Introduction
Autism spectrum disorder (ASD) is a complex neurological disorder that affects an individual’s development by impairing social interaction and communication and causes stereotypical behaviors that disrupt the anatomy and functional connectivity in the brain. Most common psychiatric comorbidities found to be associated with autism include anxiety and intellectual disability. Individuals with autism have impaired speech [1, 2] and tend to have limited social interaction mostly due to their own limitation of social skills and due to their failure to understand self-inner mental states [3]. The impairment of speech in affected individuals depends on the severity of the autism disorder as autistic individuals tend to repeat certain words or phrases they hear others say, their speech might sound more formal and they exhibit repetitive behaviors [4]. The prevalence of autism is on the rise and the global prevalence of ASD has been reported to be 1 in 160 persons, according to the World Health Organization (WHO) (2014). Based on a parent survey, the recent prevalence of ASD in the U.S. is reported to be 1 in 45 children [5]. A study conducted in 2006 in the United Kingdom reported an ASD prevalence of 38.9/10,000 in 9 to 10-year-olds [6], while another study conducted by the National Autistic Society (2014) reported that 1/100 children are affected with ASD. In Gulf Cooperation Council (GCC) countries, the prevalence of ASD was reported to range from 1.4–29 in 10,000 individuals [7]. The rise in the prevalence of ASDs is a result of many contributing factors. Some researchers believe in the biome depletion theory, which states that an overreaction of maternal immune response is an underlying factor responsible for the development of ASD in children. In addition, as our immune systems co-evolve with many types of pathogens, a lack of these pathogens within urban and developed areas can cause over-reactivity of the immune system [8]. Other factors include increased exposure to environmental toxins that can damage the genetic structure of an individual, thereby increasing genetic susceptibility (Figure 1) [9–11]. A study by Velasquez-Manoff (2012) reported that autoimmune disorders and immune dysregulation in pregnancy are also found to contribute to the rise of ASD [12]. The genetics involved in autism are of considerable importance as they help us identify various genes, proteins, and signaling pathways found in ASD. The study of genes and genetic changes found in patients with ASD can help unravel the genetic architecture underlying ASD and can aid in early diagnosis and clinical treatment. In the present study, the genomic changes that occur in ASD with different genetic variations will be discussed, and the role of ASD risk genes in gene transcription and translation regulation processes, neuronal activity modulation, synaptic plasticity, signaling pathways, and the novel candidate genes that play a significant role in the pathophysiology of ASD will be examined.
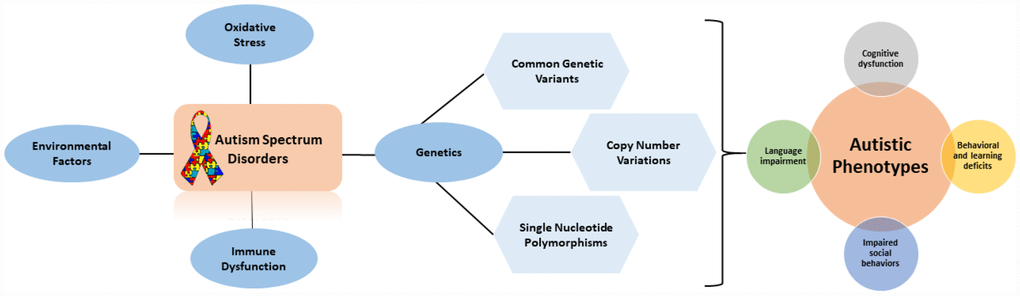
Figure 1. Flow diagram outlining the factors that contribute to autistic phenotypes.
Genetic basis of autism
Many genes associated with ASD are found in circadian entrainment, which indicates a heterogeneous genetic etiology for ASD [13]. Some rare mutations cause the development of syndromic autism. For example, about 30% of patients with Fragile-X Syndrome (FXS) have also been found to develop ASD [14]. A subgroup of patients with Cortical Dysplasia-focal Epilepsy (CDFE), Rett Syndrome and Tuberous Sclerosis Syndrome have also found to express autistic behavior [15, 16]. Copy number variations (CNVs) that involve the deletion or duplication of loci are also responsible for syndromic autism. For example, mutations in SHANK3/PROSAP2 that are found in Phelan-McDermid syndrome are associated with ASD [17]. Mutations in different genes, such as STXBP1, KCNQ4, MYH14, GJB6, COL11A1, UBE3A, KATNAL2, and THRA, have also been associated with ASD [13].
Common genetic variants
Three independent genome-wide association studies (GWAS) have reported genetic variants commonly associated with autism. Out of these 3 studies, 2 have used 0.5 million single nucleotide polymorphisms (SNPs) and have discovered that they are linked at 5p14.1 [18] and 5p15.2 [19] loci. Similarly, in a different study, other associations for rs4141463 have been found at loci 20p12.1. by using one million SNPs [20]. Another ASD risk gene, CNTNAP2, has been found to have a common genetic SNP variation caused by an alteration of functional connectivity in the frontal lobes [21]. A high contribution of these common genetic variants has been associated to autism liability, estimated to be about 40% in simplex and 60% in multiplex families. The contribution of these common variants is of great significance for better diagnosis in autism but the SNPs associated with ASD are still under research [22].
Copy number variations (CNVs)
CNVs are found to be a source of autism risk. A study conducted on autism-affected families reported excess gene duplications and deletions in affected autistic individuals compared to the normal controls [23]. Rare de novo and inherited events found in pathogenic CNVs involved genes associated with autism, such as CHD2 [24–26], HDAC4, and GDI1, SETD5, HDAC9, and MIR137 [23]. CNVs were found to be highly penetrant in females with autism and in individuals with X syndrome protein targets. It was also found that de novo CNV-affected genes converge on neuronal signaling and networks associated with the functioning of synapse and regulation of chromatin [23].
In an ASD gene study, 6 risk loci, namely 1q21.1, 3q29, 7q11.23, 16p11.2, 15q11.2-13, and 22q11.2, associated with autism disorders were reported by analyzing de novo CNVs that were tested within 2,591 families. The study found out that genes within small de novo mutations tend to overlap with high risk genes associated with ASD [27]. Most of the affected individuals were found to carry a de novo causative mutation, as well as deleterious mutations [28]. Gene disrupting mutations, such as frame-shift, splice site, and nonsense mutations, were most frequently found in individuals with ASD [28]. Three percent of the autistic individuals were found to have gene disrupting mutations that were present on both maternal and paternal chromosomes, and 2% of autistic males had a 1.5 fold increase in complete loss of function mutations for X-chromosomes, compared to males without ASD [29].
Gene aberrations associated with ASD
A study associated with the identification of novel candidate genes in ASD-associated pathways revealed several deletions and gene disruptions in many ASD cases, wherein eighteen deletions were detected at the 3p26.3, 4q12, 14q23, and 2q22.1 regions [30]. Candidate genes associated with GABAergic signaling and neural development pathways were revealed by the evidence provided by case specific CNVs. These genes include a GABA type A receptor associated protein (GABARAPL1), a postsynaptic GABA transporter protein (SLC6A11), and a GABA receptor allosteric binder known as diazepam binding inhibitor (DBI). A genetic overlap between ASD and other neurodevelopmental disorders was also reported, including genes such as GRID1, GRIK2, and GRIK4, which include glutamate receptors, NRXN3, SLC6A8, and SYN3, and are responsible for synaptic regulation. These CNVs are associated with ASD heritability and can help to uncover new etiological mechanisms underlying ASD [30].
Genetic variation in ASD
There is a substantial variation in the ASD genetic architecture and the heterogeneity of ASD is due to the genetic variability that underlies this disorder. A single mutation is enough to cause ASD and several thousand low-risk alleles can contribute to the development of ASD [31]. There are many rare variants that can contribute to the risk of developing ASD and there is extreme locus heterogeneity in ASD due to copy-number variant data and mutations involving the alteration of proteins [32]. Many of the ASD genes share a common pathway that affects neuronal and synaptic homeostasis. For example, social impairment and speech problems in ASD individuals are due to a single copy mutation SHANK3 [33]. This shows that many of the ASD associated genes are part of a large number of molecular pathways or mechanisms that are related to other neuropsychiatric conditions [34].
Novel candidates in ASD
Many mutations have been reported in CHD8, an ATP-dependent chromodomain helicase responsible for the regulation of CTNNB1 [35] and p53 pathway [36]. CHD8 has been investigated in many exome studies and is considered as a novel candidate for ASD [37–39]. In addition, the SCN2A gene, which encodes a voltage-gated sodium channel, plays an important role in the generation of action potentials in neurons. These mutations are most frequently found in Identity Disorder (ID), with some cases also showing signs of autism [40, 41]. In autism probands, three truncating mutations of GRIN2B and SYNGAP1 and TBR1 associated with dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) play an important role in excitatory signaling in ASD. While, GRIN2B is associated with learning and memory, and targeted sequencing has linked it with various neurodevelopmental disorders, including ASD [38].
An ASD study showed the differential expression of autism candidate genes in lymphoid cell lines of autistic individuals [42]. The study reported the upregulation and increased expression of protein argininosuccinate synthetase (ASS), which acts as an important brain signaling molecule and is involved in controlling the rate-limiting step in the production of nitric oxide (NO), is associated with ASD [42]. An upregulation in the mRNA expression levels of ITGA2B encoding glycoprotein αIIβ was found in ASD affected individuals. This elevated level of expression can disrupt the cellular morphology in affected individuals [43]. Another autism study that identified abnormal brain gene expression patterns in autistic brains via whole-genome analysis of mRNA levels and CNVs reported that in young autistic individuals, the highly dysregulated pathway was the adenosine A2A receptor-signaling pathway [44]. Adenosine receptors play an important role in the development and functioning of the brain, as well as the synaptic plasticity, motor, and cognitive function and neuronal stem cell proliferation regulation [44]. Many studies have reported the association between Purkinje cells (PCs) and ASD [45–47]. Additionally, many studies have reported a significant reduction in the number of PCs in the post-mortem brains of patients who were affected with ASD [48–51]. Lower expression of neurotrophins NT3 and NT4, which play an important role in the climbing fiber system development in PCs, was also reported in affected autistic individuals [52]. Downregulation of neural and muscle specific alternative splicing regulator (A2BP1/FOX1) [53] and the MOCOS gene was also reported in ASD individuals [54]. Language and speech disorders in ASD were found to be result of mutations in the FOXP2 and CNTNAP2 genes [55]. The FOXP1 gene, which is a transcription repressor and is important for normal brain development and functioning, was found to be elevated in individuals with ASD. This indicates the involvement of FOXP1, FOXP2, and CNTNAP2 genes in the pathogenesis of ASD [56]. Synaptic components such as Nav1.2 channel, which is a voltage-gated ion channel involved in action potential propagation, neuronal pacemaking and Cav1.3 channel, which is an excitability-transcription coupling were found to be mutated in autistic patients [57]. Another ASD study showed the presence of an additional copy of 22q13/SHANK3 in a boy with Asperger syndrome that caused severe social communication impairment [58]. The SHANK3 gene is involved in the enlargement of dendritic spine heads as evidenced in mice [59]. There have also been reports of X-linked mutations in NLGN3 and NLGN4 in ASD that affect synapse formation and maintenance, which is important for speech development and social communication [58]. An ASD study that identified rare variants in mGLUR signaling pathway reported rare and deleterious variants in the SHANK3, TSC1, and TSC2 genes in non-syndromic autism individuals [60]. HOMER1, which is an autism risk gene, is considered an important component of the postsynaptic density (PSD) proteins network. This network creates a link between gene products associated with autism and neuroligins [60]. Disruption of mGluR5-Homer1 interactions can cause the development of phenotypes associated with autism [61]. Mutations in SYNGAP1, which is involved in the negative regulation of the Ras/ERK pathway and synaptic transmission, have also been identified in ASD [60]. The UBE3A and GABA receptor genes, which are expressed in the central nervous system, are located at the15q11-13 locus, and are also associated with autism [62]. O’Roak, Vives, Fu, et al. (2012) also discovered that six genes (GRIN2B, TBR1, CHD8, PTEN, TBL1XR1, and DYRK1A) contribute to 1% of ASDs and were found to be sporadic. Parikshak et al. [63] found that genes associated with autism tightly bind together in modules that are responsible for human cortical development and biological functions, including transcriptional regulation and the development of synapses.
Disruption of the NRXN1 gene in autism reported that in autistic individuals, the amino acid alterations in the NRXN1 gene are not frequently present as compared to non-autistic individuals in an NRXN1 gene coding sequence scan [64]. Two missense changes seen in the residues of the leader sequence of a-NRXN1 and epidermal growth factor (EGF)-like domain suggests these changes in NRXN1 might be a contributing factor in developing autism [64].
Autistic phenotypes
To identify the genes affected by rare de novo CNVs, in autism, a network-based analysis of genetic associations (NETBAG) is used [65]. The network forming genes are associated with autism and are involved in the development of synapse, neuron motility and axon targeting. In addition to the WNT signaling pathway, which is responsible for neural circuits formation and dendrite morphogenesis regulation, there is a reelin signaling pathway that plays a significant role in neuron motility and autistic phenotypes [66, 67]. This network also includes deleted in colorectal carcinoma (DCC) protein, which is responsible for guiding the axon in autism disorder [68]. In addition, other proteins involved in the regulation of actin network that are used in axonal morphogenesis, such as p21-activated kinase (PAK) and LIM-domain containing protein kinase (LIMK) and any malfunction in these proteins influences autistic phenotypes as they play a significant role in dendrite/axon signaling [65]. Autistic individuals are also found to have increased density of spine in portions of cerebral cortex with over-connectivity in local brain regions [69, 70]. This indicates that malfunctioning of the neuronal and synaptic connectivity is central to autism, and several genes could be related to postsynaptic density and actin remodeling that could contribute to autistic phenotypes [65]. Also, higher ASD rates have been reported in individuals with low copy repeats LCR-A to B region deletion compared to individuals without deletions. This might contribute to autistic phenotypes in disorders related to 22q11.2 (also known as DiGeorge syndrome) in addition to decreased adaptive functioning [71].
There are ASD risk genes that contribute to autistic phenotypes and are associated with different lobes of the brain (Figure 2). The frontal lobe is the largest and controls different cognitive functions such as memory, behaviors, language and voluntary movements. The parietal lobe controls sensorimotor planning, language and touch. The temporal lobe is mainly involved in controlling the semantic and recognition memory and the occipital lobe, which is the smallest lobe in the brain mainly controls visual processes [72]. One of the most important ASD risk gene is CNTNAP2 and it is found to be associated with frontal and occipital lobes of the brain [70, 73]. While NRXN1 is found to be linked with parietal and frontal lobes [74]. FOXP2 is found to be linked with temporal lobes and MET in occipital and temporal lobes [75]. Genetic findings have associated cadherins as an ASD risk gene [18, 76–78] and a recent study identified the expression of cadherins CDH9 and CDH11 in the ASD-relevant areas of the cerebellum in mice and reported high expression of these genes in segregated populations of the Purkinje cells present in the cerebellum [79].
![Diagram showing ASD risk genes and autistic phenotypes associated with different lobes of the brain. CNTNAP2 is found to be associated with frontal and occipital lobes of the brain [70, 73]. NRXN1 is found to be linked with parietal and frontal lobes [74]. FOXP2 is found to be linked with temporal lobe and MET in occipital and temporal lobes [75]. Cadherins (CDH9 and CDH11) are found to be linked with the cerebellum region [37].](/article/102473/figure/f2/large)
Figure 2. Diagram showing ASD risk genes and autistic phenotypes associated with different lobes of the brain. CNTNAP2 is found to be associated with frontal and occipital lobes of the brain [70, 73]. NRXN1 is found to be linked with parietal and frontal lobes [74]. FOXP2 is found to be linked with temporal lobe and MET in occipital and temporal lobes [75]. Cadherins (CDH9 and CDH11) are found to be linked with the cerebellum region [37].
Gene regulation in ASD
DNA methylation
A global methylation profiling study in lymphoblastoid cell lines derived from autistic monozygotic twins and their non-autistic siblings revealed decreased expression of RORA and BCL-2 proteins in autistic individuals as compared to the controls [80]. The study confirms how DNA methylation in idiopathic autism affects the epigenetic regulation of gene expression and the molecular changes associated with brain pathobiology in ASD [80].
Postsynaptic translational regulation
Postsynaptic density plays a critical role in the neural transmission and maturation of synapsis and forms the basis of the etiology of ASD [81]. The encoded proteins of autism-associated genes located in the postsynaptic density are associated with FMRP, and there is a localization of mutated protein found in Fragile-X syndrome, which is responsible for the synthesis of proteins at the postsynaptic density [82]. The SHANK2 and SHANK3 genes are found in the postsynaptic density, which bind to neuroligins and are involved in the glutamatergic response in ASD, as well as in language and social cognition development [83]. Other candidate genes of autism include NF1, PTEN, MET, TSC1, TSC2, and CYFIP1, that are located in the duplication region (15q11–13) [84–87]. Mutations in ASD genes also implicate the protein metabolism at the synapse that is modulated by the ubiquitination pathways [88]. For example, UBE3A, which is the Angelman syndrome gene, has an important role in this pathway, in addition to genes such as FBXO40, RFWD2, USP7, and PARK2 [89, 90]. This indicates that remodeling and maintenance of the synapse functioning is an important determinant in the pathology of ASD [91].
Modulation of neuronal activity
Mutations in neurexin and neuroligin families are associated with the pathophysiology of ASD [92]. Together, neurexins and neuroligins are involved in the modulation of excitatory and inhibitory synaptic functions [93]. The genes of these super families that play a significant role in ASD include NRXN1, NLGN1, NLGN3, CNTN4, CNTN6, and CNTNAP2 [94]. Neuronal activity is influenced by genes such as GRIN2B, SCN1A, and SCN2A, and they are involved in the mediation of synaptic plasticity. In addition, they encode for ion channels [32]. Neuronal activity that regulates transcription factors also regulates other genes, including UBE3A, PCDH10, DIA1, and NHE9/SLC9A9 [95, 96]. Imbalances in excitation and inhibition in brain regions have been found in knockout ASD mouse models of genes, such as NRXN1, SHANK3, FMR1, CNTNAP2, and these knockout models had social interaction impairments and reduced ultrasonic vocalizations that overlapped behavioral endophenotypes relevant to ASD [91].
Synaptic plasticity
Synaptic plasticity is affected by cytogenetic abnormalities, such as the duplication of maternal allele 15q11–q13 and genetic syndromes like Rett or Fragile-X syndrome associated with ASD. In idiopathic autism, most commonly identified synaptic gene mutations include NLGN4X [96, 98] or SHANK3 [33] and NLGN3. Abnormalities in the formation of synapse and disrupted pathways, such as GTPase/Ras signaling and neurogenesis, are revealed by the analysis of genes affected by rare CNVs [99, 100]. The identification of specific ASD genes are found to be resisted by some de novo or inherited CNVs, for example, the 16p11 region, recur at the same locus in individuals who are not related [101]. This indicates a locus and allelic heterogeneity in ASD [101]. Another X-linked gene GLRA2 deletion has been identified in autism disorder [102]. This gene encodes the glycine receptors (GlyR) α2 subunits. These glycine receptors are involved in the mediation of inhibitory neurotransmission in the nervous system. Mutations in GLRA2 results in synaptic plasticity, language delay, cognitive and social impairments, as well as altered glycinergic signaling [102]. Mutations in Calcium voltage-gated channel subunit alpha1 C (CACNA1C) might contribute to NMDA-receptor independent synaptic plasticity associated with ASD [103]. A study showed that mutations in CACNA1C cause alterations in calcium homeostasis that contribute to the development of ASD [104].
Genes and brain connectivity
Genes involved in ASD are related to each other within many processes, including neuronal and synaptic development, modulation, protein synthesis, calcium signaling, oxytocin pathways, mTOR, and various transcriptional mechanisms (Figure 3) [105]. ASD potential endophenotypes include brain connectivity and morphology alterations [106]. A study by Frazier et al. (2014) on ASD subjects carrying heterogenous germline PTEN mutations showed cognitive malfunctioning and abnormalities in the white matter with reductions in PTEN protein compared to the healthy controls [117]. Alterations were also found in the gene-brain pathway based on the rs1858830 MET risk allele by differences seen in activation and deactivation patterns of fMRI in response to social stimuli, as well as the structural and functional connectivity in the temporal-parietal region of the brain in ASD subjects [108].
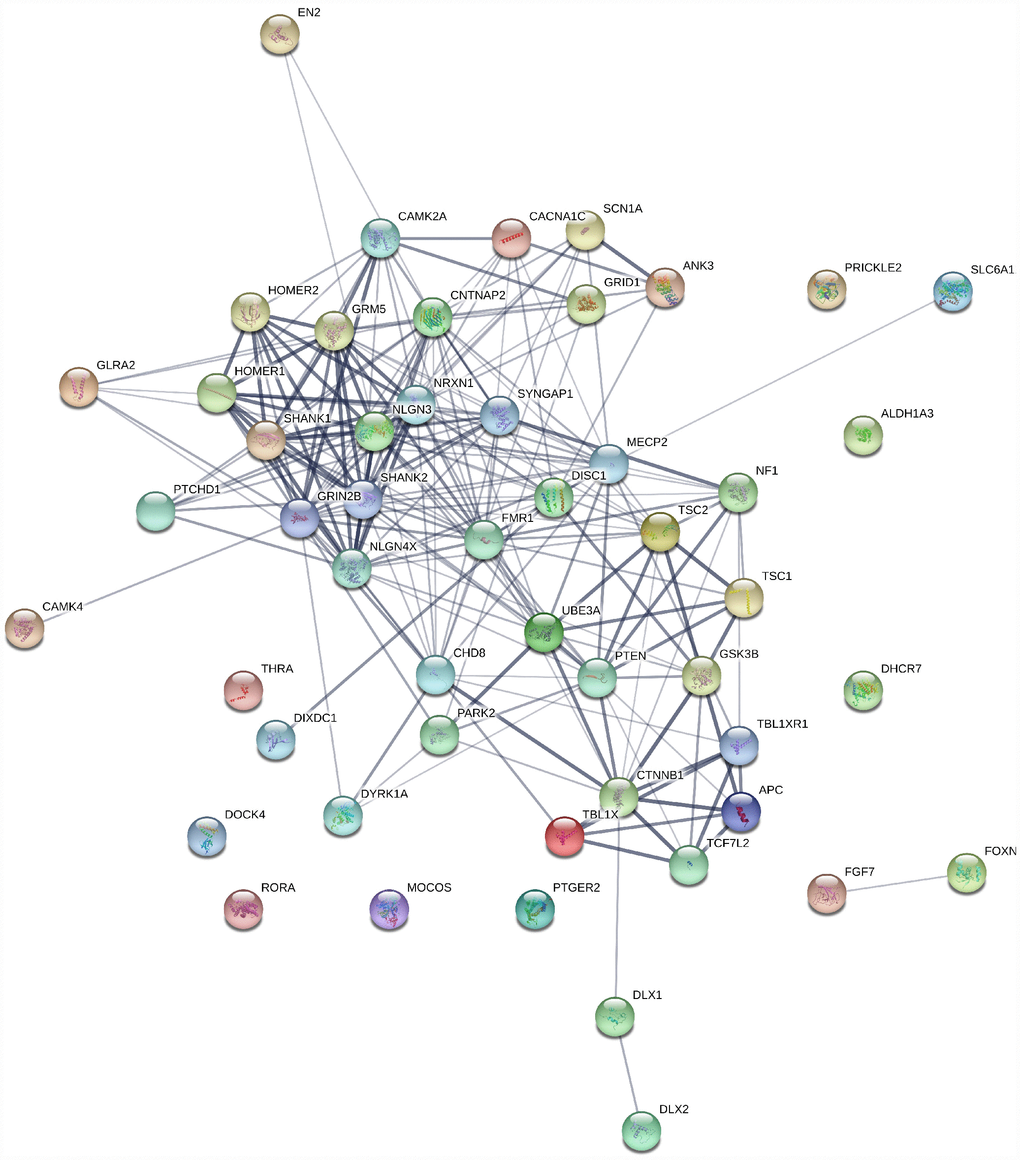
Figure 3. Gene interaction map for ASD genes generated using string1 webserver. Thickness of the line indicates the strength of the interaction between the genes. All sources are used to generate the interaction model with default medium confidence interaction score of 0.4. Ref: https://string-db.org/.
Genome-wide association study (GWAS)
Many GWASs have been performed in ASD [89, 109–111]. Within the intergenic region between CDH9 and CDH10 cell adhesion genes, a linkage disequilibrium block has been reported [18]. Another gene called Semaphorin 5A (SEMA5A), which plays an important role in axonal guidance and the development of neurons is found to be a susceptibility gene for ASD as a study has found a de novo microdeletion of SEMA5A in ASD and ID patients [112]. There was another genome-wide significance reported in the macro domain containing 2 (MACROD2) gene at an intronic SNP [113]. Replication and identification of a common variation on chromosome 5p14.1 associated with autism was reported by another GWAS study [57, 114].
Wnt-signaling in ASD
Wnt-signaling plays an important role in the differentiation and morphology of neurons, and neurotransmission [115]. This pathway is found to be dysregulated in individuals affected with ASD [116]. Dysregulation of this pathway is found to affect the cortical and spine patterning and morphology with cytoarchitecture disruption in the cortex of the affected brains of autistic individuals [117, 118]. Glycogen Synthase Kinase-3 (GSK3), which is a Wnt-signaling pathway component, plays an important role in ASD development as it was found to be hyperactive and caused impaired social interaction and increased anxiety in a GSK3 knockout mouse model [119]. Another gene, known as CHD8, which regulates the Wnt signaling pathway and promotes transcriptional factor activity in the brain, is found to be associated with ASD [120]. Knockout of CHD8 gene during cortical development resulted in the downregulation of the CHD8 gene, which lead to the reduction of the TCF/LEF transcription factor family, causing a defect in the development of the brain [121] (Figure 4). This proves that the dysregulation of the Wnt-signaling pathway affects the cortical patterning and synaptic development in individuals with ASD [122].
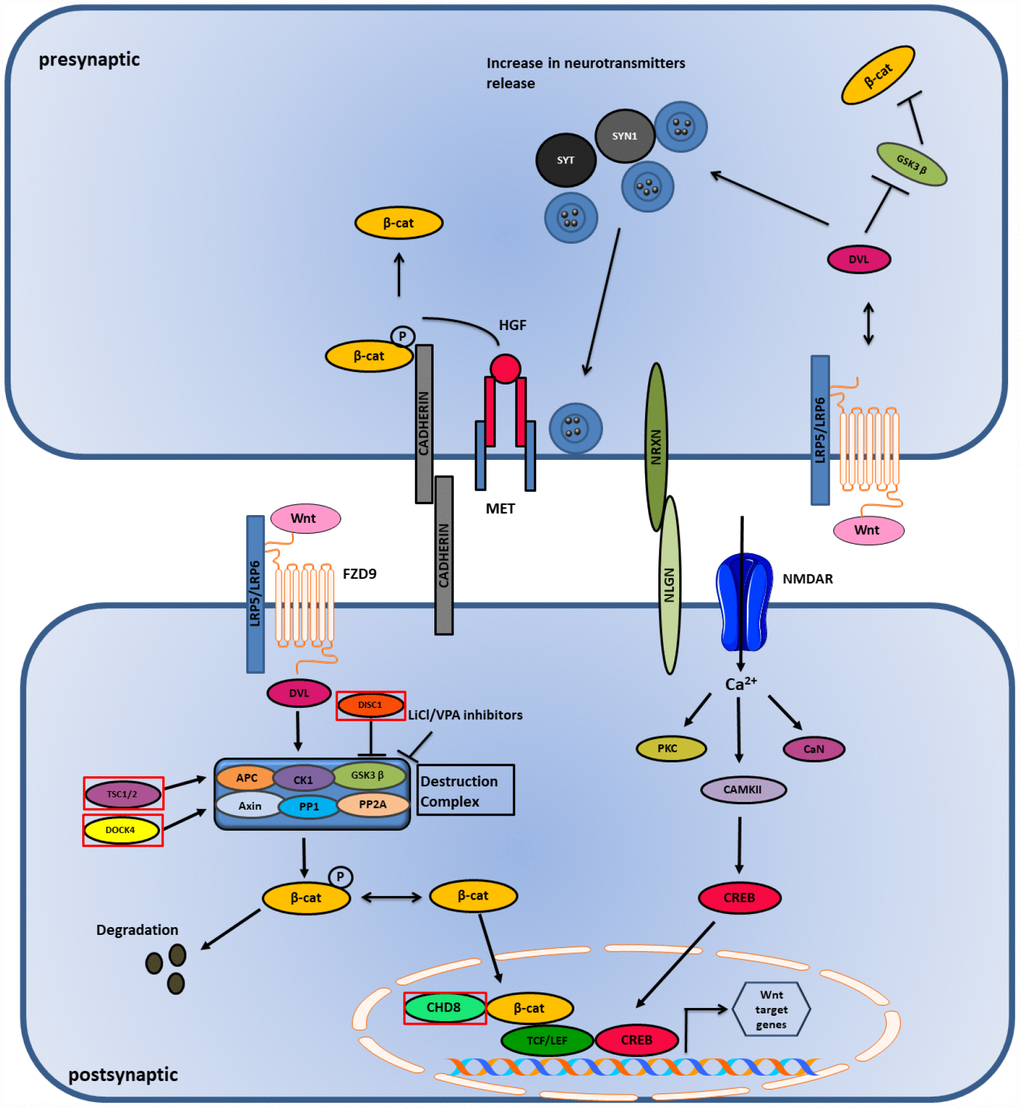
Figure 4. Wnt and Ca2+ pathway in ASD. Wnt binds to LRP5/LRP6 receptor and stabilizes β-catenin in the nucleus and cytoplasm. High influx of Ca2+ ions causes activation of CAMK and CREB genes which initiates transcription in the postsynaptic side. Genes mutated in ASD are shown in red boxes.
The DISC1 gene is associated with Wnt signaling and ASD and is involved in the inhibition of GSK3β that leads to the activation of β-catenin; thus, it acts as a positive regulator of the Wnt/β-catenin signaling [123]. Mutations in DISC1 fail to inhibit GSK3β and cause suppression of the Wnt/β-catenin signaling pathway [124]. Mutations in an intracellular Wnt/β-catenin signal pathway protein, DIX domain containing-1 (DIXDC1), displayed impairment in social behavior, coupled with anxiety and depression in mice models [125] and also showed reduction in the dendritic spines and glutamatergic synapses in brains of the experimental mice. It has been suggested that the functional sequence variants of DIXDC1 can manifest as behavioral syndromes in human due to its restricted tissue distribution property in the late developmental and postnatal central nervous system [126]. The TSC1 and TSC2 genes are also found to play a significant role in Wnt signaling [127]. These genes are associated with GSK3β and Axin and help in the degradation of β-catenin leading to the inhibition of Wnt/β-catenin mediated gene transcription. Mutations in these genes are found to increase Wnt signaling, while overexpression of these genes caused reduction in Wnt/β-catenin signaling [119, 128, 129]. Dedicator of cytokinesis 4 (DOCK4) is another gene that plays an important role in Wnt signaling [123]. DOCK4 is a part of the destruction complex and its decreased expression is found to reduce the transcriptional activity induced by Wnt signaling [123]. In autism, levels of DOCK4 are found to be diminished, which leads to reduced Wnt signaling and the growth of dendrites [123, 130]. Several genes belonging to the non-canonical Wnt signaling pathway are found to be associated with autism [119]. The Prickle2 (Pk2) gene interacts with post synaptic density 95 (PSD-95). Mouse models with Prickle2 mutations displayed altered social, learning, and behavioral abnormalities, in addition to reductions in dendrite branching. Mutations in Ankyrin-3 (ANK3) and Prostaglandin E2 (PGE2) genes are associated with ASD [131–133]. ANK3 and PGE2 genes are important in neuronal development. PGE2 is regulated by Cyclooxygenase-2 (COX2) and is the main regulator of PGE2 synthesis. Studies have shown there is an association of abnormal COX2/PGE2 signaling with ASD [133]. All the findings suggest that both activation and inhibition of Wnt signaling pathway are associated with autism risk.
Presynaptic Wnt signaling
Canonical Wnt signaling or Wnt/β-catenin signaling plays an important role in the development of synapsis in the pre- and post-synaptic terminals. The release of neurotransmitters at the presynaptic terminal is triggered by the binding of Wnt ligand to a receptor that activates Dishevelled-1 (DVL1). This binds to the pre-synaptic proteins, such as Synapsin-1 (SYN1) and Synaptotagmin (SYT), that are associated with ASD and enhance the clustering of synaptic vesicles (SV’s) and release of neurotransmitters. On the other hand, cell adhesion proteins, such as cadherins and cell adhesion complexes (NLGN/NRXN), play an important role in the modulation of presynaptic terminal activity. Their interaction regulates both excitatory and inhibitory synaptic function, disrupting the E/I balance in the postsynaptic neurons. β-catenin is bound to cadherins and their interaction is essential for the recruitment of SVs to synapses. MET, a receptor for hepatocyte growth factor (HGF), is found to have a genetic link with ASD. MET phosphorylates Tyr-142 of β-catenin and promotes its dissociation from the cadherins leading to its release in the postsynaptic terminal [119] (Figure 4). Wnt/β-catenin signaling plays a significant role in ASD as it is involved in stabilizing the synaptic structure by enhancing clustering of SVs, release of neurotransmitters and modulation of cell adhesion complexes [134].
Postsynaptic Wnt signaling
Activation of the Wnt/β-catenin signaling causes Wnt ligands to bind to the Frizzled-9 (FZD9) receptor that recruits the multi-protein destruction complex consisting of scaffolding proteins (APC and Axin), phosphatases (PP1 and PP2A), and kinases (GSK3β and CK1). This activation inhibits GSK3β and prevents the degradation of β-catenin, thus stabilizing it and translocating it to the nucleus, leading to gene transcription [123] (Figure 4). Conversely, canonical and non-canonical Wnt signaling are associated with Ca2+ signaling. Wnt ligands are found to increase the influx of Ca2+ in neurons, and voltage gated Ca2+ sensitive channels, like NMDAR, allows the entrance of Ca2+ in the postsynaptic membrane that allows long term synaptic potentiation (LTP) establishment. Both high and low responses of LTP are associated with ASD. Ca2+ causes the activation of CAMKII, which is involved in the reorganization of cytoskeleton, calcineurin (CaN), a calcium dependent protein phosphatase and protein kinase C (PKC). The activation of CAMKII leads to the activation of transcription factor CREB, causing its translocation to the nucleus, which leads to gene transcription [135].
Hyperactive pro-growth signaling pathways involved in ASD
mTOR
mTOR, a highly conserved serine/threonine kinase, is important in the regulation of cell growth, cell metabolism and cell survival processes [136]. The catalytic subunits of mTOR consist of two structurally and functionally distinct protein complexes known as mTORC1 and mTORC2, which are involved in integrating information in the brain received from various intracellular and extracellular responses [137]. As mTOR is shown to be a major role player in the regulation of various processes that control synthesis of proteins, dynamics of actin cytoskeletal, regulating energy homeostasis and metabolism, the dysregulation of these pathways can lead to disorders including ASD, and neurodegeneration [138–140]. The disruption of mTOR pathway is associated with ASD and several other disorders that are caused by genetic alterations in mTOR pathway members, such as TSC1, TSC2, and PTEN. Also, mTOR activity levels can serve as indicators of disrupted growth states in the brain [141]. Studies have shown that several mTOR substrates such as p70 ribosomal S6 kinase 1 (S6K1) and the eukaryotic translation initiation factor 4E-binding proteins (4E-BPs) are found to be associated with ASD [142]. In particular, deletion of 4E-BP2, which is also a downstream effector of mTOR results in elevated dendritic spine density, and various behavioral abnormalities that are reminiscent of ASD [143–146]. mTORC1 complex promotes the ribosome production and translation by phosphorylating S6K1 through various effectors [147]. Hyperactivation of mTORC1 is responsible for ASD symptoms as one study has showed that mTORC1 hyperactivation in cerebellar Purkinje cells resulted in autistic like behaviors in mice [148] and it can be targeted for the treatment of ASD by modulating its expression.
Brain- derived neurotrophic factor (BDNF)
Neurotrophins play an important role in the nervous system and are involved in the regulation of neuronal development, survival, morphogenesis, differentiation and synaptic plasticity [141, 149]. The most abundant neurotrophin that is found in the central nervous system (CNS) is the brain- derived neurotrophic factor (BDNF) [149]. The control of neurotrophin signaling at different epigenetic, transcriptional and translational levels are critical for the overall connectivity of neurons and other physiological functions, such as cell fate, axon and dendritic growth and synaptic pruning [150, 151]. The dysregulation of neurotrophin signaling has been reported in various neurological disorders, such as Alzheimer’s, Huntington’s disease and autism [152–154]. Under normal conditions, receptors such as FMRP, TSC1/2, and PTEN regulate the excitatory activity induced by BDNF by acting on its receptor TrkB. However, in pathological conditions such as ASD, lack of this regulation leads to disruption of synaptic functions [155]. The pro-growth signaling pathways induced by other trophic factors such as insulin-like growth factor (IGF), vascular endothelial growth factor (VEGF), glial-derived neurotrophic factor (GDNF) and ciliary neurotrophic factor (CNTF) are found to be dysregulated in ASD and used to categorize ASD into neural overgrowth or undergrowth types [141].
ERK/MAPK
ERK1/2 are members of the MAPK signaling cascade and play important role in the regulation of cell growth, proliferation, differentiation and apoptosis [156]. MAPK/ERK pathway is involved in the signal transduction from cell surface receptors to the nucleus and they respond to growth factors, oxidative stress, chemokines and cytokines [157]. The activation of ERK1/2 is essential for dendritic spines formation and stabilization [158, 159] as well as supports the development of cerebral cortex by regulating cell cycle in neural progenitor cells proliferation. It is observed that the ERK/MAPK signaling interacts with many genes and CNVs implicated in ASD [160]. Activated or blocked levels of phospho-ERK1/2 are found to be associated with autistic phenotypes [161, 162]. Thus, targeting the ERK/MAPK pathway can be used to treat cognitive and behavioral impairments implicated in ASD [160] and a recent study performed in a mouse model with 16p11.2 deletion, which is a CNV associated with autism, showed that treatment with ERK inhibitor during period of development rescued anatomical and behavioral deficits in the mice [163].
Other signaling pathways involved in ASD
There are many ASD genes that have been associated with different signaling pathways and that contribute to different ASD phenotypes (Table 1). One such pathway is the Calcium (Ca2+) and calmodulin (CaM) signaling pathway. These pathways play a significant role in the connectivity of neurons and functioning of the synapse, so dysregulation of this pathway might be responsible for the development of ASD [164, 165]. Impaired Ca2+ signaling has been found in many ASD individuals [166–168]. In skin fibroblasts derived from ASD individuals, the agonist evoked Ca2+ signaling was found to be dysfunctional [167]. Ca2+ signaling dysregulation and activity-dependent gene transcription changes were reported in a study involving induced Pluripotent Stem Cells (iPSC) that were derived from ASD individuals diagnosed with Timothy syndrome [169]. A study identified the common variants of ASD risk genes that regulated FMRP signaling showed that a SNP in the calcium/calmodulin-dependent kinase IV (CaMKIV) gene, which is a positive regulator of the FMRP transcription, causes a higher risk for the development of ASD [170]. Similarly, a de novo mutation of Glu183 to Val (E183V) in the catalytic domain of CAMKIIα increased dendritic branching and decreased synaptic transmission with reduced dendritic spine density causing ASD-related behaviors in mice [171].
Table 1. Studies showing ASD associated genes that contribute to ASD phenotypes through different signaling pathways.
| Genes in ASD | Genes affecting signaling pathways | Mutations contributing to autistic phenotypes | References |
| Calcium/Calmodulin Dependent Protein Kinase IV (CAMKIV) | CaM signaling | Deficits in learning and memory formation | [13, 119] |
| Calcium/Calmodulin Dependent Protein Kinase II (CAMKIIα) | CaM signaling | Memory impairment | [13, 171, 220] |
| Synaptic Ras GTPase Activating Protein 1 (SYNGAP1) | Excitatory/glutamatergic signaling | Non-syndromic mental retardation | [42, 221] |
| Glutamate Ionotropic Receptor NMDA Type Subunit 2B (GRIN2B) | Excitatory/glutamatergic signaling | Deficits in learning and memory | [42, 222] |
| Fibroblast Growth Factor 7 (FGF7) | FGF signaling | Epileptic seizures | [116, 184] |
| Metabotropic glutamate receptor (mGLUR5) | FGF signaling | Aberrant dendrite growth leading to cognitive abnormalities | [119, 223, 224] |
| Sodium Voltage-Gated Channel Alpha Subunit 1 (SCN1A) | GABA signaling | Cognitive and behavioral deficits | [225, 236] |
| Methyl-CpG Binding Protein 2 (MECP2) | GABA signaling | Cognitive and behavioral deficits, impaired coordination | [194] |
| Solute Carrier Family 6 Member 11 (SLC6A11) | GABA signaling | Cognitive deficits | [30, 227] |
| Neurexin 1 (NRXN1) | GABA and glutamate signaling | Cognitive impairments, behavioral and learning deficits | [228–230] |
| Glutamate Ionotropic Receptor Delta Type Subunit 1 (GRID1) | Glutamate signaling | Impaired emotional and social behaviors | [231] |
| Calcium Voltage-Gated Channel Subunit Alpha1 C (CACNA1C) | Glutamate signaling | Impaired memory, hippocampal plasticity and anxiety-related behavior | [232] |
| SH3 And Multiple Ankyrin Repeat Domains 1 (SHANK1) | Glutamate signaling | Increased anxiety, reduced long-term memory | [31] |
| SH3 And Multiple Ankyrin Repeat Domains 2 (SHANK2) | Glutamate signaling | Increased anxiety, impaired social behaviors | [31] |
| Glycine Receptor Alpha 2 (GLRA2) | Glycinergic signaling | Deficits in learning and memory | [102] |
| Tuberous Sclerosis Complex Subunit 1 and 2 (TSC1 and TSC2) | mTOR signaling pathway | Learning deficit and impaired social behavior | [233] |
| Neurofibromin 1 (NF1) | mTOR signaling pathway | Learning and attention deficits | [233] |
| Fragile X Mental Retardation 1 (FMR1) | mTOR signaling pathway | Cognitive deficits, increased anxiety | [233] |
| Contactin Associated Protein Like 2 (CNTNAP2) | mTOR signaling | Impaired social and repetitive behaviors | [234, 235] |
| Phosphatase and Tensin Homolog (PTEN) | mTOR signaling pathway | ASD like social behavior | [233] |
| Homer Homolog 1 HOMER1 | mGLUR signaling | Learning and memory deficits | [60, 236] |
| Molybdenum Cofactor Sulfurase (MOCOS) | Purine metabolism pathway | Autistic features | [54] |
| Retinoid-Related Orphan Receptor-Alpha (RORA) | Retinoic acid (RA) signaling | Language impairment | [80, 237] |
| Forkhead Box N1 (FOXN1) | Retinoic acid (RA) signaling | Brain alterations contributing to autistic features (hypothetical) | [238, 239] |
| Aldehyde Dehydrogenase 1 Family Member A3 (ALDH1A3) | Retinoic acid (RA) signaling | Autistic traits | [183] |
| Patched Domain Containing 1 (PTCHD1) | Sonic hedgehog (SHH) signaling | Cognitive alterations | [116, 175] |
| 7-Dehydrocholesterol Reductase (DHCR7) | Sonic hedgehog (SHH) signaling | Intellectual impairment | [116, 177] |
| Engrailed Homeobox 2 (EN2) | Sonic hedgehog (SHH) signaling | Deficits in social behavior | [116, 240] |
| Distal-Less Homeobox (DLX) | TGF-β/BMP signaling | Autism like behaviors | [116, 241, 242] |
| Thyroid Hormone Receptor Alpha 1 (THRA1) | Thyroid pathway | Impaired memory, anxiety, locomotor dysfunction | [243, 244] |
| Parkinsonism Associated Deglycase 2 (PARK2) | Ubiquitin pathway | Impaired speech and stereotypical behaviors | [89, 245] |
| Chromodomain Helicase DNA Binding Protein 8 (CHD8) | Wnt signaling (canonical) | Defective neural progenitor proliferation and differentiation | [121] |
| Catenin Beta 1 (CTNNB1) | Wnt signaling (canonical) | Defect in brain development | [246, 247] |
| Prickle Planar Cell Polarity Protein 2 (PRICKLE2) | Wnt signaling (non-canonical) | Abnormalities in behavior, learning and social interaction | [248] |
| Transducin Beta Like 1 X-Linked (TBL1X) | Wnt signaling | Intellectual disability and autistic features | [249] |
| SH3 And Multiple Ankyrin Repeat Domains 3 (SHANK3) | Wnt signaling | Delayed or absent speech, intellectual disability | [58, 246, 250] |
| Adenomatosis Polyposis Coli (APC) | Wnt signaling | Memory impairment, autistic behaviors | [251] |
| Ubiquitin Protein Ligase E3A (UBE3A) | Wnt signaling | Developmental delay, learning difficulties | [252, 253] |
| Glycogen Synthase Kinase 3 Beta (GSK3β) | Wnt signaling | Anxiety and impaired social interaction | [13, 254] |
| Disrupted in Schizophrenia 1 (DISC1) | Wnt signaling | Failure is establishment of long-term synaptic potentiation (LTP) causing learning and memory deficits | [119] |
| Dedicator of Cytokinesis 4 (DOCK4) | Wnt signaling | Suppression of dendrite growth causing impairments in cognitive and language abilities | [123] |
| Transcription Factor 7 Like 2 (TCF7L2) | Wnt signaling | Cognitive and sensorimotor impairments | [119, 255] |
| Neuroligin 3 and 4 (NLGN3 and NLGN4) | Wnt signaling | Failure in synapse formation resulting in impaired communication abilities | [97, 119] |
| Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1A (DYRK1A) | Wnt signaling | Head size abnormalities | [134, 256] |
| Transducin Beta Like 1 X-Linked Receptor 1 (TBL1XR1) | Wnt signaling | Delayed language development | [257] |
| DIX Domain Containing 1 (DIXDC1) | Wnt signaling | Impaired social behavior and anxiety | [116, 126] |
| Ankyrin 3 (ANK3) | Wnt signaling | Autistic features | [116, 258] |
| Prostaglandin E2 (PGE2) | Wnt signaling | Hyperactivity, repetitive behaviors and anxiety | [116, 259] |
Another signaling pathway known as Sonic hedgehog (SHH) plays a major role in the developmental processes of multicellular embryos [172]. It is also the main component involved in the regulation of neural patterning and polarity of the CNS [173]. Within the forebrain, hindbrain and spinal cord, SHH signaling paces up proliferation and axonal targeting [174]. The SHH pathway has also been implicated in ASD, and mutations have been observed in patched domain-containing 1 (PTCHD1). A deficiency of this gene in male mice caused synaptic dysfunction and abnormal neuronal excitations leading to hyperactivity and cognitive alterations [175, 176]. Mutations in genes 7-dehydrocholesterol reductase (DHCR7) [177] and engrailed2 (EN2) have also been associated with ASD [178, 179]. In addition, similar cerebellar morphological abnormalities were displayed by mouse variants of EN2 and autistic individuals [180].
Retinoic acid (RA), derived from vitamin A (retinol), is a lipophilic molecule that is essential for vertebrate development and acts as a ligand for retinoic acid receptors (RARs) and retinoid X receptors (RXRs) [181]. Vitamin A deficiency leads to a number of abnormalities and induces autistic-like behaviors in rats by suppressing the expression of CD38 in the hypothalamus of the offspring [182]. Retinoic acid-related orphan receptor alpha (RORA) variants have been found in ASD, and decreased protein expression and abnormal methylation have been found in the autistic brain [80]. RORA regulates FOXN1 and ALDH1A3, enzymes that synthesize Retinoic Acid (RA). These RA signaling genes have been found to be associated with ASD [183].
Another signaling that is found to be associated with ASD is the fibroblast growth factor (FGF) signaling [184]. FGF belongs to family of cell signaling proteins that play an important role in brain patterning and any dysregulation of FGF signaling can lead to various neurological disorders [185]. The pathological role of FGF signaling in ASD was displayed by a study that reported impairment of synapse formation in hippocampal neurons in mutant mice lacking FGF7 [184].
The TGF-β superfamily consists of TGF-β/activin and the bone morphogenetic protein (BMP)/growth that plays an important role in bone organogenesis [186]. BMPs are important in nervous system development and their signaling is dysregulated in ASD. BMPs are involved in the activation of downstream Smads proteins and also interact with other signaling pathways such as MAPK, mTOR, Notch, Hedgehog and Wnt [186]. Distal-less homeobox (DLX) genes encoding homeodomain transcription factors are found to be dysregulated in ASD that results in alteration of BMP signaling [187, 188]. DLX genes are involved in craniofacial patterning and survival of inhibitory neurons located in the forebrain [187]. One of the genes, DLX5 was found to be overexpressed in a cell line with upregulation of the BMP-binding endothelial regulator (Bmper) [189].
A common pathophysiological mechanism that is disrupted in ASD is the imbalance in excitatory and inhibitory neurotransmission. The excitatory mechanism is mediated by glutamate, while the inhibitory mechanism is mediated by GABA [190, 191]. Studies have reported abnormalities in glutamate and GABA receptors expression in the postmortem brains of individuals affected with ASD [192]. One of the genes affecting the GABA signaling is the MECP2 gene and a study showed that MECP2 transgenic mice displayed stereotypic behaviors, ataxia, motor dysfunction and seizures [193, 194].
Clinical aspects
Clinical diagnosis of ASD is primarily based on the analysis of complex behavioral and functional changes in patients during their developmental process. Genetic tests such as comparative genomic hybridization and chromosomal microarray (CMA), and G-band karyotyping can be used for the early diagnosis of ASD. CMA has been shown to have higher clinical yield and higher resolution as compared to the G-band karyotyping [195, 196]. Karyotyping is used to detect chromosomal abnormalities such as translocations or small portions of chromosomes in different disorders [197]. G-banding also known as Giemsa banding is a staining technique that is used to differentiate the chromosomal arms [198]. In ASD, G-banding is considered useful as it can help in the detection of chromosomal abnormalities in individuals affected with ASD [199]. CMA test can detect gene duplications and deletions associated with ASD [200], and proves to be more efficient in analyzing different types of variations present in ASD.
Improvement of emotional, physical and behavioral symptoms are one of the main aspects in the pharmacological treatment of ASD [201]. To treat these symptoms underlying ASD, one of the main goals is the identification of genes and biomarkers. Next generation sequencing (NGS) is a complex emerging clinical practice that is opening a new way for the identification of ASD-causing genes that includes abnormal social interaction and communication [202]. NGS helps in the identification of rare alleles, defects of single gene and variations of gene function. It includes whole-genome sequencing (WGS) and whole-exome sequencing (WES) [203, 204].
A wide range of antiepileptic drugs (AEDs) are used for effective treatment of ASD. AEDs are psychotropic drugs that modulate electrochemical activity in the brain and can induce a positive or negative affect on mood and behavior [205]. One of the reasons of using AEDs in treating ASD is high incidence of epilepsy in most of the ASD affected individuals. Research has shown that AEDs treatment improved communication and behavior in ASD affected individuals with epileptic discharges [206]. Valproic acid, lamotrigine, levetiracetam and ethosuximide are most commonly used AEDs, and they are found to reduce seizures in ASD individuals [207]. Another AED, topiramate when combined with an antipsychotic drug risperidone reduced hyperactivity, irritability and stereotypical behaviors in ASD individuals [208]. Some AEDs such as sodium valproate may have negative effect on the developing fetus if the mother takes this drug during pregnancy. Sodium valproate can lead to an abnormal brain development leading to neurological disorders such as intellectual disability and autism [209, 210]. Also AEDs such as lamotrigine can enhance or have a mood leveling effect, while AEDs such as levetiracetam are associated with side effects in behavior such as aggression, anxiety or nervousness and hostility [211, 212]. There is a limitation of the efficacy of AEDs medications in individuals affected with ASD because AEDs show improvement in only specific type of behaviors such as hyperactivity, impulsivity, mood instability, repetitive behaviors and aggression [213].
Although there are no pharmacological treatments that are able to treat the core symptoms of ASD but there are psychotropic drugs that have similar effect as AEDs in alleviating the common symptoms of ASD such as irritability, hyperactivity, lack of focus, mood dysregulation, and social withdrawal [214].
Oxytocin is another pharmacological agent currently being used to treat core ASD symptoms [215]. Controlled trial studies showed the intravenous administration of oxytocin in ASD patients improved the symptoms in various domains including social behavior [216–218]. Apart from the few limitations of oxytocin such as dosage establishment, children safety and route of administration, it can be used as a potential therapeutic agent to treat ASD [105].
Further studies are required to relate the pharmacological treatment with the genomic changes in ASD for better treatment planning. We expect that in few years there will be enough genomic data that can be used for the pharmacological analysis of patients with ASD.
Conclusions
The genetic architecture of ASD is heterogeneous and differs in every individual. The current identification of ASD is mostly based on observation of behaviors and the genetics that underlie ASD are still an active area of research. Nevertheless, advancements in the study of various molecular mechanisms encompassing the genetics of autism and the identification of many ASD risk genes have opened a new way to study the pathophysiology of ASD. We envisage that the identification of new biomarkers, risk genes and associated genetic pathways may help in the early diagnosis of ASD, and improvement in clinical and pharmacological treatments of the disorder.
Abbreviations
ASD: autism spectrum disorder;
FXS: fragile-X Syndrome;
CNV: copy number variation;
CDFE: cortical Dysplasia-focal Epilepsy;
GWAS: genome-wide association studies;
SNP: single nucleotide polymorphism;
GABARAPL1: GABA type A receptor associated protein;
DBI: Diazepam binding inhibitor;
DYRK1A: dual-specificity tyrosine phosphorylation-regulated kinase 1A;
ASS: argininosuccinate synthetase;
NO: nitric Oxide;
PC: Purkinje cells;
NT: neurotrophin;
PSD: postsynaptic density;
NETBAG: network-based analysis of genetic associations;
DCC: deleted in colorectal carcinoma;
PAK: p21-activated kinase;
LIMK: LIM-domain containing protein kinase;
LCR: low copy repeats;
CACNA1C: calcium voltage-gated channel subunit alpha1 C;
Pk2: Prickle2;
COX-2: cyclooxygenase-2;
SV: synaptic vesicle;
HGF: hepatocyte growth factor;
LTP: long term synaptic potentiation;
CaN: calcineurin;
PKC: protein kinase-C;
CaM: calmodulin;
SHH: sonic hedgehog;
CNS: central nervous system;
PTCHD: patched domain-containing 1;
EN2: engrailed-2;
DHCR7: 7-dehydrocholesterol reductase;
RORA: retinoic acid-related orphan receptor alpha;
RAR: retinoic acid receptor;
RXR: retinoid X receptor;
FGF: fibroblast growth factor;
BMP: bone morphogenetic protein;
DLX: distal-less homeobox;
Bmper: BMP-binding endothelial regulator;
CMA: chromosomal microarray;
NGS: next generation sequencing;
WGS: whole-genome sequencing;
WES: whole-exome sequencing;
GSK3: glycogen synthase kinase 3;
CK1: casein kinase 1;
APC: adenomatous polyposis coli;
DVL: Dishevelled; FZD9-Frizzled 9;
TCF: T cell factor;
MET: hepatocyte growth factor receptor;
NMDAR: N-methyl-D-aspartate receptor;
TSC1/2: Tuberous sclerosis 1 and 2;
DISC1: Disrupted in Schizophrenia 1;
NRXN: Neurexin;
NLGN: Neuroligin;
SYN1: Synapsin 1;
SYT: Synaptotagmin;
CaN: Calcineurin;
CREB: cAMP response element binding;
PP1: Protein phosphatase 1;
PP2A: Protein phosphatase 2A;
DOCK4: dedicator of cytokinesis 4;
KATNAL2: katanin catalytic Subunit A1 Like 2;
KCNQ4: potassium voltage-gated channel subfamily Q member 4;
CNTNAP2: contactin associated protein 2;
COL11A1: collagen type XI alpha 1 chain;
FMR1: fragile-X mental retardation 1;
FOXP1: forkhead box P1;
GJB6: gap junction protein beta 6;
GRIN2B: glutamate ionotropic receptor NMDA type subunit 2B;
MYH14: myosin heavy chain 14;
SCN1A: sodium voltage-gated channel alpha subunit 1;
SCN2A: sodium voltage-gated channel alpha subunit 2;
SHANK3: SH3 and multiple ankyrin repeat domains 3;
Syngap1: synaptic ras GTPase activating protein 1;
THRA: thyroid hormone receptor, alpha;
UBE3A: ubiquitin protein ligase E3A;
Wnt: wingless-type;
ANK3: Ankyrin-3;
PGE2: prostaglandin E2;
CTNNB1: catenin beta 1;
TBR1: T-box, brain 1;
MOCOS: molybdenum cofactor sulfurase;
TBL1XR1: transducin beta like 1 X-linked receptor 1;
CYFIP1: cytoplasmic FMR1 interacting protein 1;
CNTN3: contactin 3;
PCDH10: protocadherin 10;
DIA1: deleted in autism 1;
SLC9A9: solute carrier family 9 member A9;
GLRA2: glycine receptor alpha 2;
SEMA5A: semaphorin 5A;
MACROD2: mono-ADP ribosylhydrolase 2;
CHD2: chromodomain helicase DNA binding protein 2;
HDAC: histone deacetylase 4;
HDAC9: histone deacetylase 9;
GDI1: GDP dissociation inhibitor 1;
SETD5: SET domain containing 5;
MIR137: microRNA 137;
SLC6A11: solute carrier family 6 member 11;
SLC6A8: solute carrier family 6 member 8.
Author Contributions
S.N., A.B., S.H., and M.H.: Prepared the scientific material, wrote the main text, generated tables and made figures; N.S., generated figure needed for revision; S.Y., M.W., S.U., P.B., R.R.: critical revision and editing of the scientific contents. All authors read and approved the final manuscript.
Conflicts of Interest
All authors declare no conflicts of interest.
Funding
This study was supported by a PI grant from the Sidra Medicine (50610110002) to Mohammad Haris.
References
-
1.
Lidstone JS, Fernyhough C, Meins E, Whitehouse AJ. Brief report: inner speech impairment in children with autism is associated with greater nonverbal than verbal skills. J Autism Dev Disord. 2009; 39:1222–25. https://doi.org/10.1007/s10803-009-0731-6 [PubMed]
-
2.
Williams DM, Jarrold C. Brief report: Predicting inner speech use amongst children with autism spectrum disorder (ASD): the roles of verbal ability and cognitive profile. J Autism Dev Disord. 2010; 40:907–13. https://doi.org/10.1007/s10803-010-0936-8 [PubMed]
-
3.
Huang AX, Hughes TL, Sutton LR, Lawrence M, Chen X, Ji Z, Zeleke W. Understanding the Self in Individuals with Autism Spectrum Disorders (ASD): A Review of Literature. Front Psychol. 2017; 8:1422–1422. https://doi.org/10.3389/fpsyg.2017.01422 [PubMed]
-
4.
van Santen JP, Sproat RW, Hill AP. Quantifying repetitive speech in autism spectrum disorders and language impairment. Autism Res. 2013; 6:372–83. https://doi.org/10.1002/aur.1301 [PubMed]
-
5.
Zablotsky B, Black LI, Maenner MJ, Schieve LA, Blumberg SJ. Estimated Prevalence of Autism and Other Developmental Disabilities Following Questionnaire Changes in the 2014 National Health Interview Survey. Natl Health Stat Report. 2015; 13:1–20. [PubMed]
-
6.
Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, Charman T. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the Special Needs and Autism Project (SNAP). Lancet. 2006; 368:210–15. https://doi.org/10.1016/S0140-6736(06)69041-7 [PubMed]
-
7.
Salhia HO, Al-Nasser LA, Taher LS, Al-Khathaami AM, El-Metwally AA. Systemic review of the epidemiology of autism in Arab Gulf countries. Neurosciences (Riyadh). 2014; 19:291–96. [PubMed]
-
8.
Parker W, Ollerton J. Evolutionary biology and anthropology suggest biome reconstitution as a necessary approach toward dealing with immune disorders. Evol Med Public Health. 2013; 2013:89–103. https://doi.org/10.1093/emph/eot008 [PubMed]
-
9.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, Lotspeich L, Croen LA, Ozonoff S, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011; 68:1095–102. https://doi.org/10.1001/archgenpsychiatry.2011.76 [PubMed]
-
10.
Dufault R, Lukiw WJ, Crider R, Schnoll R, Wallinga D, Deth R. A macroepigenetic approach to identify factors responsible for the autism epidemic in the United States. Clin Epigenetics. 2012; 4:6. https://doi.org/10.1186/1868-7083-4-6 [PubMed]
-
11.
Grabrucker AM. Environmental factors in autism. Front Psychiatry. 2013; 3:118. https://doi.org/10.3389/fpsyt.2012.00118 [PubMed]
-
12.
Velasquez-Manoff M. An epidemic of absence: a new way of understanding allergies and autoimmune diseases. Scribner, 2012.
-
13.
Oron O, Elliott E. Delineating the Common Biological Pathways Perturbed by ASD’s Genetic Etiology: Lessons from Network-Based Studies. Int J Mol Sci. 2017; 18:828. https://doi.org/10.3390/ijms18040828 [PubMed]
-
14.
Hagerman R, Hoem G, Hagerman P. Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol Autism. 2010; 1:12–12. https://doi.org/10.1186/2040-2392-1-12 [PubMed]
-
15.
Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006; 354:1370–77. https://doi.org/10.1056/NEJMoa052773 [PubMed]
-
16.
Vignoli A, La Briola F, Peron A, Turner K, Vannicola C, Saccani M, Magnaghi E, Scornavacca GF, Canevini MP. Autism spectrum disorder in tuberous sclerosis complex: searching for risk markers. Orphanet J Rare Dis. 2015; 10:154. https://doi.org/10.1186/s13023-015-0371-1 [PubMed]
-
17.
Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003; 40:575–84. https://doi.org/10.1136/jmg.40.8.575 [PubMed]
-
18.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009; 459:528–33. https://doi.org/10.1038/nature07999 [PubMed]
-
19.
Weiss LA, Arking DE, Daly MJ, Chakravarti A, and Gene Discovery Project of Johns Hopkins and the Autism Consortium. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009; 461:802–08. https://doi.org/10.1038/nature08490 [PubMed]
-
20.
Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Sykes N, Pagnamenta AT, Almeida J, Bacchelli E, Bailey AJ, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010; 19:4072–82. https://doi.org/10.1093/hmg/ddq307 [PubMed]
-
21.
Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012; 22:229–37. https://doi.org/10.1016/j.gde.2012.03.002 [PubMed]
-
22.
Bourgeron T. Current knowledge on the genetics of autism and propositions for future research. C R Biol. 2016; 339:300–07. https://doi.org/10.1016/j.crvi.2016.05.004 [PubMed]
-
23.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, Vorstman JA, Thompson A, Regan R, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014; 94:677–94. https://doi.org/10.1016/j.ajhg.2014.03.018 [PubMed]
-
24.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012; 485:242–45. https://doi.org/10.1038/nature11011 [PubMed]
-
25.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, Stanley T, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013; 45:825–30. https://doi.org/10.1038/ng.2646 [PubMed]
-
26.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012; 380:1674–82. https://doi.org/10.1016/S0140-6736(12)61480-9 [PubMed]
-
27.
Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, Goldberg AP, Jinlu C, Keaney JF 3rd, et al, and Autism Sequencing Consortium. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015; 87:1215–33. https://doi.org/10.1016/j.neuron.2015.09.016 [PubMed]
-
28.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012; 74:285–99. https://doi.org/10.1016/j.neuron.2012.04.009 [PubMed]
-
29.
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, Leal SM, Bernier R, Eichler EE. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015; 47:582–88. https://doi.org/10.1038/ng.3303 [PubMed]
-
30.
Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, Jaworski JM, Salyakina D, Konidari I, Whitehead PL, Wright HH, Abramson RK, Williams SM, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012; 21:3513–23. https://doi.org/10.1093/hmg/dds164 [PubMed]
-
31.
Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015; 16:551–63. https://doi.org/10.1038/nrn3992 [PubMed]
-
32.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012; 485:246–50. https://doi.org/10.1038/nature10989 [PubMed]
-
33.
Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007; 81:1289–97. https://doi.org/10.1086/522590 [PubMed]
-
34.
Masi A, DeMayo MM, Glozier N, Guastella AJ. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci Bull. 2017; 33:183–93. https://doi.org/10.1007/s12264-017-0100-y [PubMed]
-
35.
Nishiyama M, Skoultchi AI, Nakayama KI. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol Cell Biol. 2012; 32:501–12. https://doi.org/10.1128/MCB.06409-11 [PubMed]
-
36.
Nishiyama M, Oshikawa K, Tsukada Y, Nakagawa T, Iemura S, Natsume T, Fan Y, Kikuchi A, Skoultchi AI, Nakayama KI. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol. 2009; 11:172–82. https://doi.org/10.1038/ncb1831 [PubMed]
-
37.
Wang J, Liu J, Gao Y, Wang K, Jiang K. Autism spectrum disorder early in development associated with CHD8 mutations among two Chinese children. BMC Pediatr. 2018; 18:338–338. https://doi.org/10.1186/s12887-018-1307-4 [PubMed]
-
38.
Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014; 37:95–105. https://doi.org/10.1016/j.tins.2013.11.005 [PubMed]
-
39.
Du X, Gao X, Liu X, Shen L, Wang K, Fan Y, Sun Y, Luo X, Liu H, Wang L, Wang Y, Gong Z, Wang J, et al. Genetic Diagnostic Evaluation of Trio-Based Whole Exome Sequencing Among Children With Diagnosed or Suspected Autism Spectrum Disorder. Front Genet. 2018; 9:594. https://doi.org/10.3389/fgene.2018.00594 [PubMed]
-
40.
Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, Makita N, Tanaka M, Fukushima K, Fujiwara T, Inoue Y, Yamakawa K. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004; 24:2690–98. https://doi.org/10.1523/JNEUROSCI.3089-03.2004 [PubMed]
-
41.
Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, Montal M, Hashikawa T, Shike T, Fujiwara T, Inoue Y, Kaneda M, Yamakawa K. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009; 73:1046–53. https://doi.org/10.1212/WNL.0b013e3181b9cebc [PubMed]
-
42.
Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics. 2006; 7:118–118. https://doi.org/10.1186/1471-2164-7-118 [PubMed]
-
43.
Kuwano Y, Kamio Y, Kawai T, Katsuura S, Inada N, Takaki A, Rokutan K. Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS One. 2011; 6:e24723. https://doi.org/10.1371/journal.pone.0024723 [PubMed]
-
44.
Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L, Fan JB, Murray S, April C, Belinson H, Fu XD, Wynshaw-Boris A, Schork NJ, Courchesne E. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012; 8:e1002592. https://doi.org/10.1371/journal.pgen.1002592 [PubMed]
-
45.
Fatemi SH, Halt AR, Realmuto G, Earle J, Kist DA, Thuras P, Merz A. Purkinje cell size is reduced in cerebellum of patients with autism. Cell Mol Neurobiol. 2002; 22:171–75. https://doi.org/10.1023/A:1019861721160 [PubMed]
-
46.
Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008; 7:406–16. https://doi.org/10.1007/s12311-008-0043-y [PubMed]
-
47.
Sudarov A. Defining the role of cerebellar Purkinje cells in autism spectrum disorders. Cerebellum. 2013; 12:950–55. https://doi.org/10.1007/s12311-013-0490-y [PubMed]
-
48.
Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Rutter M, Lantos P. A clinicopathological study of autism. Brain. 1998; 121:889–905. https://doi.org/10.1093/brain/121.5.889 [PubMed]
-
49.
Blatt GJ. The neuropathology of autism. Scientifica (Cairo). 2012; 2012:703675–703675. https://doi.org/10.6064/2012/703675 [PubMed]
-
50.
Palmen SJ, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004; 127:2572–83. https://doi.org/10.1093/brain/awh287 [PubMed]
-
51.
Kern JK, Geier DA, Sykes LK, Geier MR. Evidence of neurodegeneration in autism spectrum disorder. Transl Neurodegener. 2013; 2:17–17. https://doi.org/10.1186/2047-9158-2-17 [PubMed]
-
52.
Segura M, Pedreño C, Obiols J, Taurines R, Pàmias M, Grünblatt E, Gella A. Neurotrophin blood-based gene expression and social cognition analysis in patients with autism spectrum disorder. Neurogenetics. 2015; 16:123–31. https://doi.org/10.1007/s10048-014-0434-9 [PubMed]
-
53.
Chien WH, Gau SS, Chen CH, Tsai WC, Wu YY, Chen PH, Shang CY, Chen CH. Increased gene expression of FOXP1 in patients with autism spectrum disorders. Mol Autism. 2013; 4:23–23. https://doi.org/10.1186/2040-2392-4-23 [PubMed]
-
54.
Féron F, Gepner B, Lacassagne E, Stephan D, Mesnage B, Blanchard MP, Boulanger N, Tardif C, Devèze A, Rousseau S, Suzuki K, Izpisua Belmonte JC, Khrestchatisky M, et al. Olfactory stem cells reveal MOCOS as a new player in autism spectrum disorders. Mol Psychiatry. 2016; 21:1215–24. https://doi.org/10.1038/mp.2015.106 [PubMed]
-
55.
Han TU, Park J, Domingues CF, Moretti-Ferreira D, Paris E, Sainz E, Gutierrez J, Drayna D. A study of the role of the FOXP2 and CNTNAP2 genes in persistent developmental stuttering. Neurobiol Dis. 2014; 69:23–31. https://doi.org/10.1016/j.nbd.2014.04.019 [PubMed]
-
56.
Ansel A, Rosenzweig JP, Zisman PD, Melamed M, Gesundheit B. Variation in Gene Expression in Autism Spectrum Disorders: An Extensive Review of Transcriptomic Studies. Front Neurosci. 2017; 10:601. https://doi.org/10.3389/fnins.2016.00601 [PubMed]
-
57.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, et al, and UK10K Consortium. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014; 515:209–15. https://doi.org/10.1038/nature13772 [PubMed]
-
58.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007; 39:25–27. https://doi.org/10.1038/ng1933 [PubMed]
-
59.
Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci. 2005; 25:3560–70. https://doi.org/10.1523/JNEUROSCI.4354-04.2005 [PubMed]
-
60.
Kelleher RJ 3rd, Geigenmüller U, Hovhannisyan H, Trautman E, Pinard R, Rathmell B, Carpenter R, Margulies D. High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS One. 2012; 7:e35003. https://doi.org/10.1371/journal.pone.0035003 [PubMed]
-
61.
Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. 2012; 15:431–40, S1. https://doi.org/10.1038/nn.3033 [PubMed]
-
62.
Johnson HM, Gaitanis J, Morrow EM. Genetics in autism diagnosis: adding molecular subtypes to neurobehavioral diagnoses. Med Health R I. 2011; 94:124–26. [PubMed]
-
63.
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013; 155:1008–21. https://doi.org/10.1016/j.cell.2013.10.031 [PubMed]
-
64.
Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, Descartes M, Holt L, Braddock S, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008; 82:199–207. https://doi.org/10.1016/j.ajhg.2007.09.011 [PubMed]
-
65.
Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011; 70:898–907. https://doi.org/10.1016/j.neuron.2011.05.021 [PubMed]
-
66.
Reiner O, Sapir T. Similarities and differences between the Wnt and reelin pathways in the forming brain. Mol Neurobiol. 2005; 31:117–34. https://doi.org/10.1385/MN:31:1-3:117 [PubMed]
-
67.
Salinas PC, Zou Y. Wnt signaling in neural circuit assembly. Annu Rev Neurosci. 2008; 31:339–58. https://doi.org/10.1146/annurev.neuro.31.060407.125649 [PubMed]
-
68.
Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996; 274:1123–33. https://doi.org/10.1126/science.274.5290.1123 [PubMed]
-
69.
Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010; 1309:83–94. https://doi.org/10.1016/j.brainres.2009.09.120 [PubMed]
-
70.
Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, Bookheimer SY. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med. 2010; 2:56ra80. https://doi.org/10.1126/scitranslmed.3001344 [PubMed]
-
71.
Clements CC, Wenger TL, Zoltowski AR, Bertollo JR, Miller JS, de Marchena AB, Mitteer LM, Carey JC, Yerys BE, Zackai EH, Emanuel BS, McDonald-McGinn DM, Schultz RT. Critical region within 22q11.2 linked to higher rate of autism spectrum disorder. Mol Autism. 2017; 8:58. https://doi.org/10.1186/s13229-017-0171-7 [PubMed]
-
72.
Jawabri K, Sharma S. Physiology, Cerebral Cortex Functions. StatPearls Publishing; 2019.
-
73.
Tan GC, Doke TF, Ashburner J, Wood NW, Frackowiak RS. Normal variation in fronto-occipital circuitry and cerebellar structure with an autism-associated polymorphism of CNTNAP2. Neuroimage. 2010; 53:1030–42. https://doi.org/10.1016/j.neuroimage.2010.02.018 [PubMed]
-
74.
Voineskos AN, Lett TA, Lerch JP, Tiwari AK, Ameis SH, Rajji TK, Müller DJ, Mulsant BH, Kennedy JL. Neurexin-1 and frontal lobe white matter: an overlapping intermediate phenotype for schizophrenia and autism spectrum disorders. PLoS One. 2011; 6:e20982–20982. https://doi.org/10.1371/journal.pone.0020982 [PubMed]
-
75.
Mukamel Z, Konopka G, Wexler E, Osborn GE, Dong H, Bergman MY, Levitt P, Geschwind DH. Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J Neurosci. 2011; 31:11437–42. https://doi.org/10.1523/JNEUROSCI.0181-11.2011 [PubMed]
-
76.
Crepel A, De Wolf V, Brison N, Ceulemans B, Walleghem D, Peuteman G, Lambrechts D, Steyaert J, Noens I, Devriendt K, Peeters H. Association of CDH11 with non-syndromic ASD. Am J Med Genet B Neuropsychiatr Genet. 2014; 165B:391–98. https://doi.org/10.1002/ajmg.b.32243 [PubMed]
-
77.
Hussman JP, Chung RH, Griswold AJ, Jaworski JM, Salyakina D, Ma D, Konidari I, Whitehead PL, Vance JM, Martin ER, Cuccaro ML, Gilbert JR, Haines JL, Pericak-Vance MA. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2011; 2:1–1. https://doi.org/10.1186/2040-2392-2-1 [PubMed]
-
78.
Pagnamenta AT, Khan H, Walker S, Gerrelli D, Wing K, Bonaglia MC, Giorda R, Berney T, Mani E, Molteni M, Pinto D, Le Couteur A, Hallmayer J, et al. Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet. 2011; 48:48–54. https://doi.org/10.1136/jmg.2010.079426 [PubMed]
-
79.
Wang C, Pan YH, Wang Y, Blatt G, Yuan XB. Segregated expressions of autism risk genes Cdh11 and Cdh9 in autism-relevant regions of developing cerebellum. Mol Brain. 2019; 12:40. https://doi.org/10.1186/s13041-019-0461-4 [PubMed]
-
80.
Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010; 24:3036–51. https://doi.org/10.1096/fj.10-154484 [PubMed]
-
81.
Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011; 3:a005678. https://doi.org/10.1101/cshperspect.a005678 [PubMed]
-
82.
Kindler S, Kreienkamp HJ. The role of the postsynaptic density in the pathology of the fragile X syndrome. Results Probl Cell Differ. 2012; 54:61–80. https://doi.org/10.1007/978-3-642-21649-7_5 [PubMed]
-
83.
Miles JH. Autism spectrum disorders—a genetics review. Genet Med. 2011; 13:278–94. https://doi.org/10.1097/GIM.0b013e3181ff67ba [PubMed]
-
84.
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, Witke W, Costa-Mattioli M, Sonenberg N, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008; 134:1042–54. https://doi.org/10.1016/j.cell.2008.07.031 [PubMed]
-
85.
Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, Persico AM, Levitt P. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA. 2006; 103:16834–39. https://doi.org/10.1073/pnas.0605296103 [PubMed]
-
86.
Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008; 135:401–06. https://doi.org/10.1016/j.cell.2008.10.017 [PubMed]
-
87.
Waung MW, Huber KM. Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr Opin Neurobiol. 2009; 19:319–26. https://doi.org/10.1016/j.conb.2009.03.011 [PubMed]
-
88.
Guang S, Pang N, Deng X, Yang L, He F, Wu L, Chen C, Yin F, Peng J. Synaptopathology Involved in Autism Spectrum Disorder. Front Cell Neurosci. 2018; 12:470–470. https://doi.org/10.3389/fncel.2018.00470 [PubMed]
-
89.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009; 459:569–73. https://doi.org/10.1038/nature07953 [PubMed]
-
90.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012; 485:237–41. https://doi.org/10.1038/nature10945 [PubMed]
-
91.
Rosti RO, Sadek AA, Vaux KK, Gleeson JG. The genetic landscape of autism spectrum disorders. Dev Med Child Neurol. 2014; 56:12–18. https://doi.org/10.1111/dmcn.12278 [PubMed]
-
92.
Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008; 455:903–11. https://doi.org/10.1038/nature07456 [PubMed]
-
93.
Knight D, Xie W, Boulianne GL. Neurexins and neuroligins: recent insights from invertebrates. Mol Neurobiol. 2011; 44:426–40. https://doi.org/10.1007/s12035-011-8213-1 [PubMed]
-
94.
Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012; 13:247–247. https://doi.org/10.1186/gb-2012-13-7-247 [PubMed]
-
95.
Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008; 321:218–23. https://doi.org/10.1126/science.1157657 [PubMed]
-
96.
Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008; 60:1022–38. https://doi.org/10.1016/j.neuron.2008.11.029 [PubMed]
-
97.
Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T, and Paris Autism Research International Sibpair Study. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003; 34:27–29. https://doi.org/10.1038/ng1136 [PubMed]
-
98.
Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004; 74:552–57. https://doi.org/10.1086/382137 [PubMed]
-
99.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010; 466:368–72. https://doi.org/10.1038/nature09146 [PubMed]
-
100.
Ben-David E, Shifman S. Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet. 2012; 8:e1002556. https://doi.org/10.1371/journal.pgen.1002556 [PubMed]
-
101.
Chaste P, Leboyer M. Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin Neurosci. 2012; 14:281–92. [PubMed]
-
102.
Pilorge M, Fassier C, Le Corronc H, Potey A, Bai J, De Gois S, Delaby E, Assouline B, Guinchat V, Devillard F, Delorme R, Nygren G, Råstam M, et al. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol Psychiatry. 2016; 21:936–45. https://doi.org/10.1038/mp.2015.139 [PubMed]
-
103.
Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005; 25:9883. https://doi.org/10.1523/JNEUROSCI.1531-05.2005 [PubMed]
-
104.
Li J, Zhao L, You Y, Lu T, Jia M, Yu H, Ruan Y, Yue W, Liu J, Lu L, Zhang D, Wang L. Schizophrenia Related Variants in CACNA1C also Confer Risk of Autism. PLoS One. 2015; 10:e0133247–0133247. https://doi.org/10.1371/journal.pone.0133247 [PubMed]
-
105.
Yoo H. Genetics of Autism Spectrum Disorder: Current Status and Possible Clinical Applications. Exp Neurobiol. 2015; 24:257–72. https://doi.org/10.5607/en.2015.24.4.257 [PubMed]
-
106.
Mahajan R, Mostofsky SH. Neuroimaging endophenotypes in autism spectrum disorder. CNS Spectr. 2015; 20:412–26. https://doi.org/10.1017/S1092852915000371 [PubMed]
-
107.
Frazier TW, Embacher R, Tilot AK, Koenig K, Mester J, Eng C. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry. 2015; 20:1132–38. https://doi.org/10.1038/mp.2014.125 [PubMed]
-
108.
Rudie JD, Hernandez LM, Brown JA, Beck-Pancer D, Colich NL, Gorrindo P, Thompson PM, Geschwind DH, Bookheimer SY, Levitt P, Dapretto M. Autism-associated promoter variant in MET impacts functional and structural brain networks. Neuron. 2012; 75:904–15. https://doi.org/10.1016/j.neuron.2012.07.010 [PubMed]
-
109.
Pain O, Pocklington AJ, Holmans PA, Bray NJ, O’Brien HE, Hall LS, Pardiñas AF, O’Donovan MC, Owen MJ, Anney R. Novel Insight Into the Etiology of Autism Spectrum Disorder Gained by Integrating Expression Data With Genome-wide Association Statistics. Biol Psychiatry. 2019; 86:265–73. https://doi.org/10.1016/j.biopsych.2019.04.034 [PubMed]
-
110.
Jones RM, Cadby G, Melton PE, Abraham LJ, Whitehouse AJ, Moses EK. Genome-wide association study of autistic-like traits in a general population study of young adults. Front Hum Neurosci. 2013; 7:658. https://doi.org/10.3389/fnhum.2013.00658 [PubMed]
-
111.
Zhang L, Liu L, Wen Y, Ma M, Cheng S, Yang J, Li P, Cheng B, Du Y, Liang X, Zhao Y, Ding M, Guo X, Zhang F. Genome-wide association study and identification of chromosomal enhancer maps in multiple brain regions related to autism spectrum disorder. Autism Res. 2019; 12:26–32. https://doi.org/10.1002/aur.2001 [PubMed]
-
112.
Mosca-Boidron AL, Gueneau L, Huguet G, Goldenberg A, Henry C, Gigot N, Pallesi-Pocachard E, Falace A, Duplomb L, Thevenon J, Duffourd Y, St-Onge J, Chambon P, et al. A de novo microdeletion of SEMA5A in a boy with autism spectrum disorder and intellectual disability. Eur J Hum Genet. 2016; 24:838–43. https://doi.org/10.1038/ejhg.2015.211 [PubMed]
-
113.
Jones RM, Cadby G, Blangero J, Abraham LJ, Whitehouse AJ, Moses EK. MACROD2 gene associated with autistic-like traits in a general population sample. Psychiatr Genet. 2014; 24:241–48. https://doi.org/10.1097/YPG.0000000000000052 [PubMed]
-
114.
Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen AN, Hoffman JD, Slifer SH, Hedges DJ, Cukier HN, Griswold AJ, McCauley JL, Beecham GW, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet. 2009; 73:263–73. https://doi.org/10.1111/j.1469-1809.2009.00523.x [PubMed]
-
115.
He CW, Liao CP, Pan CL. Wnt signalling in the development of axon, dendrites and synapses. Open Biol. 2018; 8:180116. https://doi.org/10.1098/rsob.180116 [PubMed]
-
116.
Kumar S, Reynolds K, Ji Y, Gu R, Rai S, Zhou CJ. Impaired neurodevelopmental pathways in autism spectrum disorder: a review of signaling mechanisms and crosstalk. J Neurodev Disord. 2019; 11:10. https://doi.org/10.1186/s11689-019-9268-y [PubMed]
-
117.
Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, Colamarino SA, Lein ES, Courchesne E. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014; 370:1209–19. https://doi.org/10.1056/NEJMoa1307491 [PubMed]
-
118.
Casanova MF, van Kooten IA, Switala AE, van Engeland H, Heinsen H, Steinbusch HW, Hof PR, Trippe J, Stone J, Schmitz C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006; 112:287–303. https://doi.org/10.1007/s00401-006-0085-5 [PubMed]
-
119.
Caracci MO, Ávila ME, De Ferrari GV. Synaptic Wnt/GSK3β Signaling Hub in Autism. Neural Plast. 2016; 2016:9603751–9603751. https://doi.org/10.1155/2016/9603751 [PubMed]
-
120.
Kwan V, Unda BK, Singh KK. Wnt signaling networks in autism spectrum disorder and intellectual disability. J Neurodev Disord. 2016; 8:45–45. https://doi.org/10.1186/s11689-016-9176-3 [PubMed]
-
121.
Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, Tsai LH. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci. 2016; 19:1477–88. https://doi.org/10.1038/nn.4400 [PubMed]
-
122.
Viale B, Song L, Petrenko V, Wenger Combremont AL, Contestabile A, Bocchi R, Salmon P, Carleton A, An L, Vutskits L, Kiss JZ. Transient Deregulation of Canonical Wnt Signaling in Developing Pyramidal Neurons Leads to Dendritic Defects and Impaired Behavior. Cell Rep. 2019; 27:1487–1502.e6. https://doi.org/10.1016/j.celrep.2019.04.026 [PubMed]
-
123.
Kalkman HO. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol Autism. 2012; 3:10. https://doi.org/10.1186/2040-2392-3-10 [PubMed]
-
124.
Singh KK, De Rienzo G, Drane L, Mao Y, Flood Z, Madison J, Ferreira M, Bergen S, King C, Sklar P, Sive H, Tsai LH. Common DISC1 polymorphisms disrupt Wnt/GSK3β signaling and brain development. Neuron. 2011; 72:545–58. https://doi.org/10.1016/j.neuron.2011.09.030 [PubMed]
-
125.
Kwan V, Meka DP, White SH, Hung CL, Holzapfel NT, Walker S, Murtaza N, Unda BK, Schwanke B, Yuen RK, Habing K, Milsom C, Hope KJ, et al. DIXDC1 Phosphorylation and Control of Dendritic Morphology Are Impaired by Rare Genetic Variants. Cell Rep. 2016; 17:1892–904. https://doi.org/10.1016/j.celrep.2016.10.047 [PubMed]
-
126.
Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimäe S, Mulligan KA, Pirooznia M, McCombie WR, et al. DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β-catenin signaling. Mol Psychiatry. 2018; 23:467–75. https://doi.org/10.1038/mp.2016.184 [PubMed]
-
127.
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006; 126:955–68. https://doi.org/10.1016/j.cell.2006.06.055 [PubMed]
-
128.
Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS. The tuberin-hamartin complex negatively regulates β-catenin signaling activity. J Biol Chem. 2003; 278:5947–51. https://doi.org/10.1074/jbc.C200473200 [PubMed]
-
129.
Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS. Aberrant beta-catenin signaling in tuberous sclerosis. Am J Pathol. 2005; 167:107–16. https://doi.org/10.1016/S0002-9440(10)62958-6 [PubMed]
-
130.
Ueda S, Fujimoto S, Hiramoto K, Negishi M, Katoh H. Dock4 regulates dendritic development in hippocampal neurons. J Neurosci Res. 2008; 86:3052–61. https://doi.org/10.1002/jnr.21763 [PubMed]
-
131.
Iqbal Z, Vandeweyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA, Roca LT, Vulto-van Silfhout AT, Nijhof B, Kramer JM, Van der Aa N, Ansar M, Peeters H, et al. Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet. 2013; 22:1960–70. https://doi.org/10.1093/hmg/ddt043 [PubMed]
-
132.
Bi C, Wu J, Jiang T, Liu Q, Cai W, Yu P, Cai T, Zhao M, Jiang YH, Sun ZS. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat. 2012; 33:1635–38. https://doi.org/10.1002/humu.22174 [PubMed]
-
133.
Wong CT, Ussyshkin N, Ahmad E, Rai-Bhogal R, Li H, Crawford DA. Prostaglandin E2 promotes neural proliferation and differentiation and regulates Wnt target gene expression. J Neurosci Res. 2016; 94:759–75. https://doi.org/10.1002/jnr.23759 [PubMed]
-
134.
Medina MA, Andrade VM, Caracci MO, Avila ME, Verdugo DA, Vargas MF, Ugarte GD, Reyes AE, Opazo C, De Ferrari GV. Wnt/β-catenin signaling stimulates the expression and synaptic clustering of the autism-associated Neuroligin 3 gene. Transl Psychiatry. 2018; 8:45. https://doi.org/10.1038/s41398-018-0093-y [PubMed]
-
135.
Oliva CA, Vargas JY, Inestrosa NC. Wnt signaling: role in LTP, neural networks and memory. Ageing Res Rev. 2013; 12:786–800. https://doi.org/10.1016/j.arr.2013.03.006 [PubMed]
-
136.
Hall MN. mTOR-what does it do? Transplant Proc. 2008 (10 Suppl); 40:S5–8. https://doi.org/10.1016/j.transproceed.2008.10.009 [PubMed]
-
137.
Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR Activation in Autism. Annu Rev Neurosci. 2018; 41:1–23. https://doi.org/10.1146/annurev-neuro-080317-061747 [PubMed]
-
138.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21–35. https://doi.org/10.1038/nrm3025 [PubMed]
-
139.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012; 149:274–93. https://doi.org/10.1016/j.cell.2012.03.017 [PubMed]
-
140.
Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014; 7:28–28. https://doi.org/10.3389/fnmol.2014.00028 [PubMed]
-
141.
Subramanian M, Timmerman CK, Schwartz JL, Pham DL, Meffert MK. Characterizing autism spectrum disorders by key biochemical pathways. Front Neurosci. 2015; 9:313. https://doi.org/10.3389/fnins.2015.00313 [PubMed]
-
142.
Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004; 5:931–42. https://doi.org/10.1038/nrn1557 [PubMed]
-
143.
Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005; 25:9581–90. https://doi.org/10.1523/JNEUROSCI.2423-05.2005 [PubMed]
-
144.
Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006; 26:2167–73. https://doi.org/10.1523/JNEUROSCI.5196-05.2006 [PubMed]
-
145.
Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013; 493:371–77. https://doi.org/10.1038/nature11628 [PubMed]
-
146.
Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013; 493:411–15. https://doi.org/10.1038/nature11782 [PubMed]
-
147.
Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009; 10:307–18. https://doi.org/10.1038/nrm2672 [PubMed]
-
148.
Sakai Y, Kassai H, Nakayama H, Fukaya M, Maeda T, Nakao K, Hashimoto K, Sakagami H, Kano M, Aiba A. Hyperactivation of mTORC1 disrupts cellular homeostasis in cerebellar Purkinje cells. Sci Rep. 2019; 9:2799. https://doi.org/10.1038/s41598-019-38730-4 [PubMed]
-
149.
Kasarpalkar NJ, Kothari ST, Dave UP. Brain-Derived Neurotrophic Factor in children with Autism Spectrum Disorder. Ann Neurosci. 2014; 21:129–33. https://doi.org/10.5214/ans.0972.7531.210403 [PubMed]
-
150.
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006; 361:1545–64. https://doi.org/10.1098/rstb.2006.1894 [PubMed]
-
151.
Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013; 14:7–23. https://doi.org/10.1038/nrn3379 [PubMed]
-
152.
Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci (Lond). 2006; 110:167–73. https://doi.org/10.1042/CS20050163 [PubMed]
-
153.
Theoharides TC, Athanassiou M, Panagiotidou S, Doyle R. Dysregulated brain immunity and neurotrophin signaling in Rett syndrome and autism spectrum disorders. J Neuroimmunol. 2015; 279:33–38. https://doi.org/10.1016/j.jneuroim.2014.12.003 [PubMed]
-
154.
Ohja K, Gozal E, Fahnestock M, Cai L, Cai J, Freedman JH, Switala A, El-Baz A, Barnes GN. Neuroimmunologic and Neurotrophic Interactions in Autism Spectrum Disorders: relationship to Neuroinflammation. Neuromolecular Med. 2018; 20:161–73. https://doi.org/10.1007/s12017-018-8488-8 [PubMed]
-
155.
Saghazadeh A, Rezaei N. Brain-Derived Neurotrophic Factor Levels in Autism: A Systematic Review and Meta-Analysis. J Autism Dev Disord. 2017; 47:1018–29. https://doi.org/10.1007/s10803-016-3024-x [PubMed]
-
156.
Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006; 31:268–75. https://doi.org/10.1016/j.tibs.2006.03.009 [PubMed]
-
157.
Hotamisligil GS, Davis RJ. Cell Signaling and Stress Responses. Cold Spring Harb Perspect Biol. 2016; 8:a006072. https://doi.org/10.1101/cshperspect.a006072 [PubMed]
-
158.
Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004; 11:172–78. https://doi.org/10.1101/lm.67804 [PubMed]
-
159.
Medina JH, Viola H. ERK1/2: A Key Cellular Component for the Formation, Retrieval, Reconsolidation and Persistence of Memory. Front Mol Neurosci. 2018; 11:361–361. https://doi.org/10.3389/fnmol.2018.00361 [PubMed]
-
160.
Vithayathil J, Pucilowska J, Landreth GE. ERK/MAPK signaling and autism spectrum disorders. Prog Brain Res. 2018; 241:63–112. https://doi.org/10.1016/bs.pbr.2018.09.008 [PubMed]
-
161.
Yufune S, Satoh Y, Takamatsu I, Ohta H, Kobayashi Y, Takaenoki Y, Pagès G, Pouysségur J, Endo S, Kazama T. Transient Blockade of ERK Phosphorylation in the Critical Period Causes Autistic Phenotypes as an Adult in Mice. Sci Rep. 2015; 5:10252–10252. https://doi.org/10.1038/srep10252 [PubMed]
-
162.
Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, Castellani RJ, Casadesus G, Smith MA, Zhu X. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J Neurochem. 2012; 121:672–79. https://doi.org/10.1111/j.1471-4159.2012.07722.x [PubMed]
-
163.
Pucilowska J, Vithayathil J, Pagani M, Kelly C, Karlo JC, Robol C, Morella I, Gozzi A, Brambilla R, Landreth GE. Pharmacological Inhibition of ERK Signaling Rescues Pathophysiology and Behavioral Phenotype Associated with 16p11.2 Chromosomal Deletion in Mice. J Neurosci. 2018; 38:6640–52. https://doi.org/10.1523/JNEUROSCI.0515-17.2018 [PubMed]
-
164.
Yun SH, Trommer BL. Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res. 2011; 89:176–82. https://doi.org/10.1002/jnr.22546 [PubMed]
-
165.
Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006; 26:319–27. https://doi.org/10.1523/JNEUROSCI.2623-05.2006 [PubMed]
-
166.
Gargus JJ, Schmunk G. Dysregulation of Neurogenic Calcium Signaling and Autism. In: Patel VB, Preedy VR, Martin CR, editors. Comprehensive Guide to Autism. New York (NY): Springer New York; 2014. pp. 1285–312. https://doi.org/10.1007/978-1-4614-4788-7_35
-
167.
Schmunk G, Nguyen RL, Ferguson DL, Kumar K, Parker I, Gargus JJ. High-throughput screen detects calcium signaling dysfunction in typical sporadic autism spectrum disorder. Sci Rep. 2017; 7:40740–40740. https://doi.org/10.1038/srep40740 [PubMed]
-
168.
Nguyen RL, Medvedeva YV, Ayyagari TE, Schmunk G, Gargus JJ. Intracellular calcium dysregulation in autism spectrum disorder: An analysis of converging organelle signaling pathways. Biochim Biophys Acta Mol Cell Res. 2018; 1865:1718–1732. https://doi.org/10.1016/j.bbamcr.2018.08.003 [PubMed]
-
169.
Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, Bernstein JA, Hallmayer J, Geschwind DH, Dolmetsch RE. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011; 17:1657–62. https://doi.org/10.1038/nm.2576 [PubMed]
-
170.
Waltes R, Duketis E, Knapp M, Anney RJ, Huguet G, Schlitt S, Jarczok TA, Sachse M, Kämpfer LM, Kleinböck T, Poustka F, Bölte S, Schmötzer G, et al. Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum Genet. 2014; 133:781–92. https://doi.org/10.1007/s00439-013-1416-y [PubMed]
-
171.
Stephenson JR, Wang X, Perfitt TL, Parrish WP, Shonesy BC, Marks CR, Mortlock DP, Nakagawa T, Sutcliffe JS, Colbran RJ. A Novel Human CAMK2A Mutation Disrupts Dendritic Morphology and Synaptic Transmission, and Causes ASD-Related Behaviors. J Neurosci. 2017; 37:2216–33. https://doi.org/10.1523/JNEUROSCI.2068-16.2017 [PubMed]
-
172.
Simpson F, Kerr MC, Wicking C. Trafficking, development and hedgehog. Mech Dev. 2009; 126:279–88. https://doi.org/10.1016/j.mod.2009.01.007 [PubMed]
-
173.
Machold R, Fishell G. Hedgehog patterns midbrain ARChitecture. Trends Neurosci. 2002; 25:10–11. https://doi.org/10.1016/S0166-2236(00)01982-2 [PubMed]
-
174.
Álvarez-Buylla A, Ihrie RA. Sonic hedgehog signaling in the postnatal brain. Semin Cell Dev Biol. 2014; 33:105–11. https://doi.org/10.1016/j.semcdb.2014.05.008 [PubMed]
-
175.
Tora D, Gomez AM, Michaud JF, Yam PT, Charron F, Scheiffele P. Cellular Functions of the Autism Risk Factor PTCHD1 in Mice. J Neurosci. 2017; 37:11993–2005. https://doi.org/10.1523/JNEUROSCI.1393-17.2017 [PubMed]
-
176.
Ung DC, Iacono G, Méziane H, Blanchard E, Papon MA, Selten M, van Rhijn JR, Montjean R, Rucci J, Martin S, Fleet A, Birling MC, Marouillat S, et al. Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol Psychiatry. 2018; 23:1356–67. https://doi.org/10.1038/mp.2017.39 [PubMed]
-
177.
Blassberg R, Macrae JI, Briscoe J, Jacob J. Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum Mol Genet. 2016; 25:693–705. https://doi.org/10.1093/hmg/ddv507 [PubMed]
-
178.
Sen B, Singh AS, Sinha S, Chatterjee A, Ahmed S, Ghosh S, Usha R. Family-based studies indicate association of Engrailed 2 gene with autism in an Indian population. Genes Brain Behav. 2010; 9:248–55. https://doi.org/10.1111/j.1601-183X.2009.00556.x [PubMed]
-
179.
Yang P, Shu BC, Hallmayer JF, Lung FW. Intronic single nucleotide polymorphisms of engrailed homeobox 2 modulate the disease vulnerability of autism in a han chinese population. Neuropsychobiology. 2010; 62:104–15. https://doi.org/10.1159/000315441 [PubMed]
-
180.
Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol Psychiatry. 2004; 9:474–84. https://doi.org/10.1038/sj.mp.4001498 [PubMed]
-
181.
Rhinn M, Dollé P. Retinoic acid signalling during development. Development. 2012; 139:843–58. https://doi.org/10.1242/dev.065938 [PubMed]
-
182.
Lai X, Wu X, Hou N, Liu S, Li Q, Yang T, Miao J, Dong Z, Chen J, Li T, Vitamin A. Vitamin A Deficiency Induces Autistic-Like Behaviors in Rats by Regulating the RARβ-CD38-Oxytocin Axis in the Hypothalamus. Mol Nutr Food Res. 2018; 62:1700754. https://doi.org/10.1002/mnfr.201700754 [PubMed]
-
183.
Moreno-Ramos OA, Olivares AM, Haider NB, de Autismo LC, Lattig MC. Whole-Exome Sequencing in a South American Cohort Links ALDH1A3, FOXN1 and Retinoic Acid Regulation Pathways to Autism Spectrum Disorders. PLoS One. 2015; 10:e0135927–0135927. https://doi.org/10.1371/journal.pone.0135927 [PubMed]
-
184.
Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H. Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature. 2010; 465:783–87. https://doi.org/10.1038/nature09041 [PubMed]
-
185.
Turner CA, Eren-Koçak E, Inui EG, Watson SJ, Akil H. Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin Cell Dev Biol. 2016; 53:136–43. https://doi.org/10.1016/j.semcdb.2015.10.003 [PubMed]
-
186.
Rahman MS, Akhtar N, Jamil HM, Banik RS, Asaduzzaman SM. TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res. 2015; 3:15005–15005. https://doi.org/10.1038/boneres.2015.5 [PubMed]
-
187.
Hamilton SP, Woo JM, Carlson EJ, Ghanem N, Ekker M, Rubenstein JL. Analysis of four DLX homeobox genes in autistic probands. BMC Genet. 2005; 6:52. https://doi.org/10.1186/1471-2156-6-52 [PubMed]
-
188.
Lucchese G, Stahl B. Peptide Sharing Between Viruses and DLX Proteins: A Potential Cross-Reactivity Pathway to Neuropsychiatric Disorders. Front Neurosci. 2018; 12:150–150. https://doi.org/10.3389/fnins.2018.00150 [PubMed]
-
189.
Sajan SA, Rubenstein JL, Warchol ME, Lovett M. Identification of direct downstream targets of Dlx5 during early inner ear development. Hum Mol Genet. 2011; 20:1262–73. https://doi.org/10.1093/hmg/ddq567 [PubMed]
-
190.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011; 477:171–78. https://doi.org/10.1038/nature10360 [PubMed]
-
191.
Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev. 2012; 36:2044–55. https://doi.org/10.1016/j.neubiorev.2012.07.005 [PubMed]
-
192.
Blatt GJ, Fatemi SH. Alterations in GABAergic biomarkers in the autism brain: research findings and clinical implications. Anat Rec (Hoboken). 2011; 294:1646–52. https://doi.org/10.1002/ar.21252 [PubMed]
-
193.
Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004; 13:2679–89. https://doi.org/10.1093/hmg/ddh282 [PubMed]
-
194.
Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010; 468:263–69. https://doi.org/10.1038/nature09582 [PubMed]
-
195.
Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, Frazier JA, Silverstein I, Picker J, Weissman L, Raffalli P, Jeste S, et al, and Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010; 125:e727–35. https://doi.org/10.1542/peds.2009-1684 [PubMed]
-
196.
McGrew SG, Peters BR, Crittendon JA, Veenstra-Vanderweele J. Diagnostic yield of chromosomal microarray analysis in an autism primary care practice: which guidelines to implement? J Autism Dev Disord. 2012; 42:1582–91. https://doi.org/10.1007/s10803-011-1398-3 [PubMed]
-
197.
Veerabhadrappa S, Chandrappa P, Roodmal S, Shetty S, Madhu Shankari G, Mohan Kumar K. Karyotyping: current perspectives in diagnosis of chromosomal disorders. Sifa Medical Journal. 2016; 3:35–40.
-
198.
Comings DE, Avelino E, Okada TA, Wyandt HE. The mechanism of C- and G-banding of chromosomes. Exp Cell Res. 1973; 77:469–83. https://doi.org/10.1016/0014-4827(73)90601-0 [PubMed]
-
199.
Liu QJ, Ma F, Li D, Wang XW, Tian WY, Chen Y, Feng JB, Lu X, Chen DQ, Chen XN, Shen Y. Detection of chromosome aberrations in Chinese children with autism using G-banding and BAC FISH. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2005; 22:254–57. [PubMed]
-
200.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010; 86:749–64. https://doi.org/10.1016/j.ajhg.2010.04.006 [PubMed]
-
201.
Doyle CA, McDougle CJ. Pharmacotherapy to control behavioral symptoms in children with autism. Expert Opin Pharmacother. 2012; 13:1615–29. https://doi.org/10.1517/14656566.2012.674110 [PubMed]
-
202.
Sanders SJ. Next-Generation Sequencing in Autism Spectrum Disorder. Cold Spring Harb Perspect Med. 2019; 9:a026872. https://doi.org/10.1101/cshperspect.a026872 [PubMed]
-
203.
Veltman MW, Craig EE, Bolton PF. Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet. 2005; 15:243–54. https://doi.org/10.1097/00041444-200512000-00006 [PubMed]
-
204.
Buxbaum JD, Daly MJ, Devlin B, Lehner T, Roeder K, State MW; Autism Sequencing Consortium. The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron. 2012; 76:1052–6. https://doi.org/10.1016/j.neuron.2012.12.008 [PubMed]
-
205.
Nadkarni S, Devinsky O. Psychotropic effects of antiepileptic drugs. Epilepsy Curr. 2005; 5:176–81. https://doi.org/10.1111/j.1535-7511.2005.00056.x [PubMed]
-
206.
Di Martino A, Tuchman RF. Antiepileptic drugs: affective use in autism spectrum disorders. Pediatr Neurol. 2001; 25:199–207. https://doi.org/10.1016/S0887-8994(01)00276-4 [PubMed]
-
207.
Frye RE, Sreenivasula S, Adams JB. Traditional and non-traditional treatments for autism spectrum disorder with seizures: an on-line survey. BMC Pediatr. 2011; 11:37. https://doi.org/10.1186/1471-2431-11-37 [PubMed]
-
208.
Rezaei V, Mohammadi MR, Ghanizadeh A, Sahraian A, Tabrizi M, Rezazadeh SA, Akhondzadeh S. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010; 34:1269–72. https://doi.org/10.1016/j.pnpbp.2010.07.005 [PubMed]
-
209.
Meador KJ, Loring DW. Prenatal valproate exposure is associated with autism spectrum disorder and childhood autism. J Pediatr. 2013; 163:924–924. https://doi.org/10.1016/j.jpeds.2013.06.050 [PubMed]
-
210.
Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, Vestergaard M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA. 2013; 309:1696–703. https://doi.org/10.1001/jama.2013.2270 [PubMed]
-
211.
Halma E, de Louw AJ, Klinkenberg S, Aldenkamp AP, IJff DM, Majoie M. Behavioral side-effects of levetiracetam in children with epilepsy: a systematic review. Seizure. 2014; 23:685–91. https://doi.org/10.1016/j.seizure.2014.06.004 [PubMed]
-
212.
Besag FM. Epilepsy in patients with autism: links, risks and treatment challenges. Neuropsychiatr Dis Treat. 2017; 14:1–10. https://doi.org/10.2147/NDT.S120509 [PubMed]
-
213.
Tuchman R. AEDs and psychotropic drugs in children with autism and epilepsy. Ment Retard Dev Disabil Res Rev. 2004; 10:135–38. https://doi.org/10.1002/mrdd.20026 [PubMed]
-
214.
Madden JM, Lakoma MD, Lynch FL, Rusinak D, Owen-Smith AA, Coleman KJ, Quinn VP, Yau VM, Qian YX, Croen LA. Psychotropic Medication Use among Insured Children with Autism Spectrum Disorder. J Autism Dev Disord. 2017; 47:144–54. https://doi.org/10.1007/s10803-016-2946-7 [PubMed]
-
215.
Farmer C, Thurm A, Grant P. Pharmacotherapy for the core symptoms in autistic disorder: current status of the research. Drugs. 2013; 73:303–14. https://doi.org/10.1007/s40265-013-0021-7 [PubMed]
-
216.
Yatawara CJ, Einfeld SL, Hickie IB, Davenport TA, Guastella AJ. The effect of oxytocin nasal spray on social interaction deficits observed in young children with autism: a randomized clinical crossover trial. Mol Psychiatry. 2016; 21:1225–31. https://doi.org/10.1038/mp.2015.162 [PubMed]
-
217.
Watanabe T, Kuroda M, Kuwabara H, Aoki Y, Iwashiro N, Tatsunobu N, Takao H, Nippashi Y, Kawakubo Y, Kunimatsu A, Kasai K, Yamasue H. Clinical and neural effects of six-week administration of oxytocin on core symptoms of autism. Brain. 2015; 138:3400–12. https://doi.org/10.1093/brain/awv249 [PubMed]
-
218.
Guastella AJ, Hickie IB. Oxytocin Treatment, Circuitry, and Autism: A Critical Review of the Literature Placing Oxytocin Into the Autism Context. Biol Psychiatry. 2016; 79:234–42. https://doi.org/10.1016/j.biopsych.2015.06.028 [PubMed]
-
219.
Kang H, Sun LD, Atkins CM, Soderling TR, Wilson MA, Tonegawa S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell. 2001; 106:771–83. https://doi.org/10.1016/S0092-8674(01)00497-4 [PubMed]
-
220.
Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002; 3:175–90. https://doi.org/10.1038/nrn753 [PubMed]
-
221.
Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Carmant L, D’Anjou G, Fombonne E, Addington AM, et al, and Synapse to Disease Group. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009; 360:599–605. https://doi.org/10.1056/NEJMoa0805392 [PubMed]
-
222.
Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010; 42:1021–26. https://doi.org/10.1038/ng.677 [PubMed]
-
223.
Huang JY, Lu HC. mGluR5 Tunes NGF/TrkA Signaling to Orient Spiny Stellate Neuron Dendrites Toward Thalamocortical Axons During Whisker-Barrel Map Formation. Cereb Cortex. 2018; 28:1991–2006. https://doi.org/10.1093/cercor/bhx105 [PubMed]
-
224.
Gilbert J, Man HY. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front Cell Neurosci. 2017; 11:359–359. https://doi.org/10.3389/fncel.2017.00359 [PubMed]
-
225.
Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012; 489:385–90. https://doi.org/10.1038/nature11356 [PubMed]
-
226.
Nickel K, Tebartz van Elst L, Domschke K, Gläser B, Stock F, Endres D, Maier S, Riedel A. Heterozygous deletion of SCN2A and SCN3A in a patient with autism spectrum disorder and Tourette syndrome: a case report. BMC Psychiatry. 2018; 18:248–248. https://doi.org/10.1186/s12888-018-1822-8 [PubMed]
-
227.
Braat S, Kooy RF. The GABAA Receptor as a Therapeutic Target for Neurodevelopmental Disorders. Neuron. 2015; 86:1119–30. https://doi.org/10.1016/j.neuron.2015.03.042 [PubMed]
-
228.
Chen J, Yu S, Fu Y, Li X. Synaptic proteins and receptors defects in autism spectrum disorders. Front Cell Neurosci. 2014; 8:276–276. https://doi.org/10.3389/fncel.2014.00276 [PubMed]
-
229.
Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004; 119:1013–26. https://doi.org/10.1016/j.cell.2004.11.035 [PubMed]
-
230.
Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci USA. 2009; 106:17998–8003. https://doi.org/10.1073/pnas.0910297106 [PubMed]
-
231.
Yadav R, Gupta SC, Hillman BG, Bhatt JM, Stairs DJ, Dravid SM. Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One. 2012; 7:e32969. https://doi.org/10.1371/journal.pone.0032969 [PubMed]
-
232.
Dedic N, Pöhlmann ML, Richter JS, Mehta D, Czamara D, Metzger MW, Dine J, Bedenk BT, Hartmann J, Wagner KV, Jurik A, Almli LM, Lori A, et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry. 2018; 23:533–43. https://doi.org/10.1038/mp.2017.133 [PubMed]
-
233.
Sato A. mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol Disord Drug Targets. 2016; 15:533–43. https://doi.org/10.2174/1871527315666160413120638 [PubMed]
-
234.
Xing X, Zhang J, Wu K, Cao B, Li X, Jiang F, Hu Z, Xia K, Li JD. Suppression of Akt-mTOR pathway rescued the social behavior in Cntnap2-deficient mice. Sci Rep. 2019; 9:3041. https://doi.org/10.1038/s41598-019-39434-5 [PubMed]
-
235.
Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011; 147:235–46. https://doi.org/10.1016/j.cell.2011.08.040 [PubMed]
-
236.
Clifton NE, Cameron D, Trent S, Sykes LH, Thomas KL, Hall J. Hippocampal Regulation of Postsynaptic Density Homer1 by Associative Learning. Neural Plast. 2017; 2017:5959182–5959182. https://doi.org/10.1155/2017/5959182 [PubMed]
-
237.
Hu VW, Sarachana T, Kim KS, Nguyen A, Kulkarni S, Steinberg ME, Luu T, Lai Y, Lee NH. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009; 2:78–97. https://doi.org/10.1002/aur.73 [PubMed]
-
238.
Amorosi S, D’Armiento M, Calcagno G, Russo I, Adriani M, Christiano AM, Weiner L, Brissette JL, Pignata C. FOXN1 homozygous mutation associated with anencephaly and severe neural tube defect in human athymic Nude/SCID fetus. Clin Genet. 2008; 73:380–84. https://doi.org/10.1111/j.1399-0004.2008.00977.x [PubMed]
-
239.
Amorosi S, Vigliano I, Del Giudice E, Panico L, Maruotti GM, Fusco A, Quarantelli M, Ciccone C, Ursini MV, Martinelli P, Pignata C. Brain alteration in a Nude/SCID fetus carrying FOXN1 homozygous mutation. J Neurol Sci. 2010; 298:121–3. https://doi.org/10.1016/j.jns.2010.08.066 [PubMed]
-
240.
Wang L, Jia M, Yue W, Tang F, Qu M, Ruan Y, Lu T, Zhang H, Yan H, Liu J, Guo Y, Zhang J, Yang X, Zhang D. Association of the ENGRAILED 2 (EN2) gene with autism in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet. 2008; 147B:434–38. https://doi.org/10.1002/ajmg.b.30623 [PubMed]
-
241.
Gadad BS, Hewitson L, Young KA, German DC. Neuropathology and animal models of autism: genetic and environmental factors. Autism Res Treat. 2013; 2013:731935–731935. https://doi.org/10.1155/2013/731935 [PubMed]
-
242.
Liu X, Novosedlik N, Wang A, Hudson ML, Cohen IL, Chudley AE, Forster-Gibson CJ, Lewis SM, Holden JJ. The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur J Hum Genet. 2009; 17:228–35. https://doi.org/10.1038/ejhg.2008.148 [PubMed]
-
243.
Venero C, Guadaño-Ferraz A, Herrero AI, Nordström K, Manzano J, de Escobar GM, Bernal J, Vennström B. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev. 2005; 19:2152–63. https://doi.org/10.1101/gad.346105 [PubMed]
-
244.
Kalikiri MK, Mamidala MP, Rao AN, Rajesh V. Analysis and functional characterization of sequence variations in ligand binding domain of thyroid hormone receptors in autism spectrum disorder (ASD) patients. Autism Res. 2017; 10:1919–28. https://doi.org/10.1002/aur.1838 [PubMed]
-
245.
Yin CL, Chen HI, Li LH, Chien YL, Liao HM, Chou MC, Chou WJ, Tsai WC, Chiu YN, Wu YY, Lo CZ, Wu JY, Chen YT, Gau SS. Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol Autism. 2016; 7:23–23. https://doi.org/10.1186/s13229-016-0087-7 [PubMed]
-
246.
Bae SM, Hong JY. The Wnt Signaling Pathway and Related Therapeutic Drugs in Autism Spectrum Disorder. Clin Psychopharmacol Neurosci. 2018; 16:129–35. https://doi.org/10.9758/cpn.2018.16.2.129 [PubMed]
-
247.
Dong F, Jiang J, McSweeney C, Zou D, Liu L, Mao Y. Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Hum Mol Genet. 2016; 25:2738–51. https://doi.org/10.1093/hmg/ddw131 [PubMed]
-
248.
Sowers LP, Loo L, Wu Y, Campbell E, Ulrich JD, Wu S, Paemka L, Wassink T, Meyer K, Bing X, El-Shanti H, Usachev YM, Ueno N, et al. Disruption of the non-canonical Wnt gene PRICKLE2 leads to autism-like behaviors with evidence for hippocampal synaptic dysfunction. Mol Psychiatry. 2013; 18:1077–89. https://doi.org/10.1038/mp.2013.71 [PubMed]
-
249.
Chung RH, Ma D, Wang K, Hedges DJ, Jaworski JM, Gilbert JR, Cuccaro ML, Wright HH, Abramson RK, Konidari I, Whitehead PL, Schellenberg GD, Hakonarson H, et al. An X chromosome-wide association study in autism families identifies TBL1X as a novel autism spectrum disorder candidate gene in males. Mol Autism. 2011; 2:18. https://doi.org/10.1186/2040-2392-2-18 [PubMed]
-
250.
Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism. 2013; 4:17–17. https://doi.org/10.1186/2040-2392-4-17 [PubMed]
-
251.
Mohn JL, Alexander J, Pirone A, Palka CD, Lee SY, Mebane L, Haydon PG, Jacob MH. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry. 2014; 19:1133–42. https://doi.org/10.1038/mp.2014.61 [PubMed]
-
252.
Yi JJ, Paranjape SR, Walker MP, Choudhury R, Wolter JM, Fragola G, Emanuele MJ, Major MB, Zylka MJ. The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J Biol Chem. 2017; 292:12503–15. https://doi.org/10.1074/jbc.M117.788448 [PubMed]
-
253.
Noor A, Dupuis L, Mittal K, Lionel AC, Marshall CR, Scherer SW, Stockley T, Vincent JB, Mendoza-Londono R, Stavropoulos DJ. 15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with Developmental Delay and Neuropsychiatric Phenotypes. Hum Mutat. 2015; 36:689–93. https://doi.org/10.1002/humu.22800 [PubMed]
-
254.
Okerlund ND, Cheyette BN. Synaptic Wnt signaling-a contributor to major psychiatric disorders? J Neurodev Disord. 2011; 3:162–74. https://doi.org/10.1007/s11689-011-9083-6 [PubMed]
-
255.
Nair A, Treiber JM, Shukla DK, Shih P, Müller RA. Impaired thalamocortical connectivity in autism spectrum disorder: a study of functional and anatomical connectivity. Brain. 2013; 136:1942–55. https://doi.org/10.1093/brain/awt079 [PubMed]
-
256.
O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012; 338:1619–22. https://doi.org/10.1126/science.1227764 [PubMed]
-
257.
Vaqueiro AC, de Oliveira CP, Cordoba MS, Versiani BR, de Carvalho CX, Alves Rodrigues PG, de Oliveira SF, Mazzeu JF, Pic-Taylor A. Expanding the spectrum of TBL1XR1 deletion: report of a patient with brain and cardiac malformations. Eur J Med Genet. 2018; 61:29–33. https://doi.org/10.1016/j.ejmg.2017.10.008 [PubMed]
-
258.
Kloth K, Denecke J, Hempel M, Johannsen J, Strom TM, Kubisch C, Lessel D. First de novo ANK3 nonsense mutation in a boy with intellectual disability, speech impairment and autistic features. Eur J Med Genet. 2017; 60:494–498. https://doi.org/10.1016/j.ejmg.2017.07.001 [PubMed]
-
259.
Wong CT, Bestard-Lorigados I, Crawford DA. Autism-related behaviors in the cyclooxygenase-2-deficient mouse model. Genes Brain Behav. 2019; 18:e12506. https://doi.org/10.1111/gbb.12506 [PubMed]