



Introduction
Colon cancer is the third most common cancer, with increasing morbidity and mortality worldwide [1]. The morbidity of colon cancer increases along with the increase of age. In fact, more than 60% of people who developed cancer are order than 65. Despite the significant progress in tailored therapy, the high migration and invasion capabilities of this tumor have been a bottleneck in reducing the mortality, keeping its 5-year survival rate under 12% [2, 3]. It has been reported that the prognosis of colon cancer mainly depends on the clinicopathological features and the tumor stage. Nevertheless, due to the remarkable disease heterogeneity, it is difficult to determine the prognosis of patients based on these traditional factors [4]. Therefore, a better understanding of the pathogenesis and identification of new promising prognostic biomarkers are essential for development of effective therapies for colon cancer patients.
A recent study has reported that the identification of prognostic signature based on the genomic or transcriptome data can improve our understanding of cancer progression and survival rate [5]. Presently, many prognostic gene-expression signatures have been identified in various human cancers which help in predicting the cancer prognosis outcomes [6–8]. Among the various gene-expression signatures in colon cancer, the six-cluster gene expression ColoPrint [9] and Colon OncotypeDx [7] have been widely verified in retrospective studies. Recently, a study has reported that the protein-coding genes make up less than 2% of the whole genome sequences, and the noncoding genes are transcribed into noncoding RNAs, such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) [10]. Some of the recent studies have demonstrated the competing endogenous RNA (ceRNA) activity of lncRNAs, as natural miRNA decoys in human pathophysiological conditions, including cancer [11]. Therefore, systematic analysis of lncRNA-associated ceRNA network may help to explore and identify more prognostic gene-expression signatures in colon cancer.
In this study, we aimed to screen newer prognostic signatures for colon cancer through the analysis of RNA-seq expression profile and miRNA expression profiles. Based on the differentially expressed mRNAs (DEMs), lncRNA (DELs), and miRNA (DEMis) data obtained between the tumor and normal samples, a ceRNA network was constructed and DEMs involved in this network were selected for the construction of prognostic risk model. Our study presented some novel biomarkers with a potential prognostic value, and provided a preliminary bioinformatic evidence for understanding the complex mechanism of colon cancer progression.
Results
Differential analysis
The mRNA, lncRNA, and miRNA expression profiles obtained were subjected to differential expression analysis. The screening thresholds for mRNA and miRNA were adjusted P value < 0.01 and |logFC| > 2, and for lncRNA was adjusted P value < 0.05 and |logFC| > 1. A total of 1476 (323 upregulated and 1153 downregulated) DEMs, 9 (5 upregulated and 4 downregulated) DELs, and 243 (113 upregulated and 130 downregulated) DEMis were identified. Top10 genes (in ascending order according to the adjusted P value) were selected from DEMs, DELs, and DEMis to draw the heatmaps, as shown in Figure 1.
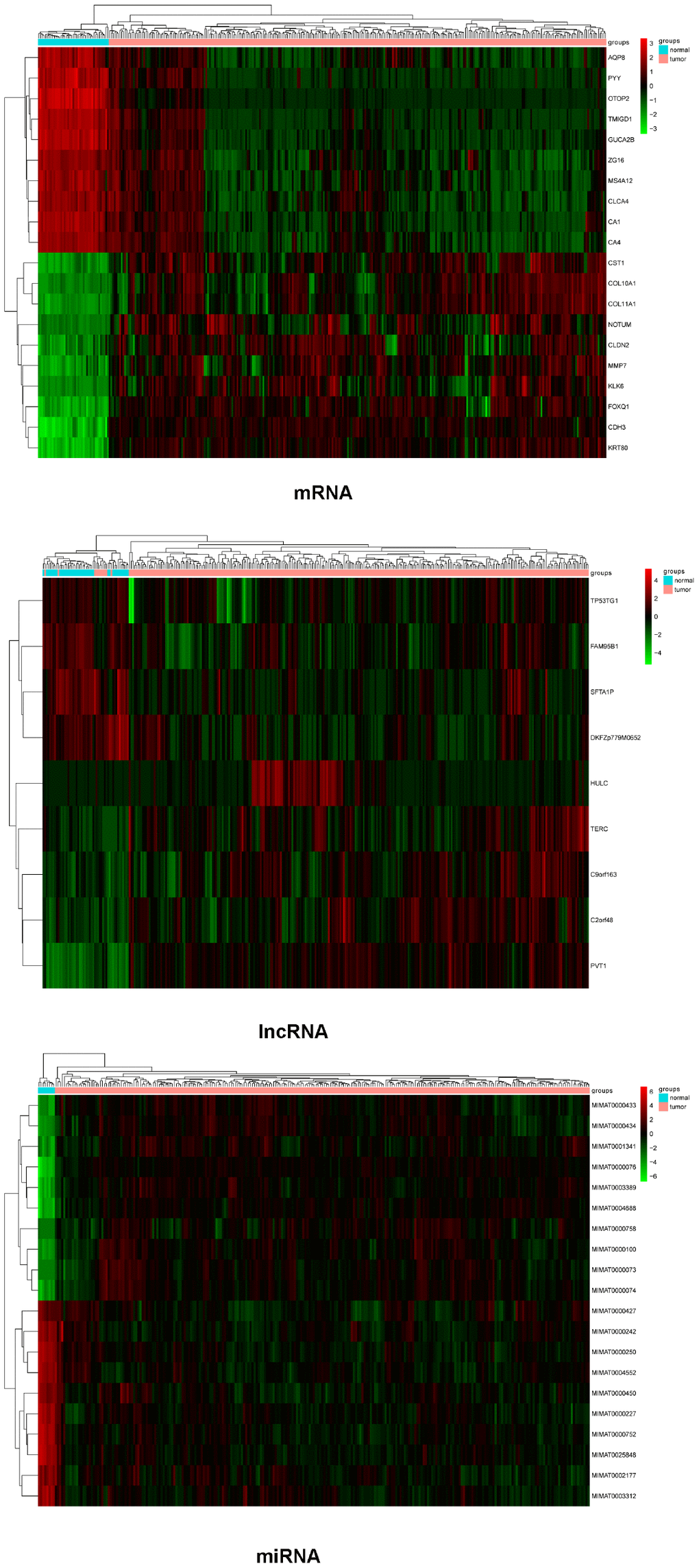
Figure 1. Heatmaps of differential expression mRNAs (top 10), lncRNAs, and miRNAs (top 10). Red represents high expression and green represents low expression.
GO enrichment analysis of DEMs
With a threshold of adjusted P value < 0.01, 13 BPs, 14 CCs, and 5 MFs were significantly enriched by the DEMs. The top 5 terms for the three categories are shown in Figure 2A. The seven most significant terms are shown in detail. According to the genes in term, the clustering of genes was carried out to construct the phylogenetic tree (Figure 2B). In terms of the involved BP terms, the DEMs were significantly enriched in chemical synaptic transmission, axon guidance, nervous system development, cell adhesion, and transport. From the perspective of CC terms, the encoding proteins were mainly located in an integral component of the plasma membrane, plasma membrane, and apical plasma membrane.
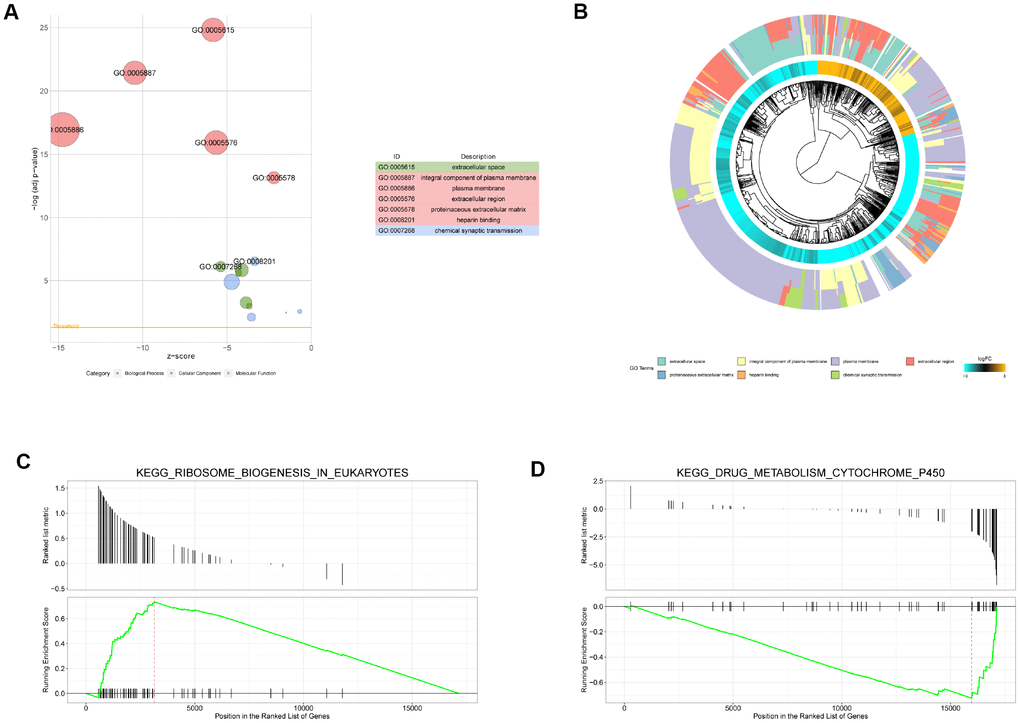
Figure 2. Gene Ontology (GO) and gene set enrichment analysis (GSEA) enrichment analysis. (A) Bubble diagram of GO enrichment analysis (top 7). The circle size indicates the number of enriched genes. Green represents biological process, red represents molecular function, and blue represents cellular component. (B) Cluster diagram of GO enrichment analysis. (C) The upregulated pathway (KEGG_RIBOSOME_BIOGENESIS_IN_EUKARYOTES) of GSEA analysis with the highest enrichment score. (D) The downregulated pathway (KEGG_DRUG_METABOLISM_CYTOCHROME_P450) with the highest enrichment score.
Pathway enrichment analysis
According to the KEGG pathway enrichment analysis of the DEMs obtained above by GSEA, a total of 67 significantly enriched pathways were identified (adjusted P value < 0.05), among which 16 were upregulated pathways (normalized enrichment score (NES) > 0) and 51 were downregulated pathways (NES < 0). KEGG ribosome biogenesis in eukaryotes had the largest NES in the significantly upregulated pathways (Figure 2C). In addition, DNA replication, RNA degradation, IL17 signaling pathway, p53 signaling pathway, and oxytocin signaling pathway were also significantly upregulated. Among the significantly downregulated pathways, drug metabolism cytochrome P450 had the largest NES (Figure 2D). Additionally, chemical carcinogenesis and mineral absorption pathways were also downregulated.
Co-expression analysis
For the screened DELs and DEMis, the correlation coefficients and statistical significance degree between them and DEMs were calculated, respectively, and the gene pairs with |r| > 0.6 and an adjusted P value < 0.01 were screened out. There were 193 nodes (199 mRNAs and 6 lncRNAs) and 229 edges in the co-expression network of mRNA-lncRNA, where lncRNA Pvt1 oncogene (PVT1) had the highest connectivity degree (Figure 3A). In the co-expression network of mRNA-miRNA, there were 184 nodes (392 mRNAs, 102 miRNAs) and 1,935 edges (Figure 3B).
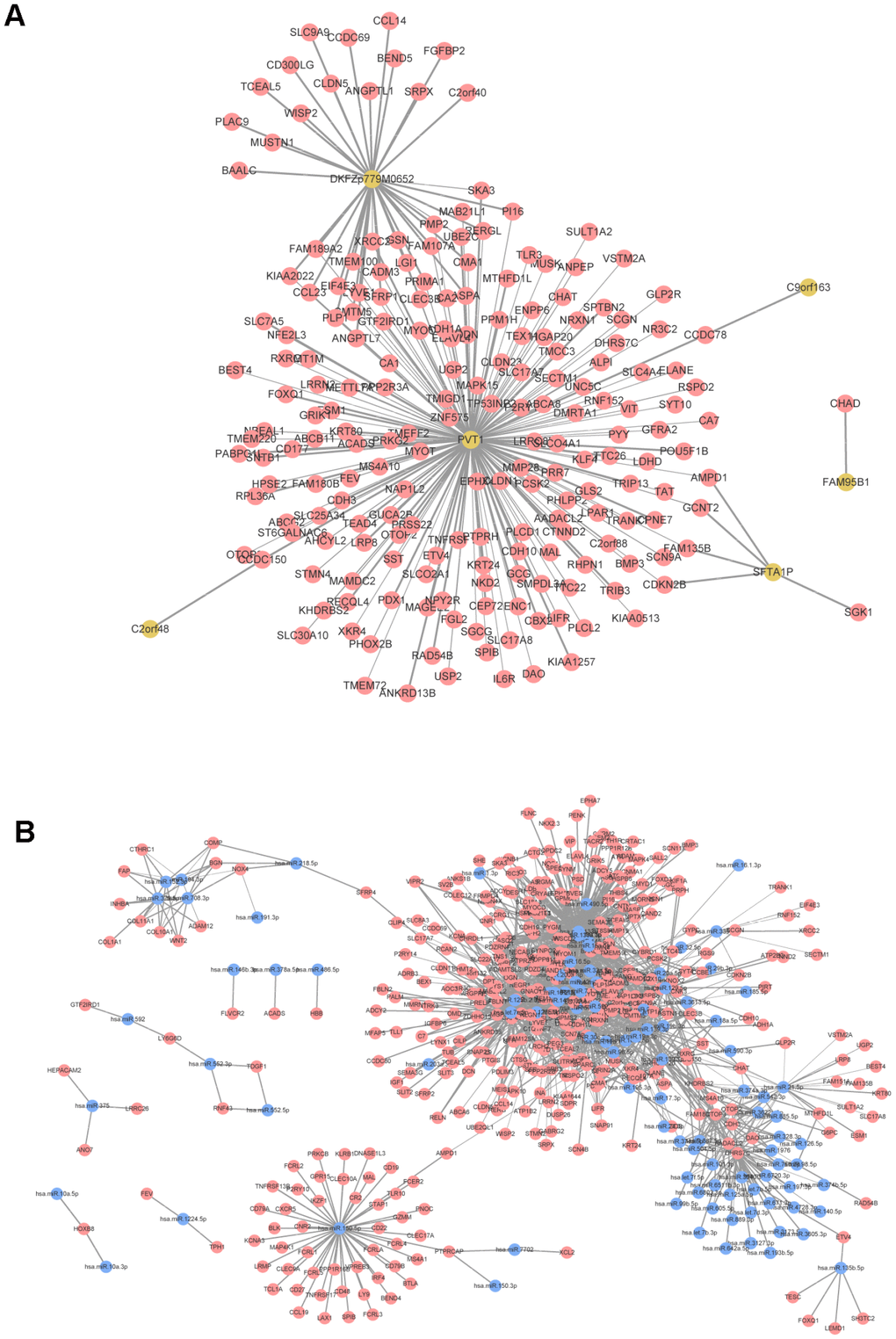
Figure 3. Co-expression network analysis. (A) mRNA-lncRNA co-expression network. (B) mRNA-miRNA co-expression network. Red node was mRNA, yellow node was lncRNA, and blue node was miRNA. The line thickness indicates the relative size of correlation coefficient.
miRNA prediction
A total of 200 lncRNA-miRNA pairs (3 lncRNAs and 141 miRNAs) were predicted, based on the lncRNA involved in lncRNA-mRNA co-expression pairs using starbase (Figure 4A). Additionally, the mRNA involved in lncRNA-mRNA co-expression pairs were subjected to mRNA-miRNA prediction, and 117,570 pairs of miRNA-mRNA (199 mRNAs and 2498 miRNAs) were obtained with Score = 1 and Position = CDS. These mRNA-miRNA pairs were intersected with the obtained mRNA-miRNA co-expression pairs, and 89 mRNA-miRNA pairs (36 mRNAs and 38 miRNAs) were further screened (Figure 4B).
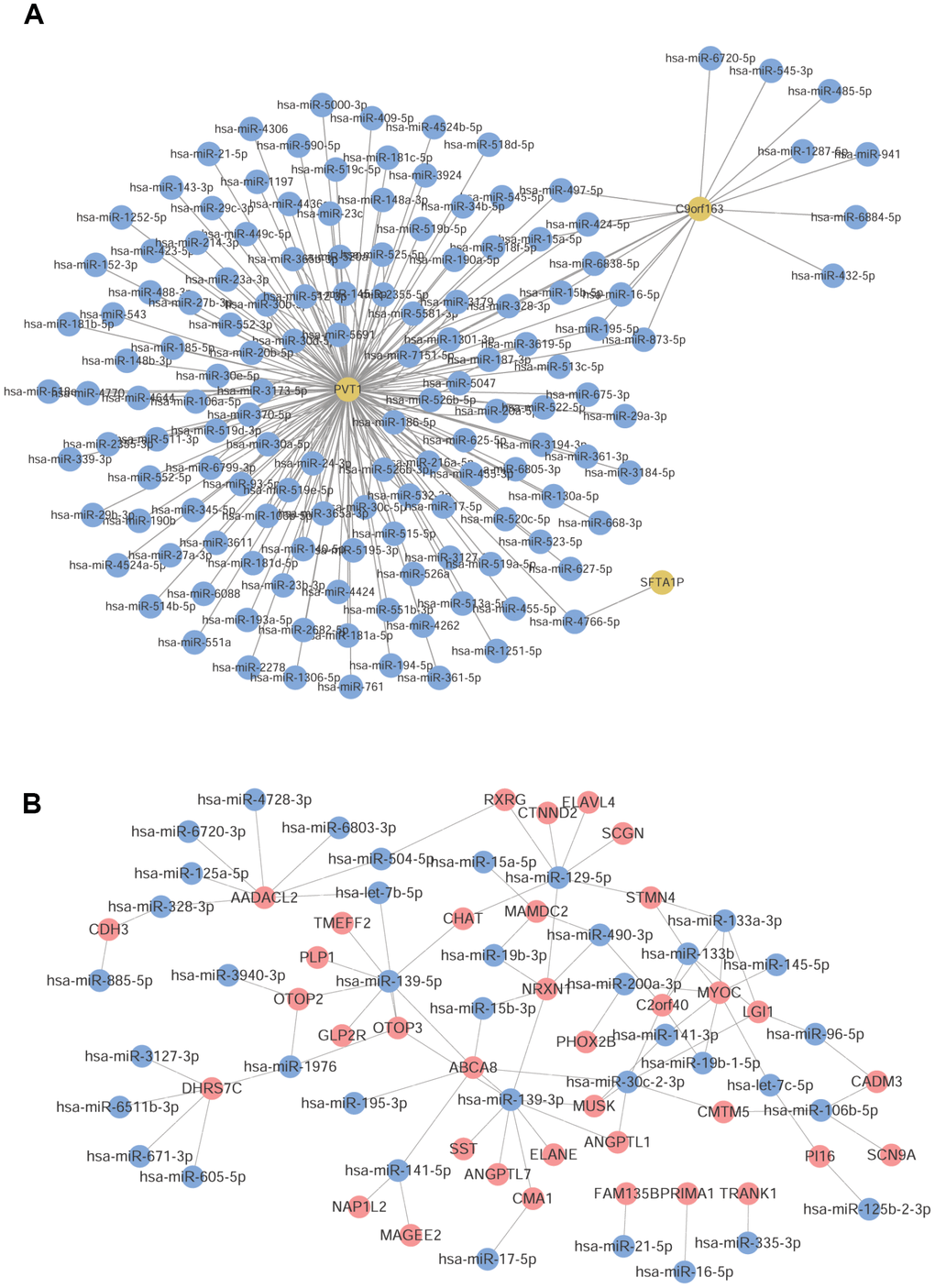
Figure 4. Prediction of common miRNAs. (A) LncRNA-miRNA pairs predicted by lncRNA. (B) The obtained common mRNA-miRNA. Red node was mRNA, yellow node was lncRNA, and blue node was miRNA.
LncRNA and miRNA pathway enrichment analyses
DKFZp779M0652 and PVT1 that had many target genes were subjected to functional enrichment. DKFZp779M0652 was associated with functions of cellular response to tumor necrosis factor and cellular response to interleukin-1. PVT1 was related to one-carbon metabolic process and reciprocal meiotic recombination.
The mRNAs in the miRNA-mRNA relation pairs obtained above were used as the target genes of miRNAs. Since there were few target genes and enrichment analysis could not be carried out, thus the functions of each target gene were inquired on the genecards to obtain the functions of all miRNA (data not shown).
ceRNA network construction
A total of 8,353 lncRNA-miRNA-mRNA pairs were obtained, involving 197 mRNAs, 135 miRNAs, and 3 lncRNAs. The constructed ceRNA network is shown in Figure 5A. After further screening of the network, only the aforementioned co-expressed mRNA-miRNA pairs were retained in the mRNAs-miRNA relation pairs. Finally, a total of 21 lncRNA-miRNA-mRNA pairs were obtained, including 7 miRNAs, 2 lncRNAs, and 10 mRNAs. The network is depicted in Figure 5B.
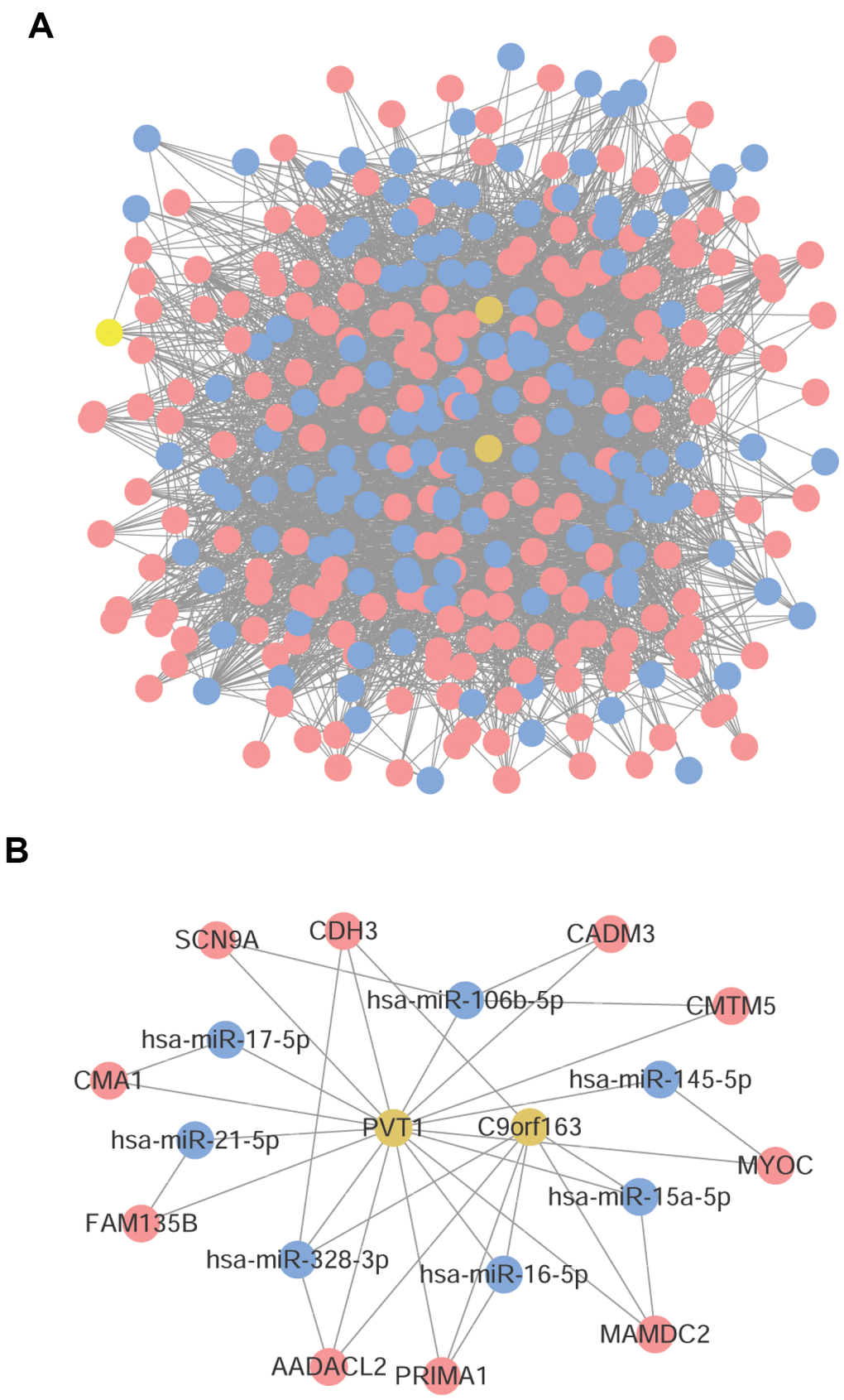
Figure 5. Construction of ceRNA network. (A) The ceRNA network constructed by 197 mRNAs, 135 miRNAs, and 3 lncRNAs. (B) The ceRNA network constructed by 7 miRNAs, 2 lncRNAs, and 10 mRNAs. Red node was mRNA, yellow node was lncRNA, and blue node was miRNA.
Establishment of mRNA prognostic risk model
A total of 277 samples (68 deceased and 209 living) with clinical information of overall survival time were selected. 197 mRNAs from ceRNA network were used as candidate mRNAs. The expression level of mRNA in the sample set was screened, and univariable Cox regression analysis was conducted based on the clinical prognosis information of the sample. The regression threshold of P value was set at 0.05 to screen the prognostic mRNAs, as shown in Table 1. The risk score models of prognostic mRNAs are shown in Table 2. The drawn survival curves are shown in Figure 6A. All models were significantly correlated, and the ROC curves were drawn based on the risk scores of all the models and survival information (Figure 6B). AUC values were calculated (Table 3), and the time periods were 1, 3, and 5 years, respectively. The largest AUC of 5-year prognosis scoring model was used as the final risk score model: Risk score = 0.58343 * exp (SGCG) + (-0.11860) * exp (CLDN23) + (-0.09726) * exp (SLC4A4) + 0.18416 * exp (CCDC78) + 0.13586 * exp (SLC17A7) + 0.40269 * exp (OTOP3) + (-0.23459) * exp (SMPDL3A). The parameters of mRNAs (SGCG, CLDN23, SLC4A4, CCDC78, SLC17A7, OTOP3, and SMPDL3A) associated with the optimal prognosis are shown in Table 4.
Table 1. Prognostic mRNAs obtained from univariate Cox regression.
| mRNA | HR (95% CI for HR) | Beta | P value |
| SGCG | 1.8 (1.3-2.4) | 0.57 | 0.00019 |
| CLDN23 | 0.73 (0.57-0.92) | -0.32 | 0.0078 |
| SLC4A4 | 0.89 (0.81-0.97) | -0.12 | 0.0085 |
| CCDC78 | 1.2 (1-1.4) | 0.2 | 0.011 |
| SLC17A7 | 1.4 (1.1-1.8) | 0.33 | 0.011 |
| OTOP3 | 1.4 (1-2) | 0.36 | 0.027 |
| SMPDL3A | 0.74 (0.56-0.97) | -0.3 | 0.029 |
| TCEAL5 | 1.3 (1-1.6) | 0.23 | 0.033 |
| MAB21L1 | 1.2 (1-1.5) | 0.22 | 0.033 |
| CDH10 | 1.7 (1-2.6) | 0.5 | 0.036 |
| CA2 | 0.88 (0.78-0.99) | -0.13 | 0.036 |
| SLC17A8 | 0.67 (0.46-0.98) | -0.4 | 0.037 |
Table 2. Risk score model.
| Score number | Risk score model |
| Score1 | 0.5700*exp(SGCG) |
| Score2 | 0.5700*exp(SGCG)+(-0.3194)*exp(CLDN23) |
| Score3 | 0.57915*exp(SGCG)+(-0.23856)*exp(CLDN23)+(-0.10073)*exp(SLC4A4) |
| Score4 | 0.66751*exp(SGCG)+(-0.16306)*exp(CLDN23)+(-0.09054)*exp(SLC4A4) +0.19449*exp(CCDC78) |
| Score5 | 0.56759*exp(SGCG)+(-0.15242)*exp(CLDN23)+(-0.10090)*exp(SLC4A4) +0.18517*exp(CCDC78)+0.18698*exp(SLC17A7) |
| Score6 | 0.57012*exp(SGCG)+(-0.13823)*exp(CLDN23)+(-0.11492)*exp(SLC4A4) +0.18897*exp(CCDC78)+0.13280*exp(SLC17A7)+0.39265*exp(OTOP3) |
| Score7 | 0.58343*exp(SGCG)+(-0.11860)*exp(CLDN23)+(-0.09726)*exp(SLC4A4) +0.18416*exp(CCDC78)+0.13586*exp(SLC17A7)+0.40269*exp(OTOP3)+(-0.23459)*exp(SMPDL3A) |
| Score8 | 0.53201*exp(SGCG)+(-0.13415)*exp(CLDN23)+(-0.10341)*exp(SLC4A4) +0.18600*exp(CCDC78)+0.09211*exp(SLC17A7)+0.39503*exp(OTOP3)+(-0.22168)*exp(SMPDL3A)+0.10713*exp(TCEAL5) |
| Score9 | 0.52245*exp(SGCG)+(-0.13418)*exp(CLDN23)+(-0.10215)*exp(SLC4A4) +0.18929*exp(CCDC78)+0.08428*exp(SLC17A7)+0.39404*exp(OTOP3)+(-0.22316)*exp(SMPDL3A)+0.09120*exp(TCEAL5)+0.03316*exp(MAB21L1) |
| Score10 | 0.50527*exp(SGCG)+(-0.14585)*exp(CLDN23)+(-0.10778)*exp(SLC4A4) +0.19778*exp(CCDC78)+0.06656*exp(SLC17A7)+0.40789*exp(OTOP3)+(-0.21657)*exp(SMPDL3A)+0.04899*exp(TCEAL5)+(-0.02491)*exp(MAB21L1)+0.41369*exp(CDH10) |
| Score11 | 0.51080*exp(SGCG)+(-0.14221)*exp(CLDN23)+(-0.09733)*exp(SLC4A4) +0.19483*exp(CCDC78)+0.07246*exp(SLC17A7)+0.41640*exp(OTOP3)+(-0.20729)*exp(SMPDL3A)+0.03954*exp(TCEAL5)+(-0.02730)*exp(MAB21L1)+0.41328*exp(CDH10)+(-0.02849)*exp(CA2) |
| Score12 | 0.46042*exp(SGCG)+(-0.08915)*exp(CLDN23)+(-0.09846)*exp(SLC4A4) +0.19869*exp(CCDC78)+0.08603*exp(SLC17A7)+0.50655*exp(OTOP3)+(-0.16637)*exp(SMPDL3A)+0.02671*exp(TCEAL5)+(-0.05120)*exp(MAB21L1)+0.54879*exp(CDH10)+(0.01309)*exp(CA2)+(-0.36081)*exp(SLC17A8) |
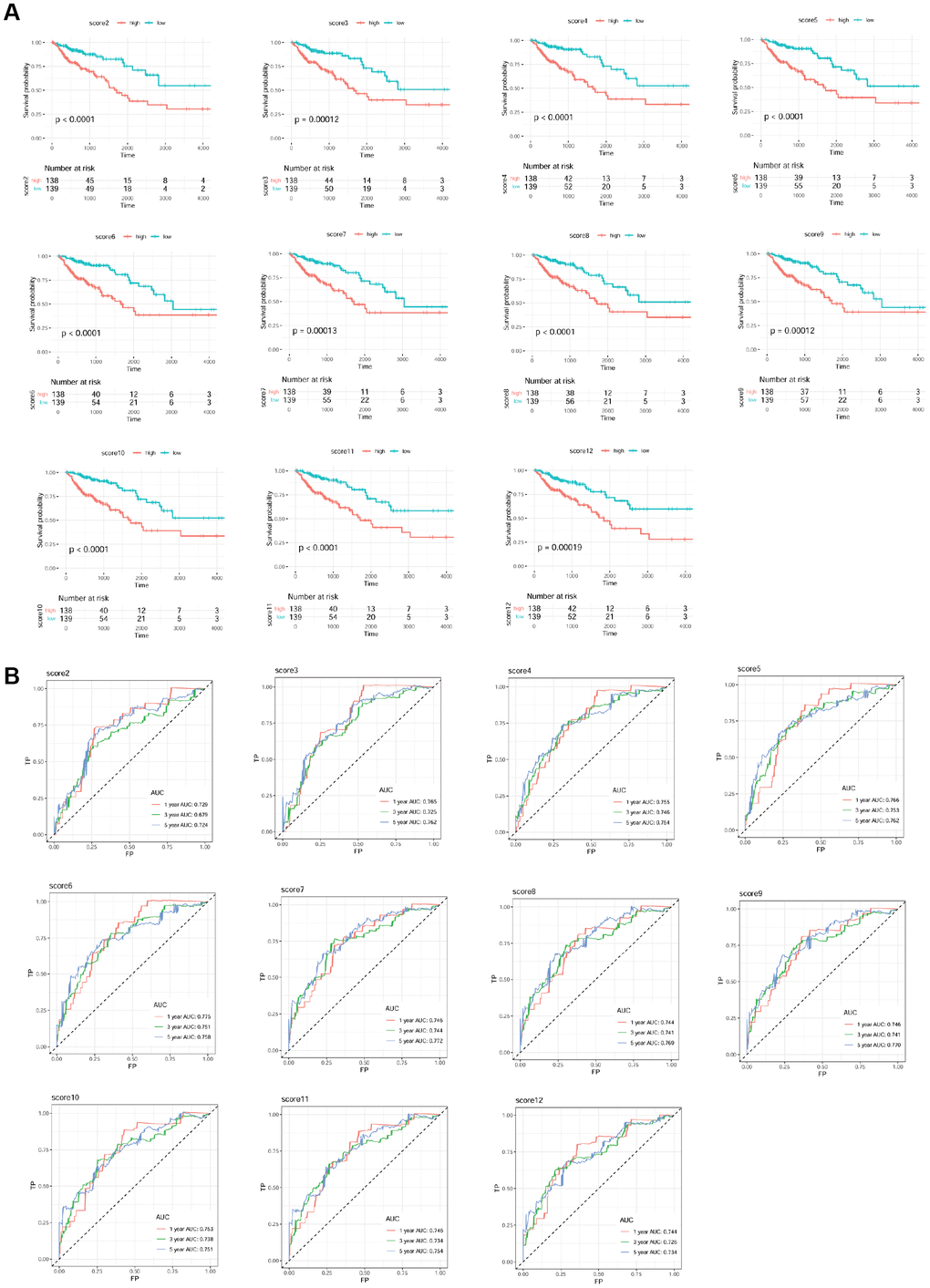
Figure 6. Survival curve (A) and receiver operation characteristic curve (B) of each scoring model.
Table 3. The Area Under The Curve (AUC) of 1 year, 3 year and 5year, with 95% confidence interval (CI).
| Score number | 1 year (95% CI) | 3 year (95% CI) | 5 year (95% CI) |
| score2 | 0.729 (0.641-0.802) | 0.679 (0.631-0.751) | 0.724 (0.652-0.805) |
| score3 | 0.765 (0.715-0.817) | 0.725 (0.679-0.790) | 0.762 (0.669-0.836) |
| score4 | 0.755 (0.690-0.820) | 0.746 (0.681-0.815) | 0.754 (0.688-0.849) |
| score5 | 0.766 (0.726-0.831) | 0.753 (0.677-0.825) | 0.762 (0.686-0.878) |
| score6 | 0.775 (0.733-0.837) | 0.751 (0.700-0.809) | 0.758 (0.685-0.886) |
| score7 | 0.746 (0.695-0.837) | 0.744 (0.720-0.808) | 0.772 (0.705-0.833) |
| score8 | 0.744 (0.689-0.839) | 0.741 (0.698-0.809) | 0.769 (0.678-0.838) |
| score9 | 0.746 (0.690-0.842) | 0.741 (0.693-0.812) | 0.77 (0.675-0.837) |
| score10 | 0.753 (0.712-0.838) | 0.738 (0.684-0.800) | 0.751 (0.671-0.825) |
| score11 | 0.746 (0.705-0.833) | 0.734 (0.691-0.792) | 0.754 (0.676-0.829) |
| score12 | 0.744 (0.701-0.833) | 0.726 (0.660-0.808) | 0.734 (0.637-0.824) |
Table 4. The parameters of mRNAs associated with the optimal prognosis
| mRNA | Coef | Hazard ratio | Pr(>|z|) | Signif. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SGCG | 0.58343 | 1.79217 | 0.00128 | ** | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CLDN23 | -0.1186 | 0.88816 | 0.37088 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SLC4A4 | -0.09726 | 0.90732 | 0.07544 | . | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CCDC78 | 0.18416 | 1.2022 | 0.02445 | * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SLC17A7 | 0.13586 | 1.14552 | 0.33808 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| OTOP3 | 0.40269 | 1.49584 | 0.02125 | * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SMPDL3A | -0.23459 | 0.7909 | 0.12177 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Coef is the coefficient value obtained by Cox-PH regression model (positive value means positive correlation with survival time, negative value means negative correlation with survival time); Hazard Ratio is the risk score, and p value is the significance threshold of the test. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Cox regression analysis of prognostic clinical factors
The clinical factors of gender, age at initial pathologic diagnosis anatomic neoplasm subdivision, and pathologic stage were selected for univariable Cox regression analysis combining with the survival information, and the influence factor of pathologic stage (P < 0.0001) was selected. The samples without pathologic stage were removed from 277 samples in the previous step, and the remaining 268 samples were used for subsequent analysis. Pathologic stage contained the following types: “Stage I”, “Stage IA”, “Stage II”, “Stage IIA”, “Stage IIB”, “Stage IIC”, “Stage III”, “Stage IIIA”, “Stage IIIB”, “Stage IIIC”, “Stage IV”, “Stage IVA”, and “Stage IVB”. These types were converted into stage “1”, “2”, “3”, and “4”, and were denoted as “stage n” which was added into the model above for multivariable Cox regression analysis (p = 0.00026) to obtain the new scoring model as follows: Risk score = 0.51396 * exp (SGCG) + (-0.16881) * exp (CLDN23) + (-0.08028) * exp (SLC4A4) + 0.11820 * exp (CCDC78) + 0.09786 * exp (SLC17A7) + 0.44172 * exp (OTOP3) + (-0.22250) * exp (SMPDL3A) + 0.66818 * stage_n. The parameters of scoring model are shown in Table 5. The survival curve and ROC curve of survival rate at 1, 3, and 5 years are shown in Figure 7.
Table 5. The parameters of scoring model.
| mRNA | Coef | Hazard ratio | Pr(>|z|) | Signif. |
| SGCG | 0.51396 | 1.67189 | 0.00706 | ** |
| CLDN23 | -0.16881 | 0.84467 | 0.20849 | |
| SLC4A4 | -0.08028 | 0.92285 | 0.14911 | |
| CCDC78 | 0.1182 | 1.12547 | 0.14434 | |
| SLC17A7 | 0.09786 | 1.10281 | 0.51499 | |
| OTOP3 | 0.44172 | 1.55538 | 0.00821 | ** |
| SMPDL3A | -0.2225 | 0.80052 | 0.14536 | |
| stage_n | 0.66818 | 1.95069 | 1.91E-05 | *** |
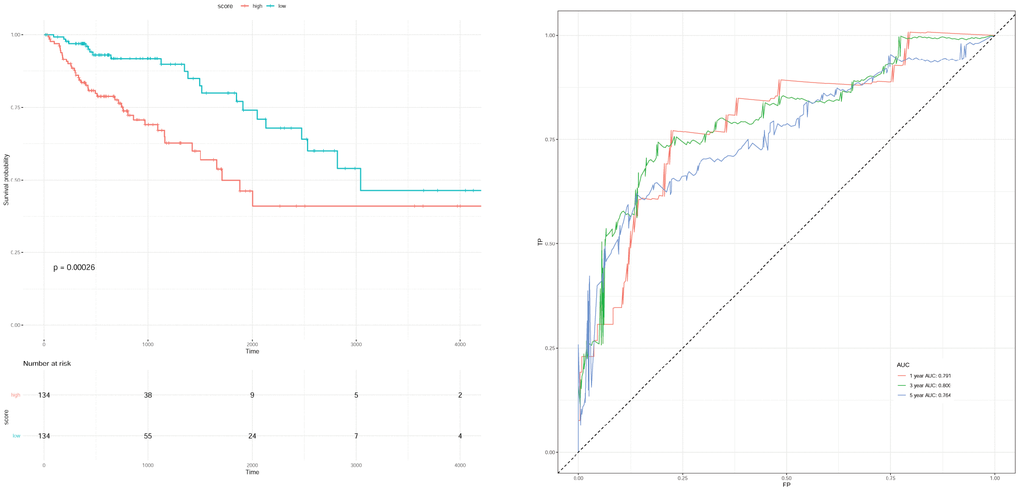
Figure 7. Survival curve (A) and receiver operation characteristic curve (B) of scoring model with clinical factor of stage.
Model validation using independent datasets
Risk score values of all the samples in two independent validation datasets were calculated based on the above risk score model. All samples were divided into high risk group and low risk group according to the median score of risk score. Kaplan-Meier survival curve analysis showed that the developed prognostic signature could well distinguish high-risk and low-risk groups; the survival time of patients in low-risk group was significantly longer than that of patients in high-risk group (P = 0.00083 and P < 0.0001) (Figure 8A and 8B). The AUCs of this prognostic signature in predicting 1/3/5 survival rate of patients in GSE17538 were 0.755, 0.799 and 0.758, respectively (Figure 8C).
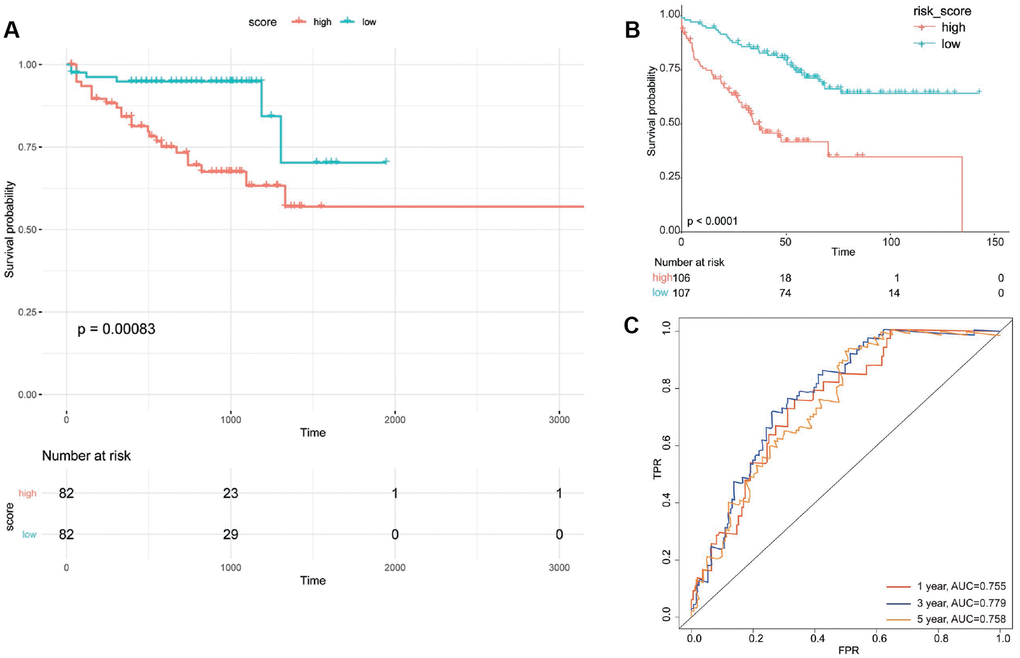
Figure 8. Validation of obtained scoring model. Survival curve of patients in high-risk group and low-risk group divided according to prognostic signature for UCSC Xene database (A) and GSE17538 (B). (C) Area under receiver operation characteristic curve (AUC) for 1/3/5 survival rate of patients in GSE17538. FPR, false positive rate; TPR, true positive rate.
Discussion
Enhanced migration and invasion potential of the colon cancer cells have led to a significant low 5-year survival rate for the colon cancer patients. Thus, an accurate prediction of prognosis is crucial for the personalized treatment of these patients. Nowadays, gene expression profiling has become a commercial adjunct to cancer therapy. For instance, a six-lncRNA expression signature is utilized as an indicator to evaluate the prognosis of colorectal cancer patients [12]. However, the prognostic tools available for patients with colon cancer usually do not include clinical factors. In this study, we identified a seven mRNAs and one clinical factor-based prognostic signature for colon cancer, which was proved to predict colon cancer reliably.
Among the seven prognosis-related mRNAs, two were from solute carrier family, including solute carrier family 4 member 4 (SLC4A4) and SLC17A7. The members of solute carrier family have been identified as tumor suppressors. For instance, methylation in the CpG islands of the SLC25A43 gene has been suggested to be a possible mechanism of gene silencing in breast cancer without loss of heterozygosity [13]. SLC5A8 is another tumor suppressor gene, which is often downregulated by promoter hypermethylation in pancreatic cancer [14]. Lin et al. [15] recently identified SLC17A7 as a tumor suppressor gene in glioblastoma, which inhibited cell proliferation and invasion of the cancer cells. SLC4A4 was found to be significantly downregulated in the clear cell renal cell carcinoma tissues, low expression of which was corelated with poor prognosis [16]. Although their roles in colon cancer have not been clearly elucidated, we speculated that the two solute carrier family genes may server as key prognostic factors of colon cancer pathogenesis.
Claudin 23 (CLDN23) belongs to the claudin family, encoding proteins with four transmembrane domains associated with the formation of tight junctions among adjacent cells [17]. Our study also revealed that CLDN23 was involved in the pathway of cell adhesion molecules and GO associated with plasma membrane, suggesting that it may play a role in the communication and interaction between cells. Previously, a study had reported that CLDN23 is downregulated in tumors of colorectal cancer and the downregulated level is correlated with the prognosis of colorectal cancer patients [18].
For the other four genes (SGCG, CCDC78, OTOP3, and SMPDL3A) in the predicted scoring model, their roles in human cancers have not yet been fully investigated. Nevertheless, the present results showed that all the seven mRNAs had interactions with PVT1. PVT1 encodes a lncRNA, which maps to chromosome 8q24 (8q24) [19]. It is known that 8q24 amplification is a frequent event in various malignant diseases, including colorectal cancer. For instance, the oncogene MYC has been mapped to 8q24 [20]. Shtivelman and Bishop et al. [21] have reported that PVT1 and MYC are co- amplified in colorectal cancer cell lines. A recent study demonstrated that PVT1 can generate antiapoptotic activity in colorectal cancer cells and abnormal expression of this gene could be a prognostic factor in colorectal cancer patients [22]. Yu et al. [23] also suggested that PVT1 functions as an oncogene to promote proliferation and metastasis of colon cancer cells in humans through the miR-30d-5p/RUNX2 axis. Taken together, we speculated that these prognosis-related mRNAs may play roles in colon cancer by interacting with PVT1.
In addition to these gene signatures, our study also identified a clinical factor (stage) associated with the prognosis of colon cancer patients. It is well known that The American Joint Committee on Cancer TNM staging system is currently the gold standard for determining the prognosis of colon cancer patients. The 5-year survival rate for patients with stage I is about 93%, for patients with stage II decreases to 80%, and for patients with stage III is only 60% [24]. Therefore, the identification of clinical prognostic factor of stage in this study further suggested the reliability of our results.
Strength of this study is that we identified a prognostic signature consisting of 7 DEMs and one clinical factor and this prognostic signature could predict the 1/3/5-year survival rate with relatively higher AUC in both training dataset and validation dataset. However, there are some limitations in this study. First, the differential expression of the 7 DEMs were identified from RNA-seq data and lack of experimental validation. Though the RNA-seq data of TCGA are of high quality, further experimental validation is still warranted. Besides, further in vitro and in vivo investigations of the functions of these 7 DEMs in colon cancer are also needed.
In conclusion, our study reveals a seven-mRNA and one-clinical factor signature that is associated with prognosis in colon cancer patients. This signature may serve as a possible candidate biomarker and therapeutic target for colon cancer patients. Pre-clinical studies followed by clinical trials are needed to validate our findings in the future.
Methods
Public data processing
RNA-seq expression profile, miRNA expression data, and clinical phenotype information of all the samples of TCGA colon adenocarcinoma were downloaded from the University of California Santa Cruz (UCSC) Xene database [16] on May 27th, 2019. RNA-seq data from 329 samples, including 288 tumor tissues and 41 normal tissues and miRNA expression data from 261 samples, including 253 tumor tissues and 8 normal tissues were downloaded. Besides, clinical information of 551 patients, including age, gender, tumor histological grading, survival time and survival status, were also downloaded.
LncRNA/mRNA re-annotation
GENCODE database is a scientific project in genome research which was used to identify and map all protein-coding genes within the ENCODE regions [24]. The gtf gene annotation file (Release 26, GRCh38.p10) provided by GENCODE database was downloaded, and the downloaded RNA-seq expression data were re-annotated with mRNA and lncRNA to obtain the mRNA expression profile and lncRNA expression profile, respectively. The gene with annotation information of "protein coding" was retained as mRNA, and with annotation information of "antisense", "sense intronic", "lincRNA", "sense overlapping", "processed transcript", "3prime overlapping ncRNA", or "non-coding" was retained as lncRNA. The clinical phenotype information corresponding to the gene expression profile of the samples were screened.
Differential analysis
The log2(count+1) data were transformed into raw count data and normalized into distribution of same mean value and equal variance for each sample using betaqn function in R package. The differences between tumor and normal samples in the three expression profiles were analyzed using the Bayesian method in limma package (version 3.40.0) [12]. Significance test was performed using the paired t-test to obtain the P values of all the genes. The P values were then adjusted for multiple test using Benjamini & Hochberg (BH) method to obtain the adjusted P values. Log2 fold change (FC) and adjusted P values were used to select the differentially expressed mRNAs (DEMs), and the final DEMs, DELs, and DEMis were screened out.
Function and pathway enrichment analyses of DEMs
Using the enrichment analysis tool DAVID [25] (version 6.8, https://david.ncifcrf.gov/), Gene Ontology (GO) (biological process (BP), cellular component (CC) and molecular function (MF)) analysis was performed, and the results were visualized using GOplot (version 1.0.2) [26]. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was conducted based on the KEGG database [27] using the gene set enrichment analysis (GSEA, version 3.0) [28].
Co-expression analysis
For DELs and DEMis, the Pearson correlation coefficients between them and with the DEMs were calculated, and correlation tests were performed using the corr.test method in R package psych [29] (ci = F, adjust = "BH"). Multiple test was performed using the BH method. The co-expression pairs were obtained according to the correlation coefficient and significance degree, and the co-expression network was constructed using Cytoscape software (version 3.7.1) [30].
Online prediction of miRNA
Using starbase (version 3.0, http://starbase.sysu.edu.cn/) [31], the lncRNAs involved in the obtained lncRNA-mRNA co-expression pairs were subjected to lncRNA-miRNA prediction, thus acquiring the lncRNA-miRNA relation pairs. Additionally, the mRNA-miRNA relation pairs were also predicted based on the mRNAs involved in the obtained lncRNA-mRNA co-expression pairs, using the online tool mirwalk (version 3.0) [32]. Moreover, the intersection of these mRNA-miRNA relation pairs with the above mRNA-miRNA co-expression pairs was screened to obtain the final mRNA-miRNA pairs.
LncRNA and miRNA pathway enrichment analyses
The mRNAs that had co-expression relations with lncRNAs were considered as the target genes of lncRNAs. The lncRNAs with the maximum target genes were subjected to pathway and function enrichment analyses using DAVID [25], and for the lncRNAs with fewer target genes, their functions were searched in genecards (https://www.genecards.org/) [33]. Furthermore, the mRNAs in the mRNA-miRNA pairs were used as the target genes of miRNAs, and the related functions of each miRNA were analyzed in the same way as described above.
ceRNA relation integration and network construction
From the lncRNA-mRNA pairs with co-expression relations, we selected the lncRNA-mRNA pairs regulated by the same miRNA, which were then integrated with miRNAs to construct the ceRNA network. Cytoscape software (version 3.7.1) [30] was used for network construction.
Establishment of mRNA prognostic risk model
The mRNAs in the ceRNA network were used as candidate mRNAs, and the univariable Cox regression analysis in R survival package (version 2.44-1.1) [34] was used to analyze the regression coefficient and P value of each candidate mRNA in relation to survival time and status. The mRNAs with P value < 0.05 were initially considered as the mRNAs related to prognosis.
To further screen the prognostic mRNAs, the calculation model of risk score was defined as follows: Risk score = β gene1*expr (gene 1) + β gene2 * expr (gene 2) + ... + β genen * expr (gene n), Where, β is the prognostic correlation coefficient beta estimated by Cox analysis which equals to log (Hazard Ratio), and expr represents the expression value of corresponding gene.
Thereafter, based on the P values of mRNAs in Cox regression analysis ranking in ascending order, the mRNA with the minimum P value was used as the starting point, followed by adding the other mRNAs (Table 2). The samples were divided into high-risk and low-risk groups according to the median value of risk core. Survival analysis for high-risk and low-risk groups was performed by log-rank test after the addition of a certain mRNA. Then, the receiver operation characteristic (ROC) curves of 1, 3, and 5 years were calculated using the R survival ROC package (version 1.0.3) [35]. The risk score model with the highest Area Under the Curve (AUC) was taken as the best scoring model.
Cox regression analysis of prognostic clinical factors
Important clinical factors (gender, age at initial pathologic diagnosis, anatomic neoplasm subdivision, and pathologic stage) were selected from the phenotypic information corresponding to the samples. Thereafter, an univariable Cox regression analysis was performed by combining the survival information, and regression coefficient, and statistical P value between each clinical factor, and survival time and state were calculated. The influencing factors with threshold of P < 0.05 were further subjected to multivariable Cox regression analysis to obtain the final risk score model.
Model validation using independent datasets
The prognostic signatures developed were further validated in two independent datasets. The first dataset was the RNA-seq expression profile of colon cancer from the UCSC Xene database. This dataset included HTSeq-FPKM data of 512 samples, clinical phenotype information of 570 samples and survival data of 546 samples. The samples in the training dataset or samples without corresponding survival information or tumor stage information were excluded. At last, 164 samples were included for the validation.
The second dataset was GSE17538 downloaded from Gene Expression Ominibus (GEO), which included 213 colon cancer samples with complete information of survival time and survival status [36, 37].
Risk score was calculated for each individual, and the samples in validation datasets were divided into high-risk group and low-risk group according to the median score of risk score. Kaplan-Meier survival curve analysis was then performed to calculate the difference in the survival prognosis time between the samples of the high and low risk groups.
Acknowledgments
We would like to thank Ms. Huaping Liu in assistance with data analysis.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Funding
This work was supported by the National Natural Science Foundation of China project (No. 81874468, No. 81403399), Shanghai Committee of Science and Technology project (No. 16401971400) and Peak Research Team Project in Shanghai University of Traditional Chinese Medicine.
References
- 1. Matusiewicz M, Kosieradzka I, Niemiec T, Grodzik M, Antushevich H, Strojny B, Gołębiewska M. In Vitro Influence of Extracts from Snail Helix aspersa Müller on the Colon Cancer Cell Line Caco-2. Int J Mol Sci. 2018; 19:1064. https://doi.org/10.3390/ijms19041064 [PubMed]
- 2. Kalyan A, Kircher S, Shah H, Mulcahy M, Benson A. Updates on immunotherapy for colorectal cancer. J Gastrointest Oncol. 2018; 9:160–69. https://doi.org/10.21037/jgo.2018.01.17 [PubMed]
- 3. Sun Y, Zheng ZP, Li H, Zhang HQ, Ma FQ. ANRIL is associated with the survival rate of patients with colorectal cancer, and affects cell migration and invasion in vitro. Mol Med Rep. 2016; 14:1714–20. https://doi.org/10.3892/mmr.2016.5409 [PubMed]
- 4. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA
Jr , Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–58. https://doi.org/10.1126/science.1235122 [PubMed] - 5. Davis-Dusenbery BN, Wu C, Hata A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol. 2011; 31:2370–77. https://doi.org/10.1161/ATVBAHA.111.226670 [PubMed]
- 6. Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007; 15:100–08. https://doi.org/10.1007/BF03085963 [PubMed]
- 7. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004; 84:767–801. https://doi.org/10.1152/physrev.00041.2003 [PubMed]
- 8. McDonald RA, Hata A, MacLean MR, Morrell NW, Baker AH. MicroRNA and vascular remodelling in acute vascular injury and pulmonary vascular remodelling. Cardiovasc Res. 2012; 93:594–604. https://doi.org/10.1093/cvr/cvr299 [PubMed]
- 9. Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995; 75:487–517. https://doi.org/10.1152/physrev.1995.75.3.487 [PubMed]
- 10. Sandison ME, Dempster J, McCarron JG. The transition of smooth muscle cells from a contractile to a migratory, phagocytic phenotype: direct demonstration of phenotypic modulation. J Physiol. 2016; 594:6189–209. https://doi.org/10.1113/JP272729 [PubMed]
- 11. Kumar KG, Trevaskis JL, Lam DD, Sutton GM, Koza RA, Chouljenko VN, Kousoulas KG, Rogers PM, Kesterson RA, Thearle M, Ferrante AW
Jr , Mynatt RL, Burris TP, et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 2008; 8:468–81. https://doi.org/10.1016/j.cmet.2008.10.011 [PubMed] - 12. Zhao J, Xu J, Shang AQ, Zhang R. A Six-LncRNA Expression Signature Associated with Prognosis of Colorectal Cancer Patients. Cell Physiol Biochem. 2018; 50:1882–90. https://doi.org/10.1159/000494868 [PubMed]
- 13. Lindqvist BM, Farkas SA, Wingren S, Nilsson TK. DNA methylation pattern of the SLC25A43 gene in breast cancer. Epigenetics. 2012; 7:300–06. https://doi.org/10.4161/epi.7.3.19064 [PubMed]
- 14. Park JY, Helm JF, Zheng W, Ly QP, Hodul PJ, Centeno BA, Malafa MP. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas. 2008; 36:e32–39. https://doi.org/10.1097/MPA.0b013e3181630ffe [PubMed]
- 15. Lin B, Lee H, Yoon JG, Madan A, Wayner E, Tonning S, Hothi P, Schroeder B, Ulasov I, Foltz G, Hood L, Cobbs C. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget. 2015; 6:5369–81. https://doi.org/10.18632/oncotarget.3030 [PubMed]
- 16. Xiao W, Wang X, Wang T, Xing J. MiR-223-3p promotes cell proliferation and metastasis by downregulating SLC4A4 in clear cell renal cell carcinoma. Aging (Albany NY). 2019; 11:615–33. https://doi.org/10.18632/aging.101763 [PubMed]
- 17. Lal-Nag M, Morin PJ. The claudins. Genome Biol. 2009; 10:235. https://doi.org/10.1186/gb-2009-10-8-235 [PubMed]
- 18. Pitule P, Vycital O, Bruha J, Novak P, Hosek P, Treska V, Hlavata I, Soucek P, Kralickova M, Liska V. Differential expression and prognostic role of selected genes in colorectal cancer patients. Anticancer Res. 2013; 33:4855–65. https://doi.org/10.1093/mutage/ger075 [PubMed]
- 19. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–27. https://doi.org/10.1038/nature07672 [PubMed]
- 20. Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, Mao JH, Yu M, Miller MA, Santos JL, Kalloger SE, Carlson JW, Ginzinger DG, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007; 13:5745–55. https://doi.org/10.1158/1078-0432.CCR-06-2882 [PubMed]
- 21. Shtivelman E, Bishop JM. The PVT gene frequently amplifies with MYC in tumor cells. Mol Cell Biol. 1989; 9:1148–54. https://doi.org/10.1128/MCB.9.3.1148 [PubMed]
- 22. Takahashi Y, Sawada G, Kurashige J, Uchi R, Matsumura T, Ueo H, Takano Y, Eguchi H, Sudo T, Sugimachi K, Yamamoto H, Doki Y, Mori M, Mimori K. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br J Cancer. 2014; 110:164–71. https://doi.org/10.1038/bjc.2013.698 [PubMed]
- 23. Yu X, Zhao J, He Y. Long non-coding RNA PVT1 functions as an oncogene in human colon cancer through miR-30d-5p/RUNX2 axis. J BUON. 2018; 23:48–54. [PubMed]
- 24. O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004; 96:1420–5. https://doi.org/10.1093/jnci/djh275 [PubMed]
- 25. Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. https://doi.org/10.1038/nprot.2008.211 [PubMed]
- 26. Walter W, Sánchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015; 31:2912–14. https://doi.org/10.1093/bioinformatics/btv300 [PubMed]
- 27. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. https://doi.org/10.1093/nar/28.1.27 [PubMed]
- 28. Damian D, Gorfine M. Statistical concerns about the GSEA procedure. Nat Genet. 2004; 36:663–64. https://doi.org/10.1038/ng0704-663a [PubMed]
- 29. Jason M. Psych issues. JEMS. 2013; 38:14. [PubMed]
- 30. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–504. https://doi.org/10.1101/gr.1239303 [PubMed]
- 31. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–97. https://doi.org/10.1093/nar/gkt1248 [PubMed]
- 32. Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: an online resource for prediction of microRNA binding sites. PLoS One. 2018; 13:e0206239. https://doi.org/10.1371/journal.pone.0206239 [PubMed]
- 33. Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016; 54:1.30.1–1.30.33. https://doi.org/10.1002/cpbi.5 [PubMed]
- 34. Therneau TM, Grambsch PM. Modeling survival data: extending the Cox model. Springer Science &; Business Media; 2013.
- 35. Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000; 56:337–44. https://doi.org/10.1111/j.0006-341X.2000.00337.x [PubMed]
- 36. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, Lu P, Johnson JC, Schmidt C, Bailey CE, Eschrich S, Kis C, Levy S, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010; 138:958–68. https://doi.org/10.1053/j.gastro.2009.11.005 [PubMed]
- 37. Freeman TJ, Smith JJ, Chen X, Washington MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG, Beauchamp RD. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology. 2012; 142:562–71.e2. https://doi.org/10.1053/j.gastro.2011.11.026 [PubMed]