



Introduction
Although ageing is readily observed at the level of the organism, our understanding of why and how this process occurs has remained speculative until normal human cells were successfully cultured outside the body, where they were found to have a finite capacity to proliferate.
Hayflick estimated that a population of human cells grown ex vivo can double approximately sixty times after which they adopt a permanent state of dormancy termed replicative senescence [1,2]. The cause of this natural limitation to proliferation was eventually found to lie in the “end-replication problem”, which if not addressed by the cell, would lead to telomere attrition at every round of DNA replication [3,4]. It was eventually demonstrated that this does indeed occur and when telomeres shorten to a critical length they trigger cells to adopt the senescent state [5,6]. The identification of telomerase, which replicates telomeres [7,8], and the fact that most adult somatic cells do not produce this enzyme, provided the last major piece of the puzzle that describes the ageing process from events beginning with molecules, proceeding to cells and culminating in the organism. Significantly, this chain of events can be prevented by ectopic expression of hTERT, the catalytic sub-unit of telomerase, which can preserve telomere length and avert senescence of some cells [9,10]. Impressively, these profound insights into the process of human ageing were acquired from careful study of ex vivo cell behaviour.
It was initially thought that the functional and physical deterioration that characterise organismal ageing are a result of insufficient replenishment of cells due to telomere-mediated restriction of cellular proliferation. Senescent cells, which accumulate increasingly in tissues in function of age, were assumed to be passive and merely a consequence of the above-described processes. This notion was short-lived when senescent cells were found to secrete molecules that are detrimental to cells and tissues; a cellular characteristic described as senescence-associated secretory phenotype (SASP) [11–13]. The role of senescent cells in actively causing age-related physical deterioration was elegantly revealed when reversal of ageing phenotype in organs and tissues was observed following the removal of senescent cells in mice [14]. As such, it would follow that if cells were prevented from becoming senescent in the first place, ageing could be avoided. Although there are external instigators such as stress and DNA damage that can also cause cells to become senescent [15], replicative senescence is particular in that it is an intrinsic feature that is part of cellular proliferation and occurs even in an ideal environment. As expression of hTERT has been repeatedly demonstrated to prevent replicative senescence of many different cell types, it is reasonable to consider ectopic expression or re-activation of endogenous hTERT expression as potential means to prevent replicative senescence, delay ageing and improve health [16].
The above proposition would be valid if senescent cells were indeed the only cause of ageing. Relatively recently, an apparently distinct form of ageing, called epigenetic ageing was described (reviewed in [17]). This discovery stems from observations that the methylation states of some specific cytosines that precede guanines (CpGs) in the human genome changed rather reliably and strictly with age [18–22]. This allowed supervised machine learning methods to be applied to DNA methylation data to generate DNA methylation-based age estimators (epigenetic clocks) of epigenetic age, which in the majority of the human population is similar with chronological age [23–27]. The difference between epigenetic age and chronological age, which reflects the rate of epigenetic aging, carries biological significance: increased epigenetic aging is associated with numerous age-related pathologies and conditions [17,28–41]. Conversely, healthy lifestyle and diet is associated with younger epigenetic age [17,42]. Furthermore, epigenetic age can be reversed or reset, as expression of Yamanaka factors in somatic adult cells reset their epigenetic ages to zero [26,43]. Hence, epigenetic age is not merely an alternative means of determining chronological age but is to some degree a measure of biological age or health; a proposition that is further supported by the impressive demonstration that acceleration of epigenetic ageing is associated with increased risk of all-cause mortality [34,39]. Collectively, the descriptions above highlight the fact that epigenetic ageing, in spite of the mathematical origins of its discovery, is not a mathematical contrivance but a genuine ageing process innate in cells.
Several DNAm-based biomarkers have been reported in the literature that differ in terms of their applicability (some were developed for specific tissues such as blood) and their biological interpretation. The pan-tissue epigenetic clock developed by Horvath [26] is applicable to almost all sources of DNA with the exception of sperm. The resulting age estimate by this clock is referred to as epigenetic age or more precisely DNAm age. Although the pan-tissue epigenetic clock is highly accurate and applicable to the vast majority of tissues in the body, it performs sub-optimally when estimating the age of fibroblasts. In response to this, we recently developed a new epigenetic age estimator, referred to as skin & blood clock that is more accurate in estimating age of different cell types including fibroblasts, keratinocytes, buccal cells, blood cells, saliva and endothelial cells [44]. Studies employing skin & blood clock and the pan-tissue epigenetic age clock revealed a startling consistency of epigenetic age across diverse tissues from the same individual, even though cellular proliferation rates and frequencies of these tissues are not the same [26,44]. This suggests that the ticking of the epigenetic clock is not a reflection of proliferation frequency, which is in stark contrast to telomere length, which enumerates cellular division. It would therefore appear that the process of epigenetic ageing is distinct from that which is driven by telomere-mediated senescence. To understand their relationship or interaction, if one indeed exists, we set out to test the impact of hTERT on epigenetic ageing. To this end we employed wild type hTERT that can prevent telomere attrition and its mutants that cannot [45], with some still able to nevertheless prolong cellular lifespan [46]. Expressing these hTERT constructs in primary cells from numerous donors, ages and cell types, we observe that while hTERT expression can indeed prevent cellular senescence, it does not prevent cells from undergoing epigenetic ageing and that extension of cellular lifespan is sufficient to support continued epigenetic ageing of the cell. These simple observations provide a very important piece to the puzzle of the ageing process because it reveals the distinctiveness of epigenetic ageing from replicative senescence-mediated ageing. They provide further empirical support to the epidemiological observation that hTERT variant that is associated with longer telomeres are also associated with greater epigenetic ageing [47].
Results
To test the effect of hTERT on epigenetic ageing, we first transduced primary neonatal foreskin fibroblasts with hTERT vectors and subjected them and the control isogenic cells, which harbour empty vectors, to continuous culture with passaging. The growth curve in Figure 1A shows that control fibroblasts from neonatal donors A and B (blue and red dots) senesced after about a hundred days in culture and having doubled approximately 50 times (Supplementary Figure 1 and 2A). Unsurprisingly, cells bearing hTERT expression vector bypassed replicative senescence. They proliferated unabated beyond 130 days and 75 cumulative population doubling. The last green and orange dots represent the time-point at which the experiments were terminated, and not the end of cellular viability. These cells have effectively become immortalised. Cellular DNA from a selection of cell passages were subjected to methylation analyses with Illumina EPIC array. The resulting data were processed using the pan-tissue clock and the new skin & blood clock. The results in Figure 1B (for Donor A) and Figure 1C (for Donor B) show control cells to age in culture and this was not perturbed by hTERT expression. Importantly, cells transduced with hTERT not only evaded replicative senescence, their epigenetic ages continued to steadily increase past the point of replicative senescence encountered by their respective isogenic control counterparts (last blue and red dots). While this behaviour is observed with results derived from both epigenetic ageing clocks, the pan-tissue clock clearly displays an off-set from the chronological age of the neonatal cells, which is zero years, as correctly indicated by the skin & blood clock. Incidentally, this systematic offset/error in accurately estimating the epigenetic age of young fibroblasts was one of the reasons for developing the skin & blood clock.
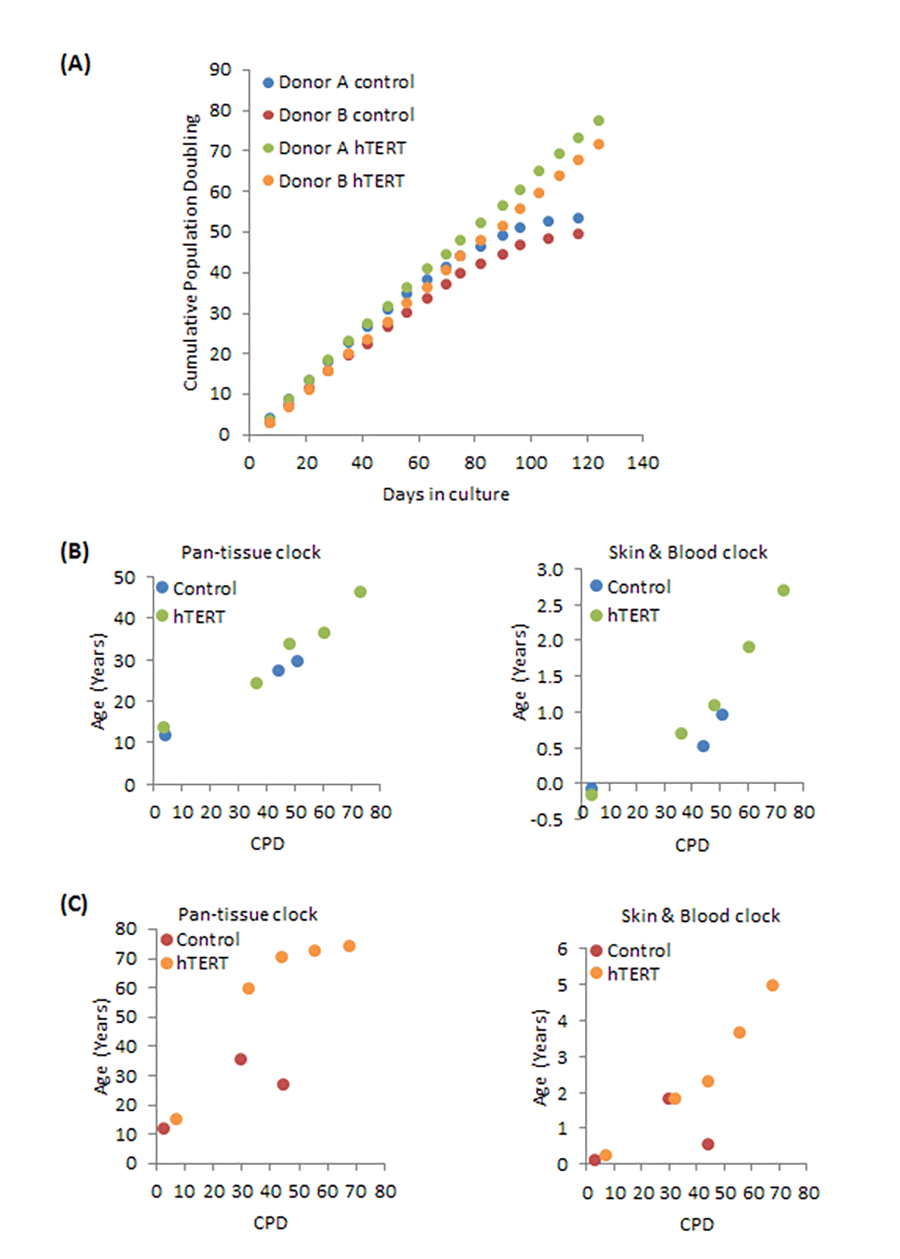
Figure 1. Effects of hTERT on growth and epigenetic ageing of human primary neonatal fibroblasts. (A) Growth dynamics of primary cells from two different donors (A and B) transduced with either empty vector (control) or hTERT expressing vector (hTERT). The ages of a selection of cell passages of donor A (B) and donor B (C) were imputed by the pan-tissue clock (left panel) or the skin & blood clock (right panel). The ages are plotted against cumulative population doubling (CPD) that corresponded to the passage of cells that were analysed.
To ascertain whether the effect of hTERT seen in neonatal foreskin fibroblasts was observable in cells from another tissue and age, we utilised human coronary artery endothelial cells (HCAEC) from adult donor (Donor C; 19 years old). The growth dynamics of these cells as shown in Figure 2A are similar in principle with those of the neonatal fibroblasts, with the difference being the earlier time-point at which the control cells senesce (Supplementary Figure 2B). This is consistent with them being adult cells and as such would have lower replicative potential. As with neonatal fibroblasts, the adult HCAEC expressing hTERT were also immortalised. A startling difference however, is apparent when the ages of these cells were estimated by the two clocks (Figure 2B). While once again the skin & blood clock showed hTERT-expressing cells, which bypassed replicative senescence, to continue ageing steadily, the epigenetic age estimates from the pan tissue clock were much higher and with no significant change in age (Figure 2B). We have observed similar pattern with HCAEC isolated from another donor (26 years old) [44].
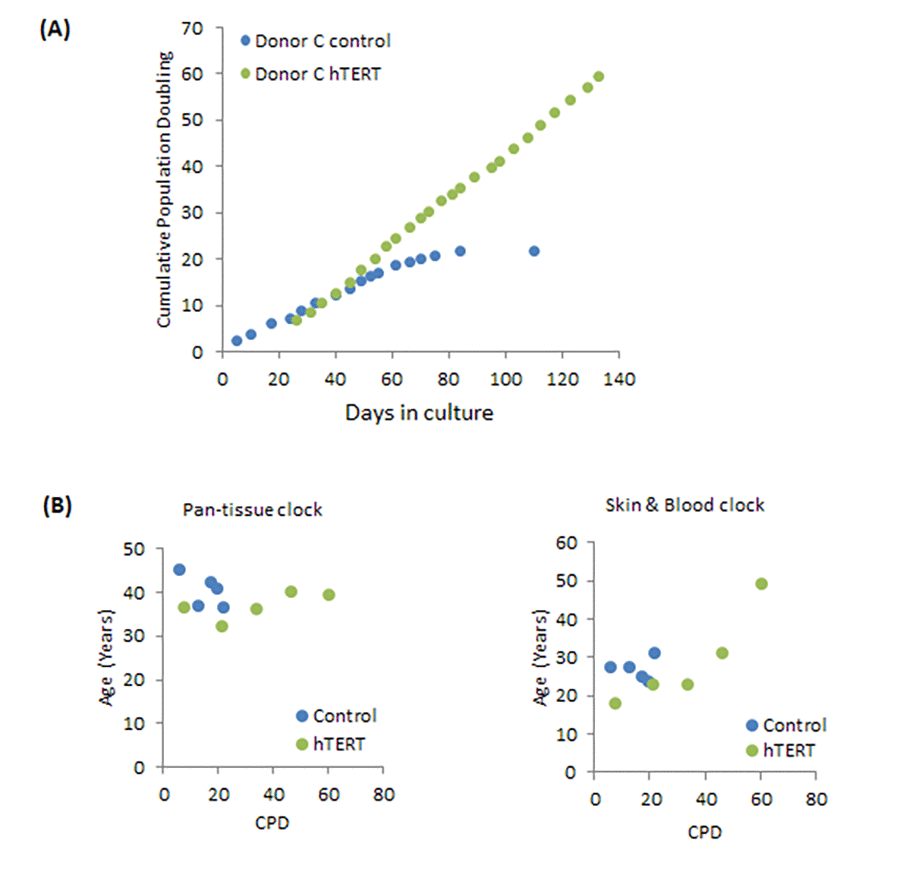
Figure 2. Effects of hTERT on growth and epigenetic ageing of adult primary human coronary artery endothelial cells. (A) Growth dynamics of primary cells from one donor (C) transduced with either empty vector (control) or hTERT expression vector (hTERT). (B) The ages of a selection of cell passages of donor C were imputed by the pan-tissue clock (left panel) or the skin & blood clock (right panel). The ages are plotted against cumulative population doubling (CPD) that corresponded to the passage of cells that were analysed.
To further investigate the relationship between hTERT and epigenetic ageing, we employed a previously validated and published panel of hTERT mutants which all possess catalytic activity but are compromised in one or several hTERT properties, namely, extension of replicative lifespan, telomere synthesis or immortalisation (Table 1 and Supplementary Figure 3) [48]. The growth characteristics of the neonatal foreskin fibroblasts transduced with these vectors confirmed that cells expressing wildtype hTERT bypassed replicative senescence (Figure 3) and aged steadily pass the point of replicative senescence encountered by the control cells (Figure 4A). Notably the hTERT IA mutant [46], which can significantly extend replicative lifespan (Figure 3) but can neither replicate telomeres nor immortalise cells, is also able to elicit steady epigenetic ageing pass the point of replicative senescence of the control cells (Figure 4B). This feature is particularly important because it shows that neither telomere synthesis nor immortalisation contribute to the steady rise in epigenetic ageing seen with hTERT-expressing cells. Instead extension of cellular lifespan appears to be the critical property associated with increased epigenetic ageing. Accordingly, the N-DAT116 mutant [46], which was reportedly also able to extend cellular lifespan of human mammary epithelial cells [46,48], but did so only very marginally with neonatal fibroblasts, did not cause a substantial rise in epigenetic ageing (Figure 4C). Likewise the N-DAT92 [46] hTERT mutant that does not increase lifespan also did not increase epigenetic ageing (Figure 4D). The patterns described above largely hold true between the two epigenetic age clocks. It is evident that age scatter plots derived from the pan-tissue clock appear more linear, as is seen in the composite plot in Figure 4E. This is not surprising as the spread of ages estimated by it is much greater than those by the skin & blood clock. Notwithstanding the age off-set that is apparent with the pan-tissue clock, and the greater noise of the skin & blood clock, their results are consistent in showing that while hTERT can prevent replicative senescence, it is ineffective in stopping epigenetic ageing.
Table 1. Mutants of hTERT used in the experiments.
| Catalytic activity | Extension of Life-span | Telomere Synthesis | Immortalisation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hTERT wt | YES | YES | YES | YES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hTERT IA | YES | YES | NO | NO | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hTERT N-DAT116 | YES | YES | NO | NO | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hTERT N-DAT92 | YES | NO | NO | NO | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The characteristics of the hTERT mutants in the left column are indicated by yes or no in regards to whether they are capable of lifespan extension, telomere synthesis or cellular immortalisation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
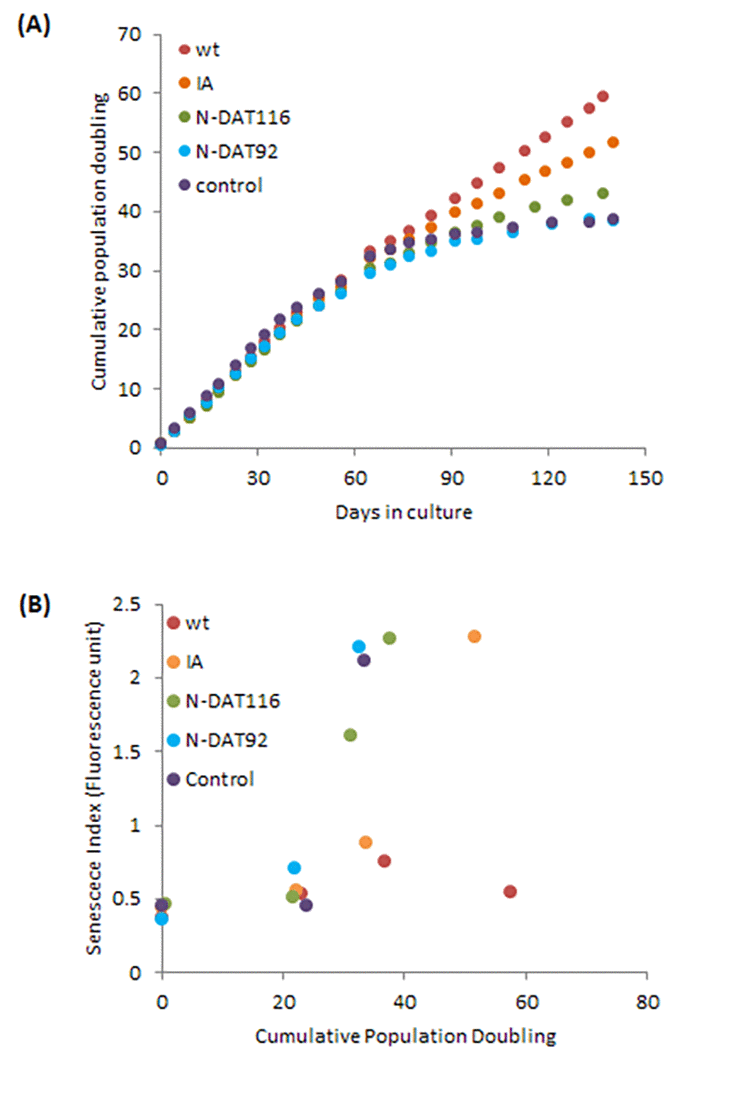
Figure 3. Effects of hTERT and its mutants on growth and senescence of human primary neonatal fibroblasts. (A) Growth dynamics of primary cells transduced with either empty vector (control), vector expressing wildtype hTERT (WT), IA mutant (IA), N-DAT116 mutant (N116) or N-DAT92 mutant (N92). (B) Cells from a selection of passages were subjected to senescence assay and the senescence index is plotted against cumulative population doubling that corresponded to the passage of cells that were analysed.
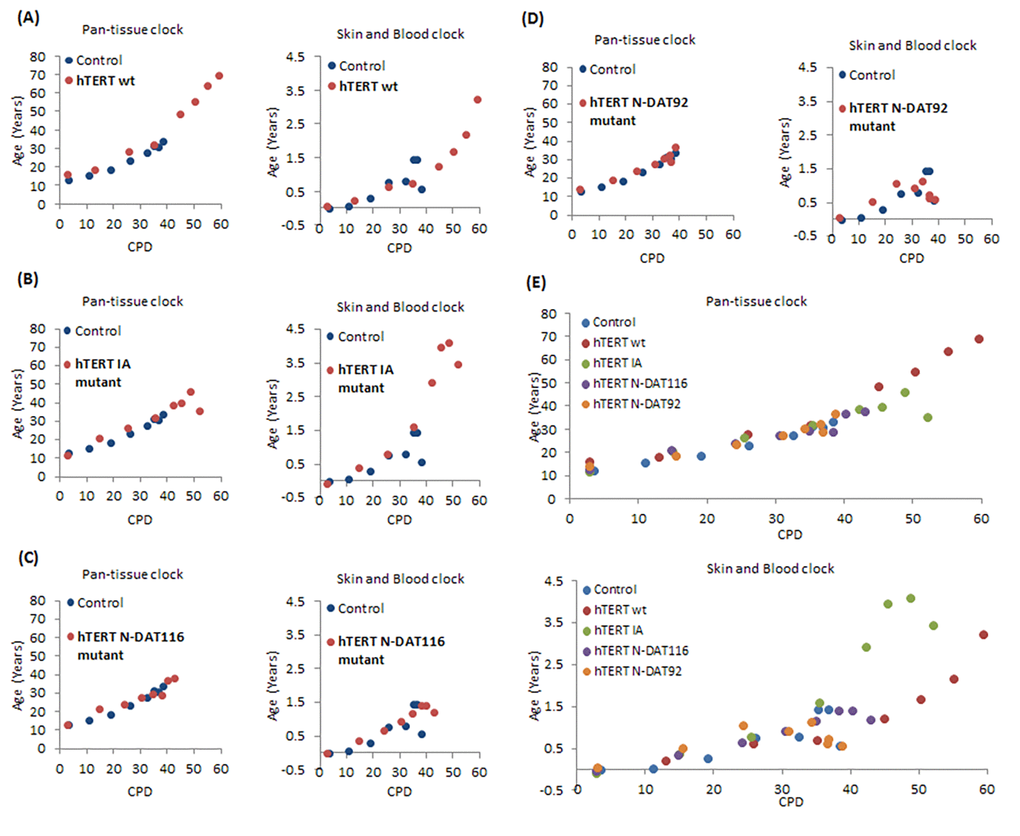
Figure 4. Effects of hTERT and its mutants on epigenetic ageing of human primary neonatal fibroblasts. Ages of primary human neonatal fibroblasts bearing empty vector (blue dots), wildtype hTERT (A), IA mutant (B), N-DAT116 mutant (C) or N-DAT92 mutant (D) were determined using the pan-tissue clock (left panel) and the skin & blood clock (right panel). (E) Composite plot of all the hTERT mutants. The ages of cells from a selection of passages are plotted against cumulative population doubling (CPD) that corresponded to the passages of cells that were analysed.
Discussion
We carried out these simple but very long-drawn out experiments to interrogate the connection, if there was one, between replicative senescence (as mediated by telomeres) and epigenetic ageing. In order to interpret these findings correctly, it is necessary to be reminded that epigenetic age, as imputed by the epigenetic clocks is neither a measure of cellular proliferation rate nor a measure of proliferation or passage number. This is evident from the fact that epigenetic age of isogenic tissues (from the same individual) with high and low turn-over rates (blood and heart for example) are similar [26,44]. Epigenetic ageing is also not a measure of senescent cells, as is evident from this study where epigenetic age continues to rise inexorably in hTERT-expressing cells, which do not senesce. These characteristics underline the distinctiveness of epigenetic ageing from replicative senescence-mediated ageing, which supports our previous findings [49] and three genome-wide association studies (GWAS) which did not detect a relationship between telomere length and epigenetic ageing [50–53].
Do these two different ageing processes interact? Since hTERT-expressing cells (subsequently referred to as hTERT cells) exhibit greater age, it would appear that hTERT promotes epigenetic ageing. This however is not the case because hTERT cells do not exhibit higher ages than control cells prior to the point of replicative senescence of the latter. This is evident from the similar gradient of age increase between control and hTERT cells. The acquisition of greater age by hTERT cells after senescence of the control cells is a smooth continuum of the ageing gradient. As such, observations from these experiments do not support the proposition that hTERT stimulates epigenetic ageing. Instead, by causing cells to bypass replicative senescence, hTERT allows the inherent process of epigenetic ageing, which occurs regardless of its presence, to continue. Put simply, while hTERT may appear on the surface, to exacerbate the epigenetic ageing of cells, in truth hTERT, by preventing telomere attrition, prolonged the lifespan of the cells, allowing them growing older (as measured by the epigenetic clocks).
Accordingly, telomere synthesis and immortalisation are not necessary for the acquisition of greater age; a point that is clearly made by hTERT IA mutant, which increased epigenetic age in spite of its inability to maintain telomere length or immortalise cells, but is still able to extend lifespan [46]. It is interesting that although both IA and N-DAT116 mutants are reportedly able to increase life-span of human mammary epithelial cells [46,48], the magnitude of their effect in human neonatal fibroblasts is very different. The hTERT IA mutant, which is far more effective in this regard, is also highly effective in increasing epigenetic age. The hTERT N-DAT116 mutant on the other hand induces only a marginal increase in lifespan and accordingly, no age increase beyond the control cells is evident (measured by the skin and blood clock) and a correspondingly small increase as measured by the pan-tissue clock. These observations are consistent and they point to increased epigenetic ageing in hTERT cells as a result of extension of lifespan.
This conclusion is also consistent with the recently reported genome-wide association study (GWAS) that identified an variant of hTERT that is associated with increased epigenetic ageing [47]. Interestingly, this allele is also associated with longer telomeres. This observation appeared counter-intuitive in the first instance because short telomeres are unequivocally associated with greater age. As such hTERT variants that generate short telomeres would be expected to be associated with increased epigenetic ageing. The apparent paradox disappears when it is realised that while telomere length undoubtedly records the proliferative history of the cell, it also indicates its proliferative or lifespan potential. As such, cells with longer telomeres have longer lifespan, and as empirically demonstrated here, longer lifespan is accompanied by greater epigenetic ageing. In other words, ectopic expression of hTERT (in this study) and expression of a natural hTERT locus variant associated with longer telomeres in vivo (suggested by GWAS) would increase cellular lifespan, with the consequence of greater epigenetic ageing.
The distinctiveness and independence of epigenetic ageing from replicative senescence, exerts a serious impact on ageing intervention strategies. It is likely that re-activation of hTERT expression or elimination of senescent cells will go some way to mitigate the effects of ageing. These measures however, are unlikely to be sufficient to halt ageing altogether since they will not prevent epigenetic ageing. Interventions that prevent or eliminate senescent cells hold great promise for extending human healthspan. Our study suggests that these interventions might not arrest epigenetic aging, which is disconcerting considering the over-whelming evidence that point to the association between accelerated epigenetic ageing and a host of disparate diseases and conditions [17,28–41]. To what extent epigenetic aging of various cells causes the decline in organ function remains an area of active research, but it is arguable that to maximise healthspan there may be a need to develop compounds that target epigenetic ageing as well. In this regard the new skin & blood clock can form the basis of an assay to test for such interventions. This clock out-performs the pan-tissue clock which is already highly accurate for most tissues and cells in the body, but for unknown reasons exhibit a considerable age off-set when used on some cells cultured in vitro. Furthermore, the pan tissue clock also differed substantially from the skin & blood clock when applied to adult human coronary artery cells: unlike the skin & blood clock, it led to a substantial offset and did not reveal the increase of epigenetic age in function of cell growth. We have at present, no explanation for this curious observation. Overall, it appears that the skin & blood clock is more suitable for cultured cells, which is particularly important because the ability to accurately measure cellular age in vitro will allow the yet unknown mechanism of the epigenetic clock to be elucidated more easily. Gratifyingly, the compatibility of the skin & blood clock with cells in vitro does not come with any loss of compatibility with cells in vivo, as was recently described [44].
In summary, these experiments greatly advance our understanding of the connection between epigenetic ageing and senescence-mediated ageing and on this, they have successfully provided empirical evidence that these two mechanisms of ageing are distinct and uncoupled. With the tools that are available (epigenetic clock and primary cells) and the realisation of the difference between these two ageing processes, we are in much improved position to proceed towards understanding the mechanism of epigenetic ageing and its role in human pathology.
Materials and Methods
Isolation and culture of primary cells
Primary human neonatal fibroblasts were isolated from circumcised foreskins. Informed consent was obtained prior to collection of human skin samples with approval from the Oxford Research Ethics Committee; reference 10/H0605/1. The tissue was cut into small pieces and digested overnight at 4 °C with 0.5 mg/ml Liberase DH in CnT-07 keratinocyte medium (CellnTech) supplemented with penicillin/streptomycin (Sigma) and gentamycin/amphotericin (Life Tech). Following digestion, the epidermis was peeled off from the tissue pieces. The de-epidermised tissue pieces were placed faced down on plastic cell culture plates and allowed partially dry before addition of DMEM supplemented with 10% FBS and antibiotics. After several days incubation in a 37 °C, 5% CO2 humidified environment, fibroblasts can be seen to migrate out from the tissue pieces and when their growth reached confluence, they were trypsinised, counted and seeded into fresh plates for experiments. Adult human coronary artery endothelial cells (HCAEC) were purchased from Cell Applications (USA) and cultured in MesoEndo Cell Growth Medium (Sigma) at 37 °C humidified incubator with 5% CO2.
Neonatal foreskin fibroblasts
100,000 cells were seeded into a 10cm plate and cultured as described above. Upon confluence the cells were harvested with trypsin digestion followed by neutralisation with soybean trypsin inhibitor. The number of cells was ascertained and 100,000 was taken and seeded into a fresh plate. The remaining cells were used for DNA extraction. Population doubling was calculated with the following formula: [log(number of cells harvested) – log(number of cells seeded)] x 3.32. Cumulative population doubling was obtained by addition of population doubling of each passage.
Adult human coronary artery endothelial cells
500,000 cells were seeded into a fibronectin-coated 75cm2 flask and cultured as described above. The procedure of passing the cells, counting and ascertaining population doubling is similar to those described for neonatal foreskin fibroblasts above.
DNA extraction
DNA was extracted from cells using the Zymo Quick DNA mini-prep plus kit (D4069) according to the manufacturer’s instructions and DNA methylation levels were measured on Illumina 850 EPIC arrays according to the manufacturer’s instructions.
Cellular senescence assay
Cells were trypsinised, neutralised and counted. After centrifugation at 8,000 revolutions per minute in a micro-centrifuge, cell pellet was resuspended in 200 μl reaction buffer (Cell Signaling, Senescence β-Galactosidase Staining Kit #9860) containing 10 mM FDG and 0.2% of TritonX-100. After an hour of incubation at 37oC, measurements of fluorescence were made with a plate reader with emission at 488 nm. The fluorescence reading is divided by the cell number to obtain the fluorescence index.
DNA methylation data
The Illumina BeadChips (EPIC or 450K) measures bisulfite-conversion-based, single-CpG resolution DNAm levels at different CpG sites in the human genome. These data were generated by following the standard protocol of Illumina methylation assays, which quantifies methylation levels by the β value using the ratio of intensities between methylated and un-methylated alleles. Specifically, the β value is calculated from the intensity of the methylated (M corresponding to signal A) and un-methylated (U corresponding to signal B) alleles, as the ratio of fluorescent signals β = Max(M,0)/[Max(M,0)+ Max(U,0)+100]. Thus, β values range from 0 (completely un-methylated) to 1 (completely methylated). We used the "noob" normalization method, which is implemented in the "minfi" R package. Both pan-tissue clock and Skin & blood clock algorithms were previously published [26,44].
Western blot analysis
30 μg of protein lysates were separated on 6% polyacrylamide gel and transferred onto PVDF membrane (Bio-Rad, cat. 170-4156) using semidry Trans-Blot® Turbo™ Transfer System (Bio-Rad) and high molecular weight standard protocol. Membranes were blocked in 5% TBS-T milk at room temperature for a minimum of 1 hour with gentle rocking and incubated overnight at room temperature with the following primary antibodies: hTERT (Rockland, cat. 600-401-252, 1:10 000 dilution) and GAPDH (Santa Cruz, cat. sc-25778, 1:30 000 dilution).
Telomerase catalytic activity (TRAP) assay
Telomerase catalytic activity (TRAP) was measured using TRAPEZE® RT Telomerase Detection Kit (Millipore, cat. S7710) according to manufacturer’s instructions. Briefly, for each sample 1 million cells was resuspended in CHAPS buffer supplemented with 0.2U/μl of Superaze in inhibitor and incubated on ice for 30 min. Cellular debris were pelleted for 20 min at 12 000 g at 4 °C, supernatant aliquoted into fresh low protein binding tubes and stored at -80°C until further use; 5μl of the lysates was used for protein concentration measurement. Real-time quantitative PCR was performed using RotorGene Q. All reactions were run in triplicate using master mix provided with the kit and AccuStart II Taq DNA polymerase (Quanta Bioscience, cat. 733-2258). Equivalent of 2500 cells was used per reaction. Cycling parameters were 30 min at 30°C, then 2.0 min at 95 °C, followed by 45 cycles of 15 sec at 94 °C, 60 sec at 59 °C and 10 sec at 45 °C where fluorescence readings were taken.
Data was collected and analysed by RotorGene Q analysis software using standard curve prepared from artificial template provided with the kit. The following controls were run with the samples, positive and negative hTERT control, non-template control and heat inactivated control for each sample analysed.
Author Contributions
SK and KR performed the experiments. SH carried out the statistical analysis. HC provided skin tissue samples. KR, SH and SK conceived the study, KR wrote the manuscript.
Acknowledgments
We would like to acknowledge the support from Simon Bouffler and all members of the Raj lab.
Conflicts of Interest
UC Regents, the employer of SH, has filed patent applications directed at the epigenetic biomarkers mentioned in the article. The other authors declare no conflict of interest.
Funding
The experimental research was funded by the National Institute for Health Research (NIHR) through Health Protection Research Unit (Health Impact of Environmental Hazards with King’s College London) in partnership with Public Health England (PHE). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the Department of Health or Public Health England.
References
- 1. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37:614–36. https://doi.org/10.1016/0014-4827(65)90211-9 [PubMed]
- 2. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961; 25:585–621. https://doi.org/10.1016/0014-4827(61)90192-6 [PubMed]
- 3. Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972; 239:197–201. https://doi.org/10.1038/newbio239197a0 [PubMed]
- 4. Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973; 41:181–90. https://doi.org/10.1016/0022-5193(73)90198-7 [PubMed]
- 5. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345:458–60. https://doi.org/10.1038/345458a0 [PubMed]
- 6. Harley CB, Vaziri H, Counter CM, Allsopp RC. The telomere hypothesis of cellular aging. Exp Gerontol. 1992; 27:375–82. https://doi.org/10.1016/0531-5565(92)90068-B [PubMed]
- 7. Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987; 51:887–98. https://doi.org/10.1016/0092-8674(87)90576-9 [PubMed]
- 8. Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006; 12:1133–38. https://doi.org/10.1038/nm1006-1133 [PubMed]
- 9. Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998; 8:279–82. https://doi.org/10.1016/S0960-9822(98)70109-5 [PubMed]
- 10. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279:349–52. https://doi.org/10.1126/science.279.5349.349 [PubMed]
- 11. Young AR, Narita M. SASP reflects senescence. EMBO Rep. 2009; 10:228–30. https://doi.org/10.1038/embor.2009.22 [PubMed]
- 12. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8:729–40. https://doi.org/10.1038/nrm2233 [PubMed]
- 13. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009; 9:81–94. https://doi.org/10.1038/nrc2560 [PubMed]
- 14. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 15. Regulski MJ. Cellular Senescence: What, Why, and How. Wounds. 2017; 29:168–74. [PubMed]
- 16. Boccardi V, Herbig U. Telomerase gene therapy: a novel approach to combat aging. EMBO Mol Med. 2012; 4:685–87. https://doi.org/10.1002/emmm.201200246 [PubMed]
- 17. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018; 19:371–84. https://doi.org/10.1038/s41576-018-0004-3 [PubMed]
- 18. Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010; 20:440–46. https://doi.org/10.1101/gr.103606.109 [PubMed]
- 19. Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor BJ, Singleton AB. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011; 20:1164–72. https://doi.org/10.1093/hmg/ddq561 [PubMed]
- 20. Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010; 20:434–39. https://doi.org/10.1101/gr.103101.109 [PubMed]
- 21. Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, Shin SY, Dempster EL, Murray RM, et al, and MuTHER Consortium. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012; 8:e1002629. https://doi.org/10.1371/journal.pgen.1002629 [PubMed]
- 22. Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, Colantuoni C, Weinberger DR, Kleinman JE, Lipska BK. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012; 90:260–72. https://doi.org/10.1016/j.ajhg.2011.12.020 [PubMed]
- 23. Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS One. 2011; 6:e14821. https://doi.org/10.1371/journal.pone.0014821 [PubMed]
- 24. Florath I, Butterbach K, Müller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014; 23:1186–201. https://doi.org/10.1093/hmg/ddt531 [PubMed]
- 25. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013; 49:359–67. https://doi.org/10.1016/j.molcel.2012.10.016 [PubMed]
- 26. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. https://doi.org/10.1186/gb-2013-14-10-r115 [PubMed]
- 27. Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jöckel KH, Erbel R, Mühleisen TW, Zenke M, Brümmendorf TH, Wagner W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014; 15:R24. https://doi.org/10.1186/gb-2014-15-2-r24 [PubMed]
- 28. Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci USA. 2014; 111:15538–43. https://doi.org/10.1073/pnas.1412759111 [PubMed]
- 29. Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015; 14:491–95. https://doi.org/10.1111/acel.12325 [PubMed]
- 30. Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, Ritz BR, Chen B, Lu AT, Rickabaugh TM, Jamieson BD, Sun D, Li S, et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016; 17:171. https://doi.org/10.1186/s13059-016-1030-0 [PubMed]
- 31. Horvath S, Langfelder P, Kwak S, Aaronson J, Rosinski J, Vogt TF, Eszes M, Faull RL, Curtis MA, Waldvogel HJ, Choi OW, Tung S, Vinters HV, et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY). 2016; 8:1485–512. https://doi.org/10.18632/aging.101005 [PubMed]
- 32. Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015; 212:1563–73. https://doi.org/10.1093/infdis/jiv277 [PubMed]
- 33. Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015; 7:1130–42. https://doi.org/10.18632/aging.100859 [PubMed]
- 34. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015; 16:25. https://doi.org/10.1186/s13059-015-0584-6 [PubMed]
- 35. Levine AJ, Quach A, Moore DJ, Achim CL, Soontornniyomkij V, Masliah E, Singer EJ, Gelman B, Nemanim N, Horvath S. Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J Neurovirol. 2016; 22:366–75. https://doi.org/10.1007/s13365-015-0406-3 [PubMed]
- 36. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015; 7:1198–211. https://doi.org/10.18632/aging.100864 [PubMed]
- 37. Levine ME, Lu AT, Chen BH, Hernandez DG, Singleton AB, Ferrucci L, Bandinelli S, Salfati E, Manson JE, Quach A, Kusters CD, Kuh D, Wong A, et al. Menopause accelerates biological aging. Proc Natl Acad Sci USA. 2016; 113:9327–32. https://doi.org/10.1073/pnas.1604558113 [PubMed]
- 38. Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY). 2015; 7:690–700. https://doi.org/10.18632/aging.100809 [PubMed]
- 39. Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, Roetker NS, Just AC, Demerath EW, Guan W, Bressler J, Fornage M, Studenski S, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY). 2016; 8:1844–65. https://doi.org/10.18632/aging.101020 [PubMed]
- 40. Dugué PA, Bassett JK, Joo JE, Jung CH, Ming Wong E, Moreno-Betancur M, Schmidt D, Makalic E, Li S, Severi G, Hodge AM, Buchanan DD, English DR, et al. DNA methylation-based biological aging and cancer risk and survival: pooled analysis of seven prospective studies. Int J Cancer. 2018; 142:1611–19. https://doi.org/10.1002/ijc.31189 [PubMed]
- 41. Maierhofer A, Flunkert J, Oshima J, Martin GM, Haaf T, Horvath S. Accelerated epigenetic aging in Werner syndrome. Aging (Albany NY). 2017; 9:1143–52. https://doi.org/10.18632/aging.101217 [PubMed]
- 42. Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, Ritz B, Bandinelli S, Neuhouser ML, Beasley JM, Snetselaar L, Wallace RB, Tsao PS, et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY). 2017; 9:419–46. https://doi.org/10.18632/aging.101168 [PubMed]
- 43. Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, Gladyshev VN. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017; 25:954–960.e6. https://doi.org/10.1016/j.cmet.2017.03.016 [PubMed]
- 44. Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, Felton S, Matsuyama M, Lowe D, Kabacik S, Wilson JG, Reiner AP, Maierhofer A, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY). 2018; 10:1758–75. https://doi.org/10.18632/aging.101508 [PubMed]
- 45. Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, Sedivy JM, Weinberg RA. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA. 1998; 95:14723–28. https://doi.org/10.1073/pnas.95.25.14723 [PubMed]
- 46. Armbruster BN, Banik SS, Guo C, Smith AC, Counter CM. N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol Cell Biol. 2001; 21:7775–86. https://doi.org/10.1128/MCB.21.22.7775-7786.2001 [PubMed]
- 47. Lu AT, Xue L, Salfati EL, Chen BH, Ferrucci L, Levy D, Joehanes R, Murabito JM, Kiel DP, Tsai PC, Yet I, Bell JT, Mangino M, et al. GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat Commun. 2018; 9:387. https://doi.org/10.1038/s41467-017-02697-5 [PubMed]
- 48. Mukherjee S, Firpo EJ, Wang Y, Roberts JM. Separation of telomerase functions by reverse genetics. Proc Natl Acad Sci USA. 2011; 108:E1363–71. https://doi.org/10.1073/pnas.1112414108 [PubMed]
- 49. Lowe D, Horvath S, Raj K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget. 2016; 7:8524–31. https://doi.org/10.18632/oncotarget.7383 [PubMed]
- 50. Breitling LP, Saum KU, Perna L, Schöttker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016; 8:21. https://doi.org/10.1186/s13148-016-0186-5 [PubMed]
- 51. Nwanaji-Enwerem JC, Colicino E, Trevisi L, Kloog I, Just AC, Shen J, Brennan K, Dereix A, Hou L, Vokonas P, Schwartz J, Baccarelli AA. Long-term ambient particle exposures and blood DNA methylation age: findings from the VA normative aging study. Environ Epigenet. 2016; 2. https://doi.org/10.1093/eep/dvw006 [PubMed]
- 52. Marioni RE, Harris SE, Shah S, McRae AF, von Zglinicki T, Martin-Ruiz C, Wray NR, Visscher PM, Deary IJ. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol. 2016; 45: 424–32. https://doi.org/10.1093/ije/dyw041 [PubMed]
- 53. Belsky DW, Moffitt TE, Cohen AA, Corcoran DL, Levine ME, Prinz JA, Schaefer J, Sugden K, Williams B, Poulton R, Caspi A. Eleven telomere, epigenetic clock, and biomarker-composite quantifications of biological aging: do they measure the same thing? Am J Epidemiol. 2018; 187:1220–30. https://doi.org/10.1093/aje/kwx346 [PubMed]