown that mortality had a significant increase per 10 kb of CNV length increase in long-lived individuals [6]. We hypothesize that the long-lived CNVs are not hazard, for example, the deletions of carcinogenesis-related regions may help people improve lifespan.
In this study, we identified 11 CNVRs associated with long-lived Han Chinese including 2 duplications and 9 deletions. These CNVRs might affect the expressions of 19 known genes (Table 2), some of these genes were known to regulate cellular aging process.
For example, several studies have shown that the variant rs8105767 (ZNF208), which locates 20kb away from our 19p12 CNV region, led to several diseases by shortening the length of telomeres. A population study [28] demonstrated that the mutation of this gene could cause neuroblastoma. A genome-wide meta-analysis identified seven loci affecting telomere, and one of which was the rs8105767. This variant was also shown to be involved in shortened telomeres in leucocytes and increased the risk of coronary artery disease in the European descent population [29]. Thus, these results indicated that this CNV may affect telomere, therefore, the human longevity.
ARHGEF10 in 8p23.3 has been shown to play a key role in the RhoA signaling; an animal research demonstrated that the inactivation of ARHGEF10 could inhibit the platelet aggregation and protect mice from thrombus formation [30]. A genome-wide association study indicated that the variant rs7862362 A>T significantly decreased the cutaneous melanoma-specific survival [31]. Interestingly, one research showed that the deletion of 8p23.3 could induce intellectual disability and delay developmental processes [32], specifically in the Chinese population. These results suggest the deletion of ARHGEF10 may promote healthy aging by improving vascular function and suppress cancer occurrence.
The LAMA5 gene locates in 20q13.33. An Italian GWAS study has associated its rs4925386 T allele with longevity (p = 0.001) and shorter stature (p = 0.01) [18]. LAMA5 was reported to mediate cellular adhesion, migration, and organization [18]. Overexpression of LAMA5 can induce colorectal cancer through KRAS, while the genetic inactivation of LAMA5 impairs the adhesion of KRAS-mutant colorectal cancer cells [33]. It was showed in a Chinese population that the alterations of the duplication DNVR in 20q13.33 could increase the risk of ovarian endometriosis and glioma [34,35]. Thus, the deletion of this region may reduce the risk of the carcinogenesis and extend lifespan for these long-lived individuals.
Among all the 19 CNVR related genes, FOXA1 might have the strongest effects on longevity. The FOXA1 transcription factor network plays a key role in DNA repairing, therefore, maintaining the integrity of the genome. It has been shown that inhibiting the expression of FOXA1 can extend the lifespan in C.elegans [20]. Some studies suggested that FOXA1 was potentially an oncogene because overexpression or increased copy numbers of FOXA1 were found in a variety of cancers [36,37]. In our samples, 1.53% of cases had deletions in FOXA1, which may potentially reduce the risk of cancer, leading to a healthier lifespan. It has been hypothesized that some people live longer were due to the reduced copy numbers in certain oncogene regions.
Besides their direct effects on gene structures, SNPs in CNVs can also change gene expression. We showed, in three of our identified CNVRs, that eQTLs resulting from multiple SNPs could affect the expression of nearby genes. For example, the SNPs in the 21q22.13 CNVR correlated significantly with the expression of the PIGP and TTC3 genes in brain tissue. PIGP regulates glycosylphosphatidylinositol-anchor biosynthesis in animals and has also been related to Down syndrome [38]. Variants of this gene could lead to age-dependent Alzheimer’s disease [39]. Another example is that the 20q13.33 deletion region also contains multiple SNPs that could affect the expression of LAMA5 and CABLES2 genes. A study involving a SNP from the 11p15.4 deletion region led to the discovery that SLC22A18 in this region may function as a tumor suppressor [40]. Our analyses showed that most of our identified CNVRs contain multiple SNPs, thus affecting gene expression.
In conclusion, we analyzed 4007 samples in a genome-wide association study and identified 11 CNVRs including 9 deletions and 2 duplications that were strongly associated with long-lived Han Chinese. Four of them were also found in the similar chromosomal regions in a Danish or a U.S. long-lived populations, suggesting these might be commonly shared in long-lived human. The other seven were first identified in this study. We also found that the number of deletions increased significantly with longevity. Based on our gene and pathway analysis results, we conclude that some of our identified CNVRs encode cancer-causing or aging-related genes, and the deletions of these regions may extend healthy lifespan for long-lived individuals. Future research on these CNVRs will shed light on the regulatory mechanisms controlling human longevity.
Significance
In this study, we analyzed copy number variations in a longevity cohort of Han Chinese. A total of 4,007 samples, obtained from 1950 long-lived and 2057 middle-aged healthy individuals, were investigated. By comparing these two age-groups, we identified 126 CNV regions (CNVRs) that significantly increased in the long-lived populations. Eleven among these 126 CNVRs were enriched significantly among long-lived individuals with multiple test p-values less than 3.97×10-4 and population frequency larger than 1%. Strikingly, four among these eleven CNVRs were found to partially overlap with the CNVRs identified in American ‘Wellderly’ and Danish longevity cohort studies. Annotation of these CNVRs revealed nineteen genes in these regions; some of them have been previously shown to function in the pathways regulating aging process. Our findings suggested that the genomic structure changes such as CNVs might contribute to the complex traits of longevity by altering gene expression.
Methods
The population studied
Our study included 1950 long-lived cases and 2057 middle-age controls from the Chinese Longitudinal Healthy Longevity Survey (CLHLS), which represented 22 provinces of China, following the method described previously [5,41; http://web5.pku.edu.cn/ageing/html/datadownload.html).
These people were divided, based on their geographical locations, into two population cohorts, representing the discovery and replicate samples. The former was composed of 1000 long-lived cases and 1215 middle-age controls from 11 provinces in the north of China, while the latter consisted of 950 long-lived and 842 middle-aged individuals from 11 provinces in the south of China.
The whole genome genotyping
Genotyping was carried out following the standard protocols of the Illumina HumanOmniZhongHua-8 BeadChips (Illumina Inc.). A beadchip contains 900,015 SNPs markers with a mean and median spacing of 3.3 kb and1.7kb, respectively. Quality control (QC) was conducted using plink 1.06 and call rate > 0.95; 339 samples from the replicate was excluded. Principal component analysis (PCA) was also subsequently performed using plink [42].
CNV detection
CNVs were detected using the software PennCNV 1.04 [43]. In the software, a hidden Markov model (HMM) determined the CNVs with the total signal intensity Log R ratio (LRR) and B allele frequency (BAF) for each SNP marker. The LRR and BAF values can be downloaded from the Illumina Genomestudio2011. The population frequency of the B allele (PFB) was calculated using the BAF values collected from the discovery or replicate study. The GC file was obtained from UCSC genome bioinformatics (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/gc5Base.txt.gz). The final CNVs were obtained after excluding those that met these conditions: LRR SD (standard deviation) > 0.3, GC wave factor > 0.05, CNV number >100, CNV length < 10kb, consecutive SNPs < 10, and confidence <10.
CNVs encoded gene annotation and pathway analysis
The centenarian specific CNVs were annotated using human genome reference version 19 (hg19) within the range of 100kb. Genome ontology (GO) enrichment was performed using Go Ontology (http://geneontology.org/) [23]. Functional enrichment and interaction network analysis of genes and proteins were performed using The Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/home.jsp) [24] and Functional Enrichment analysis tool (FunRich) 3.1.3 [25].
Determination of the CNVRs associated with longevity
We compared the long-lived to the middle-aged individuals to identify CNVs associated with longevity using the CNVR searching software, ParseCNV20 [17]. The CNVs were called based on p values calculated by Fisher’s exact test. The SNPs in the CNVRs were also compared among the old and middle-aged groups with Fisher’s test.
The identified CNVRs were then annotated based on the hg19 reference genome. Those CNVs overlapping with telomeres were discarded.
We then compared our identified CNVs with a Danish study (personal communications with Dr. Nygaard) and a U.S. study (personal communications with Dr. Ali Torkamani).
The validations of CNVRs
Quantitative polymerase chain reaction (qPCR) was used to validate CNVRs. Considering the long lived DNA are valuable, limited samples could be used for the validation. In addition, we still need samples for proceeding the next stage of GWAS experiments, so the top six CNVRs (with p-value less than 10-5) were chose to do the quantitative PCR and confirm the accuracy of genotyping and CNV detecting methods. Primers sets were designed for each of selected CNVRs (Supplementary Table 3). The qPCR conditions followed the ABI Step One (Life technologies Inc.) in 96-well plates: 95°C for 10 min to denature DNA samples, followed by 40 cycles (95°C for 15 s and then 58°C for 1 min). Each sample was repeated three times. The relative copy number (RCN) was calculated using CT values (2–∆∆Ct). RCN was then adjusted by setting the no-copy-number-change as “1”. Larger than “1.2” was regarded as duplication, while smaller than “0.8” indicated deletion.
Expression Quantitative Trait Loci (eQTL) analysis
Haploreg V4.1, a web-based tool for annotating noncoding genomic regions for variations was used to examine the effects of SNPs on gene expression and determine eQTLs (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php). These eQTLs from Haploreg were also identified from the GTEx Project (https://www.gtexportal.org/home/) and Geuvadis Data Browser (https://www.ebi.ac.uk/Tools/geuvadis-das/).
Statistical analysis of CNV burden
To determine if the CNV numbers or total added lengths of CNVs were statistically different between the long-lived and middle-aged cohorts, a Bonferroni correction independent students’ t test was applied. The Spearman correlation was calculated between CNV numbers and CNV lengths. Since genetic association studies are often confronted by the problem of complex population structure that can result in false positive results, in this study, we used an adjusted p-value of 3.97×10-4 (0.05/126), based on multiple testing, as a cutoff to identify statistically significant in long-lived specific CNVRs. The t test analyses were performed using SPSS Statistics 19.0 software (SPSS, Inc.) and p<0.05 was regarded as significant. Data correlation analyses were performed using a R software from the R Project for Statistical Computing (3.1.4). Figure 1 and supplement figures were constructed with Origin 8.5, a graphing software (Origin Lab Inc.), and Figure 2 was by Circos 0.69, a software package for visualizing genomic data and information in circular layout (http://circos.ca/).
Author Contributions
XJ, XL, YH, TN, WT, JG, LB, WG, XT, HY, JW, XX, MWL, XG suggest this study; CN and YZ designed experiments; LL and JS carried out experiments; XZ, XL, AZ, HC, RY, ZC and MN analyzed experimental results. XZ, XL, AZ, QH, WL, QAL, JM, YZ, LB and CN wrote the manuscript.
Acknowledgements
We thank Dr. Ali Torkamani from the Scripps Research Institute for helping our data validation by providing the CLHLS GWAS and ‘Wellderly’ sequencing data. We also would like to thank Drs. Qihua Tan and Kaare Christensen from the University of Southern Denmark for providing us the CLHLS GWAS and ‘Danish’ array data for comparison. This study followed the guidelines in the Declaration of Helsinki and was approved by the Ethical Review Committees of BGI (IRB13062), Peking University, and Duke University.
Conflicts of Interest
The authors declare no competing financial interests.
Funding
For this study, the Chinese Longitudinal Healthy Longevity Study (CLHLS) was supported by National Natural Sciences Foundation of China (71490732, to YZ); U.S. National Institute of Aging ( 3P01AG031719-07S1, to YZ), and United Nations Fund for Population Activities (to YZ).
References
- 1. Herskind AM, McGue M, Holm NV, Sørensen TI, Harvald B, Vaupel JW. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870-1900. Hum Genet. 1996; 97:319–23. https://doi.org/10.1007/BF02185763 [PubMed]
- 2. Murabito JM, Yuan R, Lunetta KL. The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. J Gerontol A Biol Sci Med Sci. 2012; 67:470–79. https://doi.org/10.1093/gerona/gls089 [PubMed]
- 3. Deelen J, Beekman M, Uh HW, Broer L, Ayers KL, Tan Q, Kamatani Y, Bennet AM, Tamm R, Trompet S, Guðbjartsson DF, Flachsbart F, Rose G, et al. Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum Mol Genet. 2014; 23:4420–32. https://doi.org/10.1093/hmg/ddu139 [PubMed]
- 4. Shadyab AH, LaCroix AZ. Genetic factors associated with longevity: a review of recent findings. Ageing Res Rev. 2015; 19:1–7. https://doi.org/10.1016/j.arr.2014.10.005 [PubMed]
- 5. Zeng Y, Nie C, Min J, Liu X, Li M, Chen H, Xu H, Wang M, Ni T, Li Y, Yan H, Zhang JP, Song C, et al. Novel loci and pathways significantly associated with longevity. Sci Rep. 2016; 6:21243. https://doi.org/10.1038/srep21243 [PubMed]
- 6. Nygaard M, Debrabant B, Tan Q, Deelen J, Andersen-Ranberg K, de Craen AJ, Beekman M, Jeune B, Slagboom PE, Christensen K, Christiansen L. Copy number variation associates with mortality in long-lived individuals: a genome-wide assessment. Aging Cell. 2016; 15:49–55. https://doi.org/10.1111/acel.12407 [PubMed]
- 7. Kuningas M, Estrada K, Hsu YH, Nandakumar K, Uitterlinden AG, Lunetta KL, van Duijn CM, Karasik D, Hofman A, Murabito J, Rivadeneira F, Kiel DP, Tiemeier H. Large common deletions associate with mortality at old age. Hum Mol Genet. 2011; 20:4290–96. https://doi.org/10.1093/hmg/ddr340 [PubMed]
- 8. Glessner JT, Smith AV, Panossian S, Kim CE, Takahashi N, Thomas KA, Wang F, Seidler K, Harris TB, Launer LJ, Keating B, Connolly J, Sleiman PM, et al. Copy number variations in alternative splicing gene networks impact lifespan. PLoS One. 2013; 8:e53846. https://doi.org/10.1371/journal.pone.0053846 [PubMed]
- 9. Iakoubov L, Mossakowska M, Szwed M, Duan Z, Sesti F, Puzianowska-Kuznicka M. A common copy number variation (CNV) polymorphism in the CNTNAP4 gene: association with aging in females. PLoS One. 2013; 8:e79790. https://doi.org/10.1371/journal.pone.0079790 [PubMed]
- 10. Iakoubov L, Mossakowska M, Szwed M, Puzianowska-Kuznicka M. A common copy number variation polymorphism in the CNTNAP2 gene: sexual dimorphism in association with healthy aging and disease. Gerontology. 2015; 61:24–31. https://doi.org/10.1159/000363320 [PubMed]
- 11. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006; 7:85–97. https://doi.org/10.1038/nrg1767 [PubMed]
- 12. Duyzend MH, Nuttle X, Coe BP, Baker C, Nickerson DA, Bernier R, Eichler EE. Maternal Modifiers and Parent-of-Origin Bias of the Autism-Associated 16p11.2 CNV. Am J Hum Genet. 2016; 98:45–57. https://doi.org/10.1016/j.ajhg.2015.11.017 [PubMed]
- 13. Zanda M, Onengut-Gumuscu S, Walker N, Shtir C, Gallo D, Wallace C, Smyth D, Todd JA, Hurles ME, Plagnol V, Rich SS. A genome-wide assessment of the role of untagged copy number variants in type 1 diabetes. PLoS Genet. 2014; 10:e1004367. https://doi.org/10.1371/journal.pgen.1004367 [PubMed]
- 14. Pelttari LM, Shimelis H, Toiminen H, Kvist A, Törngren T, Borg Å, Blomqvist C, Bützow R, Couch F, Aittomäki K, Nevanlinna H. Gene-panel testing of breast and ovarian cancer patients identifies a recurrent RAD51C duplication. Clin Genet. 2018; 93:595–602. https://doi.org/10.1111/cge.13123 [PubMed]
- 15. Habibi M, Mirfakhraie R, Khani M, Rakhshan A, Azargashb E, Pouresmaeili F. Genetic variations in UGT2B28, UGT2B17, UGT2B15 genes and the risk of prostate cancer: A case-control study. Gene. 2017; 634:47–52. https://doi.org/10.1016/j.gene.2017.08.038 [PubMed]
- 16. Zhou C, Zhang W, Chen W, Yin Y, Atyah M, Liu S, Guo L, Shi Y, Ye Q, Dong Q, Ren N. Integrated Analysis of Copy Number Variations and Gene Expression Profiling in Hepatocellular carcinoma. Sci Rep. 2017; 7:10570. https://doi.org/10.1038/s41598-017-11029-y [PubMed]
- 17. Glessner JT, Li J, Hakonarson H. ParseCNV integrative copy number variation association software with quality tracking. Nucleic Acids Res. 2013; 41:e64. https://doi.org/10.1093/nar/gks1346 [PubMed]
- 18. De Luca M, Crocco P, De Rango F, Passarino G, Rose G. Association of the Laminin, Alpha 5 (LAMA5) rs4925386 with height and longevity in an elderly population from Southern Italy. Mech Ageing Dev. 2016; 155:55–59. https://doi.org/10.1016/j.mad.2016.03.003 [PubMed]
- 19. Al-Shami A, Wilkins C, Crisostomo J, Seshasayee D, Martin F, Xu N, Suwanichkul A, Anderson SJ, Oravecz T. The adaptor protein Sh2d3c is critical for marginal zone B cell development and function. J Immunol. 2010; 185:327–34. https://doi.org/10.4049/jimmunol.1000096 [PubMed]
- 20. Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007; 447:550–55. https://doi.org/10.1038/nature05837 [PubMed]
- 21. Molinari E, Bar H, Pyle AM, Patrizio P. Transcriptome analysis of human cumulus cells reveals hypoxia as the main determinant of follicular senescence. Mol Hum Reprod. 2016; 22:866–76. https://doi.org/10.1093/molehr/gaw038 [PubMed]
- 22. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017; 45:D183–89. https://doi.org/10.1093/nar/gkw1138 [PubMed]
- 23. Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007 (suppl_2); 35:W169-75. https://doi.org/10.1093/nar/gkm415 [PubMed]
- 24. Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, Bacic A, Hill AF, Stroud DA, et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015; 15:2597–601. https://doi.org/10.1002/pmic.201400515 [PubMed]
- 25. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012; 40:D930–34. https://doi.org/10.1093/nar/gkr917 [PubMed]
- 26. Erikson GA, Bodian DL, Rueda M, Molparia B, Scott ER, Scott-Van Zeeland AA, Topol SE, Wineinger NE, Niederhuber JE, Topol EJ, Torkamani A. Whole-Genome Sequencing of a Healthy Aging Cohort. Cell. 2016; 165:1002–11. https://doi.org/10.1016/j.cell.2016.03.022 [PubMed]
- 27. Forsberg LA, Rasi C, Razzaghian HR, Pakalapati G, Waite L, Thilbeault KS, Ronowicz A, Wineinger NE, Tiwari HK, Boomsma D, Westerman MP, Harris JR, Lyle R, et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet. 2012; 90:217–28. https://doi.org/10.1016/j.ajhg.2011.12.009 [PubMed]
- 28. Walsh KM, Whitehead TP, de Smith AJ, Smirnov IV, Park M, Endicott AA, Francis SS, Codd V, Samani NJ, Metayer C, Wiemels JL, Wiemels JL, and ENGAGE Consortium Telomere Group. Common genetic variants associated with telomere length confer risk for neuroblastoma and other childhood cancers. Carcinogenesis. 2016; 37:576–82. https://doi.org/10.1093/carcin/bgw037 [PubMed]
- 29. Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, Broer L, Nyholt DR, Mateo Leach I, et al, and CARDIoGRAM consortium. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013; 45:422–27, e1–2. https://doi.org/10.1038/ng.2528 [PubMed]
- 30. Lu DH, Hsu CC, Huang SW, Tu HJ, Huang TF, Liou HC, Liao HM, Chen CH, Fu WM, Gau SS. ARHGEF10 knockout inhibits platelet aggregation and protects mice from thrombus formation. J Thromb Haemost. 2017; 15:2053–64. https://doi.org/10.1111/jth.13799 [PubMed]
- 31. Liu S, Wang Y, Xue W, Liu H, Xu Y, Shi Q, Wu W, Zhu D, Amos CI, Fang S, Lee JE, Hyslop T, Li Y, et al. Genetic variants in the genes encoding rho GTPases and related regulators predict cutaneous melanoma-specific survival. Int J Cancer. 2017; 141:721–30. https://doi.org/10.1002/ijc.30785 [PubMed]
- 32. Wu Y, Ji T, Wang J, Xiao J, Wang H, Li J, Gao Z, Yang Y, Cai B, Wang L, Zhou Z, Tian L, Wang X, et al. Submicroscopic subtelomeric aberrations in Chinese patients with unexplained developmental delay/mental retardation. BMC Med Genet. 2010; 11:72. https://doi.org/10.1186/1471-2350-11-72 [PubMed]
- 33. Bartolini A, Cardaci S, Lamba S, Oddo D, Marchiò C, Cassoni P, Amoreo CA, Corti G, Testori A, Bussolino F, Pasqualini R, Arap W, Corà D, et al. BCAM and LAMA5 Mediate the Recognition between Tumor Cells and the Endothelium in the Metastatic Spreading of KRAS-Mutant Colorectal Cancer. Clin Cancer Res. 2016; 22:4923–33. https://doi.org/10.1158/1078-0432.CCR-15-2664 [PubMed]
- 34. Song X, Zhou K, Zhao Y, Huai C, Zhao Y, Yu H, Chen Y, Chen G, Chen H, Fan W, Mao Y, Lu D. Fine mapping analysis of a region of 20q13.33 identified five independent susceptibility loci for glioma in a Chinese Han population. Carcinogenesis. 2012; 33:1065–71. https://doi.org/10.1093/carcin/bgs117 [PubMed]
- 35. Yang W, Zhang Y, Fu F, Li R. High-resolution array-comparative genomic hybridization profiling reveals 20q13.33 alterations associated with ovarian endometriosis. Gynecol Endocrinol. 2013; 29:603–07. https://doi.org/10.3109/09513590.2013.788632 [PubMed]
- 36. Lin L, Miller CT, Contreras JI, Prescott MS, Dagenais SL, Wu R, Yee J, Orringer MB, Misek DE, Hanash SM, Glover TW, Beer DG. The hepatocyte nuclear factor 3 alpha gene, HNF3alpha (FOXA1), on chromosome band 14q13 is amplified and overexpressed in esophageal and lung adenocarcinomas. Cancer Res. 2002; 62:5273–79. [PubMed]
- 37. Nucera C, Eeckhoute J, Finn S, Carroll JS, Ligon AH, Priolo C, Fadda G, Toner M, Sheils O, Attard M, Pontecorvi A, Nose V, Loda M, Brown M. FOXA1 is a potential oncogene in anaplastic thyroid carcinoma. Clin Cancer Res. 2009; 15:3680–89. https://doi.org/10.1158/1078-0432.CCR-08-3155 [PubMed]
- 38. Piłsyk S, Paszewski A. The Aspergillus nidulans pigP gene encodes a subunit of GPI-N-acetylglucosaminyltransferase which influences filamentation and protein secretion. Curr Genet. 2009; 55:301–09. https://doi.org/10.1007/s00294-009-0246-x [PubMed]
- 39. Kohli MA, Cukier HN, Hamilton-Nelson KL, Rolati S, Kunkle BW, Whitehead PL, Züchner SL, Farrer LA, Martin ER, Beecham GW, Haines JL, Vance JM, Cuccaro ML, et al. Segregation of a rare TTC3 variant in an extended family with late-onset Alzheimer disease. Neurol Genet. 2016; 2:e41. https://doi.org/10.1212/NXG.0000000000000041 [PubMed]
- 40. Jung Y, Jun Y, Lee HY, Kim S, Jung Y, Keum J, Lee YS, Cho YB, Lee S, Kim J. Characterization of SLC22A18 as a tumor suppressor and novel biomarker in colorectal cancer. Oncotarget. 2015; 6:25368–80. https://doi.org/10.18632/oncotarget.4681 [PubMed]
- 41. Yi Z. Introduction to the chinese longitudinal healthy longevity survey (CLHLS). Healthy longevity in China: Springer), 2008; pp. 23-38.
- 42. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81:559–75. https://doi.org/10.1086/519795 [PubMed]
- 43. Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007; 17:1665–74. https://doi.org/10.1101/gr.6861907 [PubMed]