



Introduction
Cardiovascular disease (CVD) is still the leading cause of death worldwide due to its high morbidity and mortality; it not only reduces human life span but also places a heavy burden to the national health care system according to the latest authoritative statistics [1]. Atherosclerosis, a major inducer of CVD, is a chronic inflammatory disease arising from an imbalance in lipid metabolism and a maladaptive immune response driven by the accumulation of cholesterol-laden macrophages in the arterial wall [2]. During atherosclerotic lesion formation, macrophage polarization, which leads to diverse phenotypes, is a critical process that depends on various stimuli [3]. Notably, it has been concluded that the anti-inflammatory M2 macrophage phenotype exerts atheroprotective effects, although anti-inflammatory CD163+ macrophages also promote angiogenesis and vascular permeability [4]. Oxidized low-density lipoprotein (ox-LDL), which contributes directly to macrophage polarization, induces foam cell formation, ultimately promoting plaque formation [5]. Previous opinions on atherosclerosis therapy have mainly focused on lipid-lowering, antithrombotic, antioxidative and anti-inflammatory strategies, and have ignored macrophage polarization [6]. Thus, the pharmacological targeting of macrophage polarization represents a promising therapeutic strategy for atherosclerosis.
Increasing literature has reported the significant role of autophagy, a lysosome-mediated conserved cellular pathway that controls protein and organelle degradation in human health and disease [7]. As expected, autophagy is an emerging therapeutic target for atherosclerosis that has been summarized recently [8, 9]. According to the recent literature, macrophage autophagy induction prevents atherosclerosis [10], whereas impaired macrophage autophagy increases atherosclerotic plaque formation [11]. Silent information regulator 1 (Sirt1), a member of the sirtuin class of proteins, is widely studied and shows atheroprotective effects in macrophages [12], endothelial cells [13], and vascular smooth muscle cells [14]. Thus, screening for potent selective Sirt1 activators has been the focus of research in the antiatherosclerosis drug development field, and more research into the mechanism by which Sirt1 activation affects atherosclerosis is imperative.
Clinically, although lipid-lowering statins and anti-inflammatory canakinumab have been used for atherosclerosis treatment, they also have inevitable side effects [15–17] and do not meet clinical needs. In recent decades, strategies involving natural products have focused on cell autophagy for atherosclerosis prophylaxis and treatment, and many natural compounds have been indicated to exhibit excellent antiatherosclerotic properties [18]. Araloside C (AsC, Figure 1), a bioactive triterpenoid, is the major active constituent of Aralia elata (Miq.) Seem, which has been widely used in traditional Chinese medicine [19]. Recently, our group reported that AsC alleviated hypoxia/reoxygenation-induced cardiomyocyte apoptosis in vitro [20] and ex vivo [21] studies. Moreover, we found that total saponins of Aralia elata (Miq.) (TASAES) protected against endothelial cell injury and atherosclerosis in ApoE-/- mice [22, 23]. According to the emerging reports of the cardioprotective effects of AsC and the endothelial protective effects of TASAES, we believe that the antiatherosclerotic effects of AsC and its possible molecular mechanism need to be elucidated.
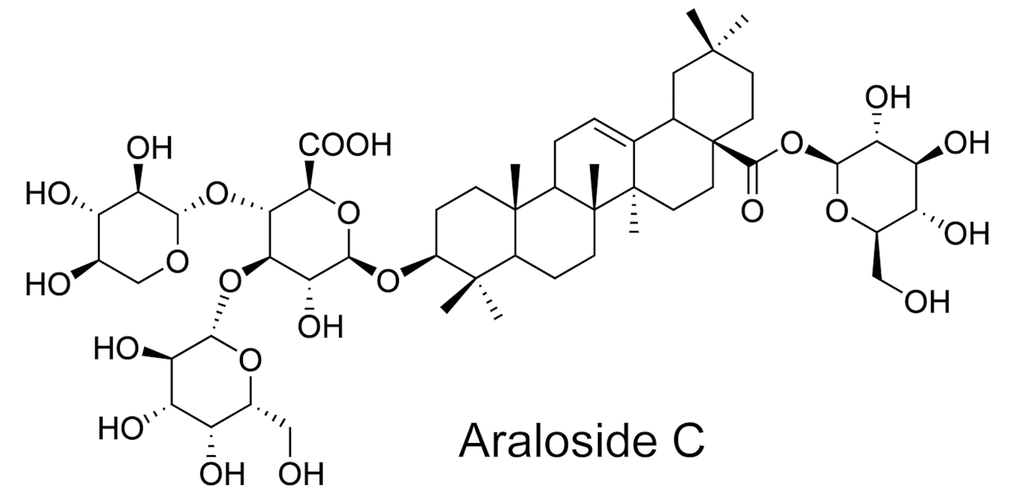
Figure 1. The chemical structure of Araloside C (AsC).
Based on our previous research, this study is the first to investigate the antiatherosclerotic effects and underlying mechanism of AsC on ox-LDL-induced foam cell formation. Additionally, we speculate that AsC attenuates foam cell formation and lessens atherosclerosis by modulating macrophage polarization via Sirt1-mediated autophagy.
Results
AsC attenuated atherosclerosis in HFD-induced ApoE-/- mice and reduced foam cell formation in vitro
To test whether AsC exerts an antiatherosclerotic effect, we first measured the weight, blood lipid levels and atherosclerotic area at the aortic root of high-fat diet (HFD)-treated ApoE-/- mice according to our previous method [24]. Unexpectedly, no significant differences in weight, blood lipid levels (Figure 2B and 2C), fat and lean proportion, or necrotic core (Supplementary Figure 1) were observed upon AsC treatment. Moreover, as shown in Figure 2D and 2E, the mice in the AsC group developed significantly smaller plaque areas in the aortic root than those in the model group after 4 weeks of treatment. We further examined the effects of AsC on serum lipid profiles. These data suggest that the antiatherosclerotic effect of AsC was not dependent on lipid level regulation.
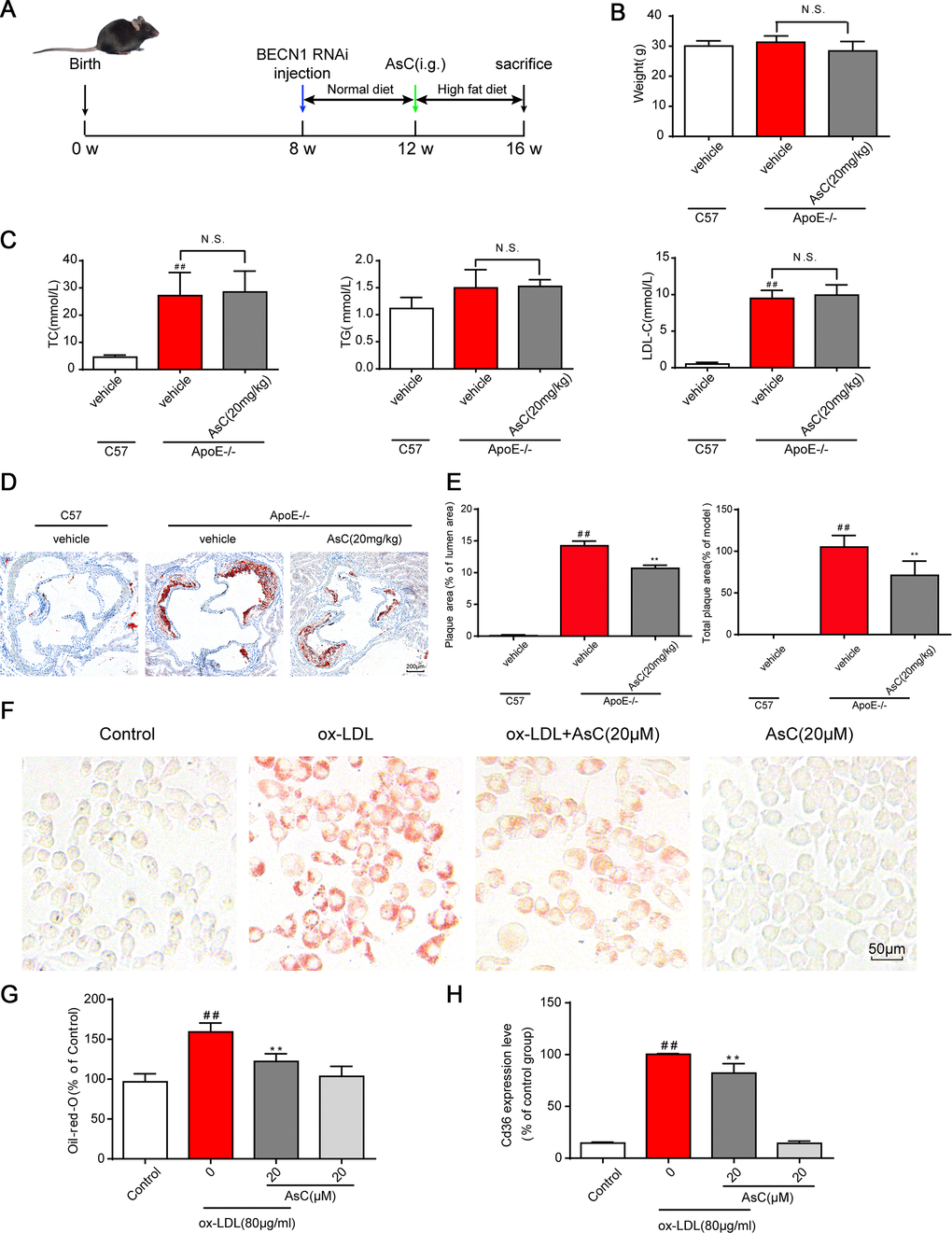
Figure 2. AsC attenuated atherosclerosis in HFD-fed ApoE-/- mice and reduced foam cell formation in vitro. All mice were fed a HFD in the presence or absence of AsC (20 mg·kg-1·day-1, i.g.) for 4 weeks. In the in vitro assay, RAW264.7 cells were pretreated with AsC (20 μM) for 12 h, and then exposed to ox-LDL for another 24 h. (A) Experimental protocol of the in vivo study. (B) Body weight. (C) Blood lipid levels. (D) Representative images of oil red O staining of the aortic root. (E) Quantification of the plaque area by oil red O staining. (F) Representative images of oil red O staining in ox-LDL-treated RAW264.7 cells. (G) Quantification of oil red O staining, as detected by a microplate reader. (H) Cd36 expression level in ox-LDL-treated RAW264.7 cells, as determined by flow cytometry. The data are presented as the means ± SDs (n = 5). ##P < 0.01 vs. the control group, **P < 0.01 vs. the model group; N.S. means no significance.
Macrophage-derived foam cells play an important role in atherosclerosis formation. Next, we analyzed the effects of AsC on ox-LDL-induced foam cells in vitro, and the results showed that AsC remarkably decreased foam cell formation (Figure 2F and 2G). Meanwhile, compared with ox-LDL group, it ameliorated Cd36 expression (Figure 2H, Supplementary Figure 2), which strengthened our conclusion. Collectively, these findings confirm that AsC had antiatherosclerotic effects and reduced foam cell formation.
AsC polarized macrophages to an M2-like phenotype
Mounting evidence points to a key role of macrophage polarization in plaque progression and vulnerability [5].
We thus detected the expression of macrophage polarization markers in the aortic root in control and AsC-treated mice. Immunostaining analysis showed that the expression of Cd86 was significantly decreased in the AsC group, whereas the expression of Arg1 was significantly increased (Figure 3A and 3B). A similar tendency was observed in the ox-LDL-induced macrophage model by flow cytometry analysis (Figure 3C, Supplementary Figure 3). Arginase activity is also a macrophage polarization marker, as indicated in Figure 3D. AsC treatment obviously increased arginase activity. Moreover, we also detected the mRNA and protein expression levels of M1 and M2 macrophage markers. As shown in Figure 3E–3G, AsC significantly downregulated Nos2, Il1b, and Cd86 expression, and upregulated Arg1 and Mrc1 expression, which was consistent with a previous study [25]. These results indicate that AsC pretreatment was able to polarize macrophages to an M2-like phenotype.
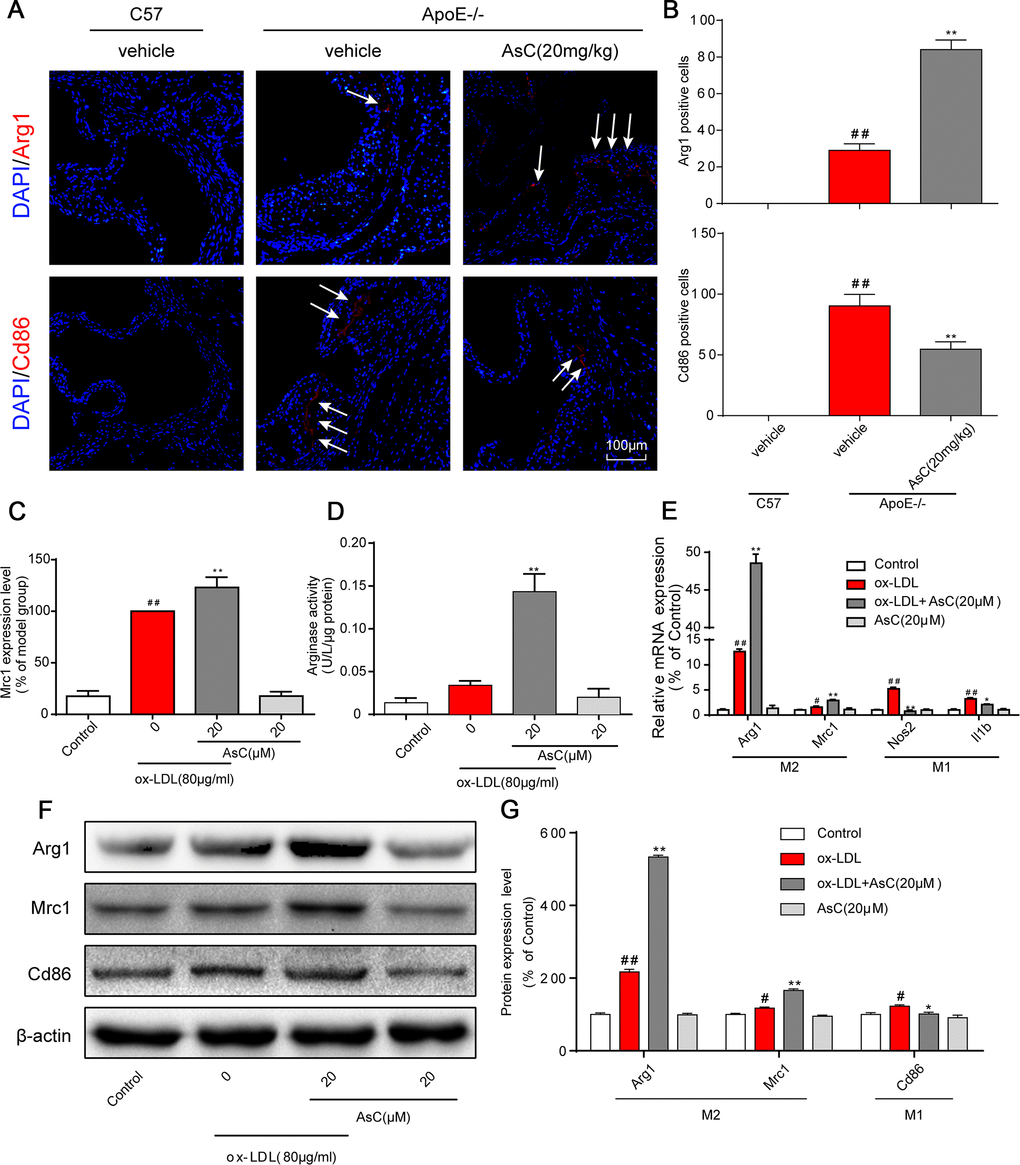
Figure 3. AsC polarized macrophages to an M2-like phenotype. All mice were fed a HFD in the presence or absence of AsC (20 mg·kg-1·day-1, i.g.) for 4 weeks. In the in vitro assay, RAW264.7 cells were pretreated with AsC (20 μM) for 12 h, and then exposed to ox-LDL for another 24 h. (A) Dual immunofluorescence staining for Arg1 (red) or Cd86 (red) and DAPI (blue) in lesions in the aortic root. (B) Quantification of the relative fluorescence intensity. (C) The Mrc1 expression level in ox-LDL-treated macrophages, as determined by flow cytometry. (D) Arginase activity was measured as described in the Methods section. (E) mRNA levels of Arg1, Mrc1, Nos2 and Il1b in macrophages, as quantified by real-time PCR. (F) Representative photographs of Mrc1, Cd86 and Arg1 expression in ox-LDL-treated macrophages, as evaluated by western blot analysis. (G) Statistical results of Mrc1, Cd86 and Arg1 expression levels compared with those in the control group. The data are presented as the means ± SDs (n = 5). #P < 0.05, ##P < 0.01 vs. the control group, **P < 0.01 vs. the model group.
AsC induced macrophage autophagy
Ongoing laboratory studies have demonstrated that autophagy is a therapeutic target for atherosclerosis [8]. To determine whether AsC regulates autophagy, we first investigated autophagosomes by TEM, the most valid method for both qualitative and quantitative analysis of autophagy [26]. The results showed that AsC pretreatment increased the number of autophagosomes in ox-LDL-treated macrophages, but that the number of autophagosomes decreased when the cells were pretreated with the autophagy inhibitor 3-MA (Figure 4A and 4B). Cyto-ID® and flow cytometric assays demonstrated that AsC treatment increased autophagic vacuoles and flux (Figure 4C), further confirming AsC-induced autophagy in ox-LDL-stimulated macrophages. Next, we determined the level of LC3II, one of the gold standard markers of autophagosome formation [27]. Our data indicated that AsC dramatically elevated LC3II expression levels, suggesting that autophagic flux was increased, and these levels were also blocked by 3-MA (Figure 4D and 4E). To further confirm the role of AsC in the modulation of autophagic flux, we measured the expression levels of autophagy-related proteins. As shown in Figure 4F and 4G, AsC significantly increased the ratio of LC3II/LC3I, which is considered an accurate indicator of autophagy, upregulated BECN1 and ATG5 expression levels, and reduced the P62 expression level. Similar results were confirmed in aortic lysates (Figure 4H and 4I). Taken together, these findings strongly indicate that pretreatment with AsC enhanced ox-LDL-induced macrophage autophagy level.
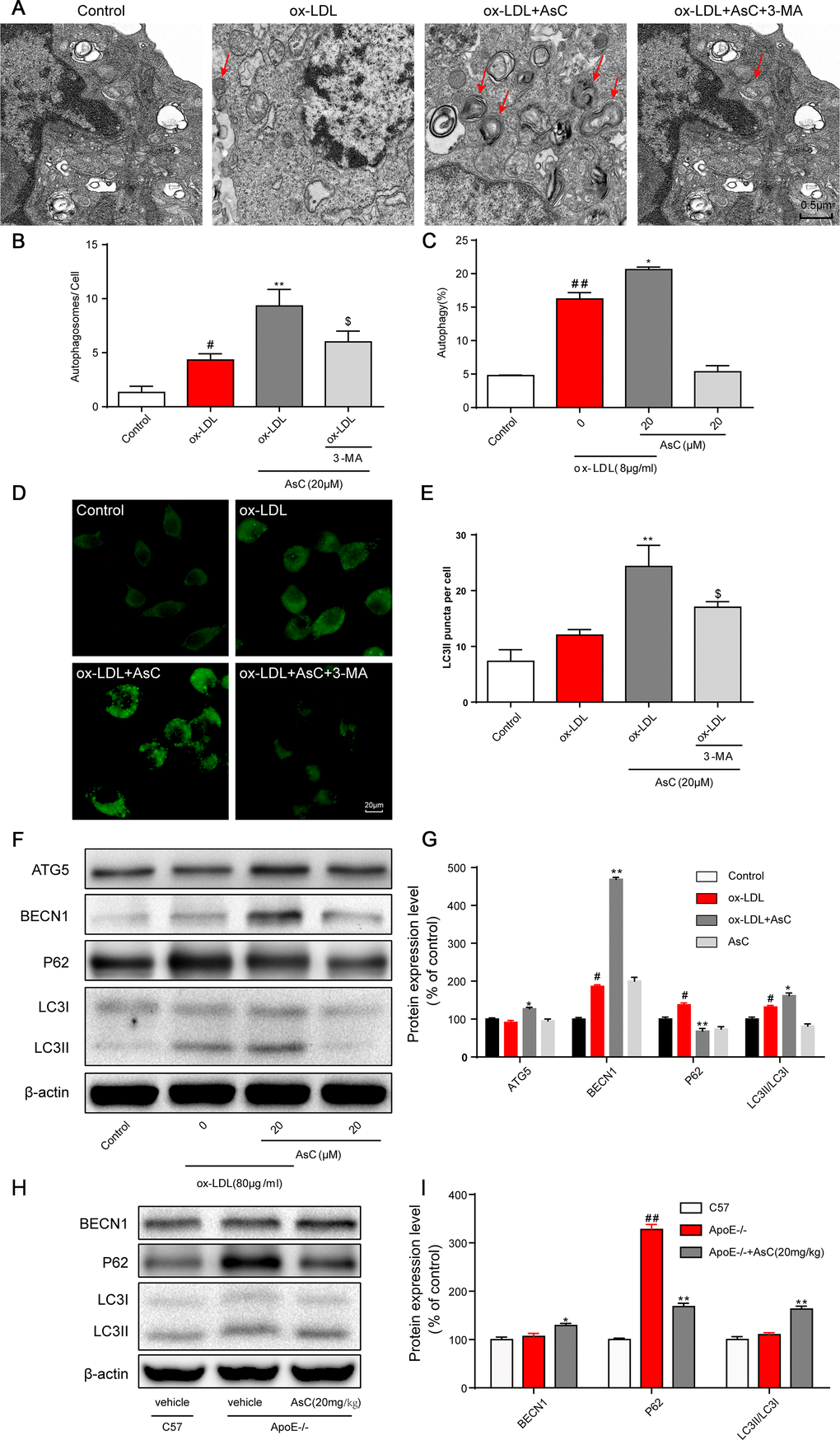
Figure 4. AsC induced macrophage autophagy. RAW264.7 cells were pretreated with 3-MA (5 mM) for 2 h, treated with AsC (20 μM) for 12 h, and then exposed to ox-LDL for another 24 h. (A) Representative photographs of autophagosomes (red arrows) examined using a JEOL JEM1230 electron microscope. (B) Statistical results of autophagosomes. (C) Summarized data showing the percentage of cells that were positive for CytoID fluorescence, as detected by flow cytometry analysis. (D) Representative photographs of LC3II staining. (E) Statistical results of LC3II-positive cells. (F) Representative photographs of ATG5, BECN1, P62, LC3 and β-actin expression in ox-LDL-treated macrophages, as evaluated by western blot analysis. (G) Statistical results of ATG5, BECN1, P62, LC3II/LC3I expression levels compared with those in the control group. (H) Representative photographs of BECN1, P62, LC3 and β-actin expression in aortic lysates. (I) Statistical results of BECN1, P62, LC3II/LC3I expression levels compared to those in the control group. The data are presented as the means ± SDs (n = 5). #P < 0.05, ##P < 0.01 vs. the control group, *P < 0.05, **P < 0.01 vs. the model group; $P < 0.05 vs. the ox-LDL and AsC group.
Autophagy inhibition blocked AsC-mediated antiatherosclerotic effects and macrophage polarization
Based on our above research results, we next investigated the effects of AsC-mediated autophagy on atherosclerosis and macrophage polarization. First, our in vivo data showed that the reduction in plaque area by AsC was significantly reversed by the autophagy inhibition (Figure 5A and 5D). Similarly, the increase in Arg1 expression by AsC was significantly inhibited by BECN1 knockdown (Figure 5B and 5E). Moreover, the inhibitory effect of AsC on ox-LDL-induced foam cell formation was also abrogated by 3-MA in RAW264.7 cells (Figure 5C and 5F). In addition, AsC-mediated macrophage polarization was also abolished by BECN1 siRNA in vitro (Figure 5G and 5H). Together, these data suggest that AsC mitigated atherosclerosis and promoted macrophage polarization through the promotion of macrophage autophagy.
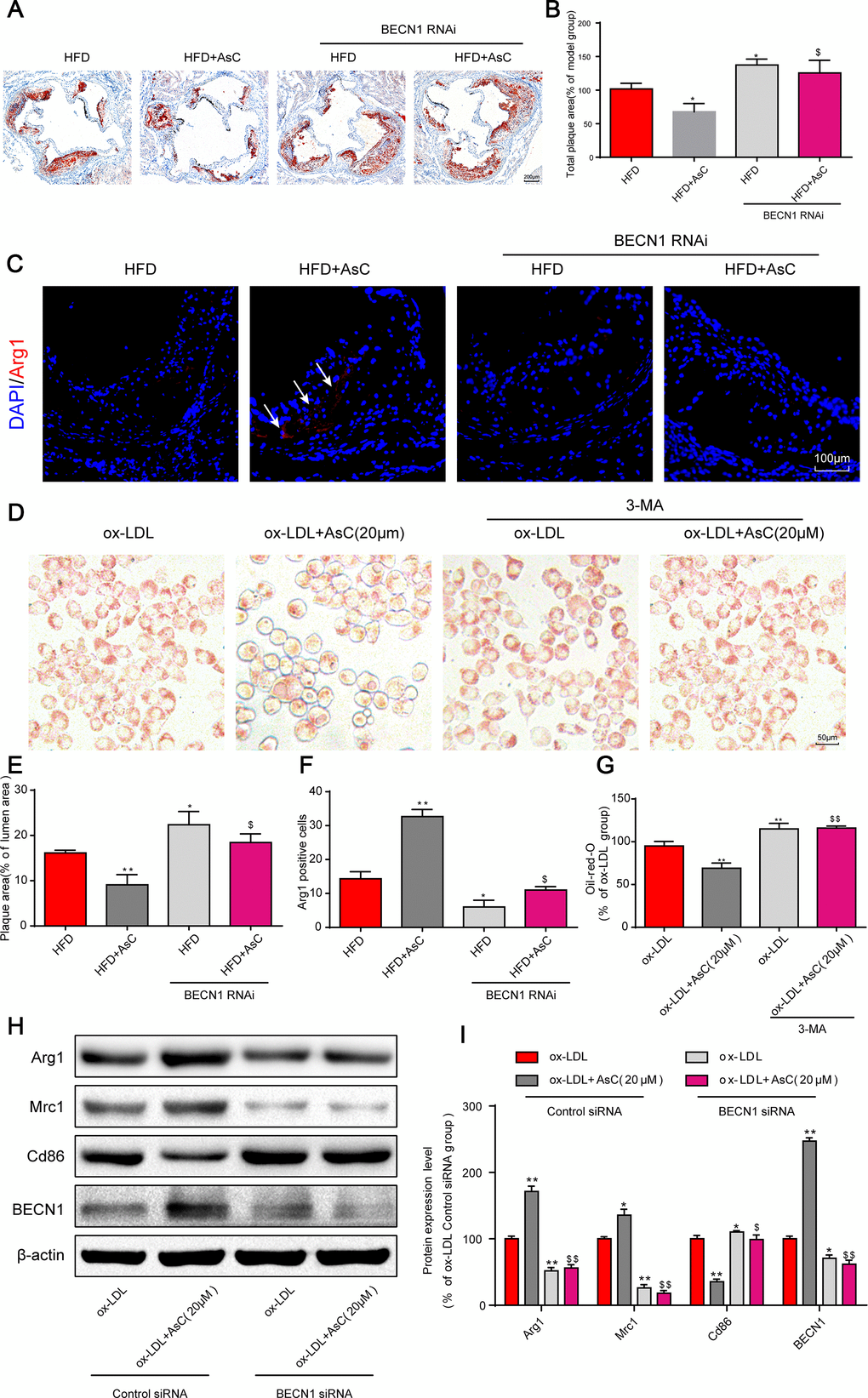
Figure 5. Autophagy inhibition abolished AsC-mediated antiatherosclerotic effects and macrophage polarization. All mice were fed a HFD in the presence or absence of AsC (20 mg·kg-1·day-1, i.g.) for 4 weeks. In the in vitro assay, RAW264.7 cells were pretreated with 3-MA (5 mM) for 2 h, treated with AsC (20 μM) for 12 h, and then exposed to ox-LDL for another 24 h. (A) Representative images of oil red O staining of the aortic root. (B) Quantification of the total plaque area. (C) Dual immunofluorescence staining forArg1 (red) and DAPI (blue) in lesions in the aortic root. (D) Representative images of oil red O staining of ox-LDL-treated RAW264.7 cells. (E) The percentage of plaque area relative to lumen area. (F) Quantification of relative fluorescence intensity. (G) Quantification of oil red O staining, as detected by a microplate reader. (H) Representative photographs of Arg1, Mrc1, Cd86 and BECN1 expression, as evaluated by western blot analysis. (I) Statistical results of Mrc1, Cd86 and Arg1 expression levels compared with those in the ox-LDL-treated group. The data are presented as the means ± SDs (n = 5). *P < 0.05, **P < 0.01 vs. the model group; $P < 0.05, $$P < 0.01 vs. the ox-LDL and AsC group.
AsC promoted Sirt1 expression in macrophages
Next, we explored the mechanism of AsC-mediated autophagy in macrophages. Sirt1 is a well-known autophagy regulator [28]; therefore, we determined the Sirt1 expression level in macrophages in the aortic root and ox-LDL-activated macrophages. Immunofluorescence colocalization assays indicated that compared with vehicle, AsC notably increased Sirt1 expression in atherosclerotic macrophages (Figure 6A and 6B). Moreover, our results showed that Sirt1 levels increased in response to AsC pretreatment in a dose- and time-dependent manner (Figure 6C and 6D).
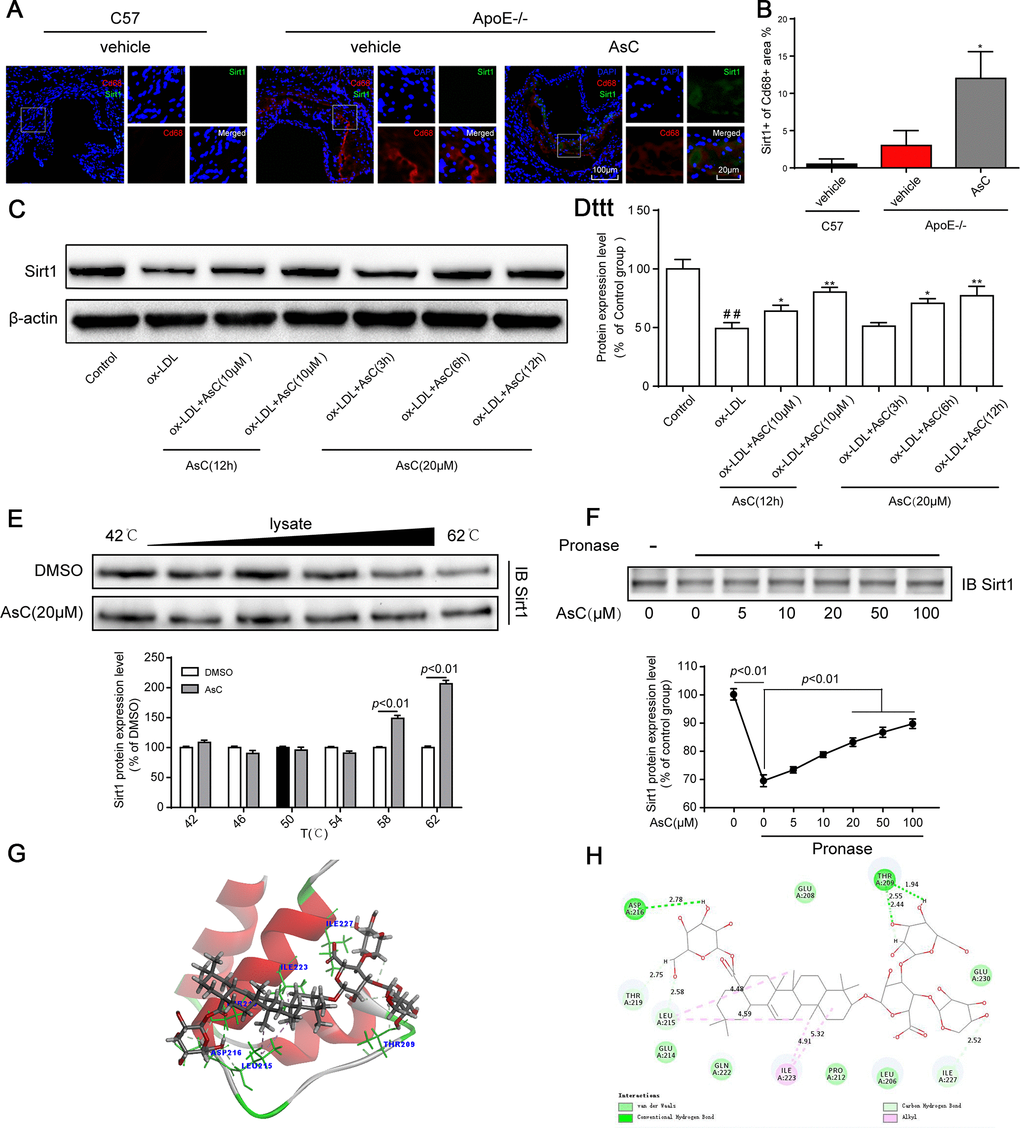
Figure 6. AsC promoted Sirt1 expression in macrophages. All mice were fed a HFD in the presence or absence of AsC (20 mg·kg-1·day-1, i.g.) for 4 weeks. In the in vitro assay, RAW264.7 cells were pretreated with AsC (20 μM) for 12 h, and then exposed to ox-LDL for another 24 h. (A) Aortic roots from ApoE−/− mice were stained for the macrophage marker Cd68 and coprobed with antibodies against Sirt1. (B) Quantification of Sirt1 expression in aortic root lesions. (C) Representative photographs of Sirt1 expression, as evaluated by western blot analysis. (D) Statistical results of the Sirt1 expression level compared with that in the control group. (E) Cellular thermal shift assay (CETSA) using macrophage lysates, which were exposed to AsC (20 μM). (F) AsC promoted the resistance of its target protein Sirt1 to proteases (DARTS). (G) Three-dimensional modeling of the binding of AsC to the binding domain of Sirt1. (H) Two-dimensional ligand interaction diagram of AsC and SIRT1. The data are presented as the means ± SDs (n = 5). ##P < 0.01 vs. the control group; *P < 0.05, **P < 0.01 vs. the model group.
Meanwhile, we explored the interaction between AsC and Sirt1 by CETSA and a DARTS assay. As shown in Figure 6E and 6F, compared with the control, treatment with AsC significantly inhibited Sirt1 degradation induced by temperature and pronase, which was in accordance with a previous study [29]. Next, we further examined the potential interaction between AsC and Sirt1 protein through molecular docking. As shown in Figure 6G, the N-terminus of Sirt1 forms an independently folded three α-helix bundle, and AsC binds to the helix-turn-helix motif. Meanwhile, it was observed that AsC interacted extensively with several important amino acids in the N-terminus: it formed hydrogen bonds with THR 209, LEU 215, ASP 216, THR 219 and ILE 227, van der Waals interactions with LEU 206, GLU 208, PRO 212, GLU 214, GLN 222 and GLU 230, and alkyl interactions with ILE 223 (Figure 6H). This result is basically the same as that reported by other researchers [30].
The in vivo, in vitro and molecular docking results, suggest that AsC prevented foam cell and atherosclerosis formation by targeting and upregulating Sirt1 expression.
AsC-mediated polarization and autophagy in ox-LDL-treated macrophages were Sirt1-dependent
Previous studies have shown that Sirt1 activation is necessary for autophagic flux activation [31]. It has been demonstrated that Sirt1 could be an ideal target for drugs [32]. We hypothesized that Sirt1 is involved in autophagic flux activation by AsC. Thus, the Sirt1 inhibitor EX527 and Sirt1 siRNA were utilized to investigate whether Sirt1 activation was essential for AsC-induced autophagic flux in our study. After treatment with EX527, the modulation of Mrc1 and ATG5 by AsC was not detected in macrophages, demonstrating that the activation of polarization and autophagy were abolished by Sirt1 inhibition (Figure 7A–7D). Sirt1 siRNA had similar effects, and these results collectively demonstrated that AsC enhanced autophagy through SIRT1 signaling (Figure 7E and 7F).
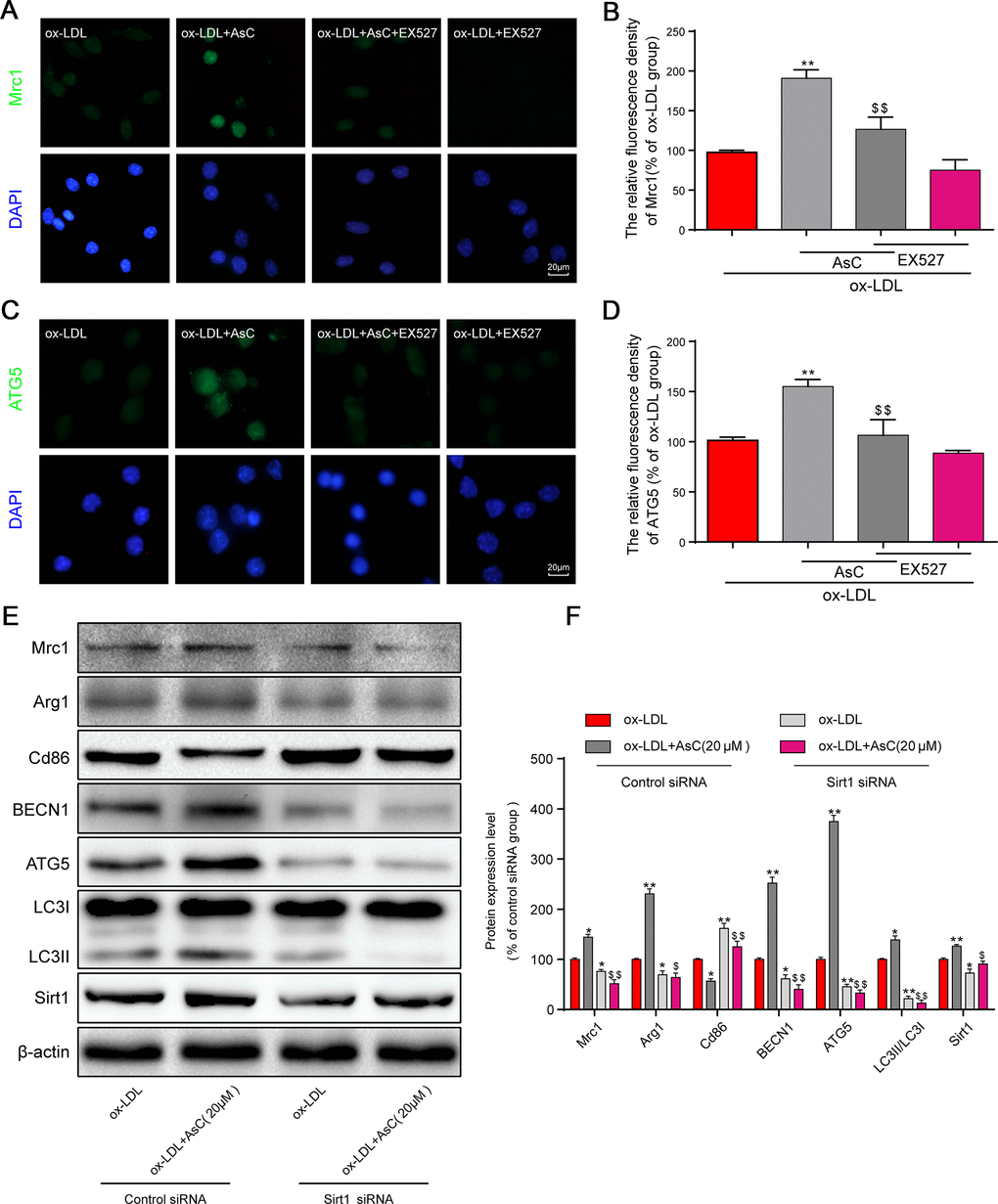
Figure 7. AsC-induced autophagy and M2 phenotype polarization were Sirt1-dependent in ox-LDL-treated macrophages. Sirt1 was inhibited by 10 μM EX527 for 6 h or knocked down by siRNA, as described in the Materials and Methods section. Twenty-four hours posttransfection, cells were treated with AsC (20 μM) for 12 h and then incubated with ox-LDL (80 μg/mL) for an additional 24 h. (A, C) Representative immunofluorescence images showing Mrc1 and ATG5 expression in RAW264.7 cells. (B, D) The relative quantitative analysis of Mrc1 and ATG5 fluorescence in RAW264.7 cells. (E) Representative western blot analysis of Mrc1, Arg1, Cd86, BECN1, ATG5, LC3, Sirt1 and β-actin in macrophages. (F) Quantification of the expression of Mrc1, Arg1, Cd86, BECN1, ATG5, LC3, and Sirt1. The data are presented as the means ± SDs (n = 5). *P < 0.05, **P < 0.01 vs. the ox-LDL group; $P < 0.05, $$P < 0.01 vs. the ox-LDL and AsC group.
Together, these results suggest that decreased Sirt1 expression is involved in the impairment of autophagy in macrophages and that the effects of AsC in promoting autophagy may occur through Sirt1 activation (Figure 8).
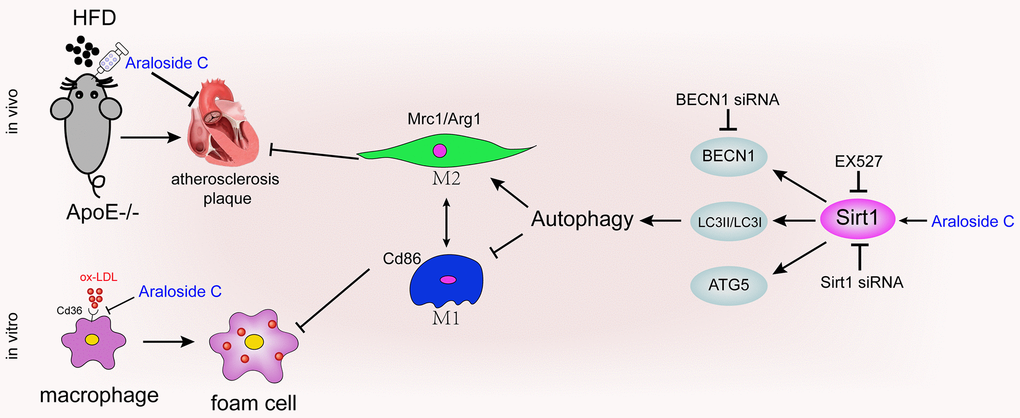
Figure 8. Diagram of the proposed molecular mechanism of the antiatherosclerotic effect of AsC.
Discussion
Although atherosclerosis has been effectively treated through lipid-reducing and anti-inflammatory drugs, the adverse effects associated with muscle symptoms and glucose homeostasis [33] compelled us to find effective and less toxic drugs to improve patient prognosis. In this study, we demonstrated that AsC, a naturally derived saponin, exerted a potent antiatherosclerotic effect. We further confirmed the beneficial pharmacological effect of AsC on macrophage autophagy and phenotype switching. Mechanistically, the administration of AsC potently targeted and activated Sirt1 signaling, which subsequently mediated macrophage autophagy and polarization.
Recently, many natural products have been studied to determine their impact on atherosclerosis [18]. Our group and several others have reported that TASAES exerts therapeutic effects on myocardial damage [34, 35] and atherosclerosis [22, 23]. AsC, isolated from TASAES, was also reported to have powerful cardioprotective effects against ischemia-reperfusion injury in our previous study [20, 21]. Moreover, we demonstrated that another AsC analogue can protect against ox-LDL-damaged endothelial cell injury via autophagy induction [36], which prompted us to explore whether AsC has antiatherosclerotic effects and determine its protective mechanism. Expectedly, our data demonstrated that AsC ameliorated plaque area in atherosclerosis in HFD-fed ApoE-/- mice. Foam cell formation is a key event in the development of atherosclerotic plaques. Furthermore, we found that AsC reduced ox-LDL-induced foam cell formation in vitro through classical oil red O staining and Cd36 detection. In addition, AsC had no obvious effects on blood lipid levels, body weight or the fat or lean to body weight ratio, which implied that the antiatherosclerotic effects of AsC are different from statins. The above results led us to understand how AsC executed its antiatherosclerosis ability.
In past decades, research on antiatherosclerosis drugs mainly focused on lipid-reducing, antiinflammatory, and antioxidative strategies, endothelial protection and foam cell inhibition. Few studies have focused on macrophage polarization, which is also a promising target for atherosclerosis therapy [37, 38]. Generally, the anti-inflammatory macrophage phenotype is considered atheroprotective, although CD163+ macrophages promote atherosclerosis [3, 4]. In this study, we observed a notable change in macrophage polarization. AsC treatment elevated Arg1 expression and reduced Cd86 expression in atherosclerosis, suggesting phenotypic switch to anti-inflammatory M2 macrophages. These results in addition to our oil red O staining results, revealed that the AsC-mediated polarization of macrophages to the M2 was correlated with the dynamics of atherosclerotic plaque regression. Furthermore, the changes in gene or protein expression of macrophages Arg1, Mrc1, Cd86, Nos2 and Il1b were in accordance with the in vivo results. Taken together, our results indicate that AsC specifically regulated macrophage polarization both in vitro and in vivo.
It has been widely reported that macrophage autophagy can reduce the accumulation of foam cells and inhibit the formation and development of plaques [39, 40]. Here, we have analyzed autophagic flux in macrophages. Our results showed that AsC significantly increased the number of macrophage autophagosomes, which were blocked by 3-MA. Meanwhile, the immunofluorescence data showed that the increase in LC3II-positive macrophages by AsC was dramatically decreased by 3-MA. Similar results were also observed at the protein level in ox-LDL-treated macrophages. These results suggest that AsC observably promoted macrophage autophagy.
There is increasing evidence indicating that autophagy is important for the induction of M2 macrophages. Many natural products have been found to mediate the crosstalk of polarization and autophagy in macrophages for disease therapy [41–43]. To study the relationship between ability of AsC to increase autophagy and the macrophage polarization, we constructed BECN1 RNAi mice for use in ApoE-/- mice [44] and BECN1 siRNA for use in macrophages. In the current study, we found that the decrease in plaque area and increase in Arg1 expression by AsC were markedly inhibited by BECN1 RNAi. Moreover, AsC-mediated foam cell inhibition and macrophage phenotype switching were also abolished by 3-MA and BECN1 siRNA, respectively. Therefore, these data strongly support that AsC can enhance autophagy, which contributes to M2 phenotype polarization.
Next, we investigated the molecular mechanism by which AsC protects against atherosclerosis. Sirt1, a nicotinamide adenine dinucleotide-dependent protein deacetylase, has gained attention for its protective effects against atherosclerosis [45, 46]. Excitingly, Sirt1 can modulate macrophage polarization [47] and autophagy [48].
Our present results showed that compared with vehicle, AsC increased the expression of Sirt1 in vivo and in vitro. Moreover, AsC treatment elevates the temperature and enzyme stability of Sirt1, which indicated that Sirt1 may be a target of AsC. Molecular docking results confirmed this conclusion. To confirm the crucial roles of Sirt1 in AsC-mediated macrophage autophagy and polarization, a Sirt1 inhibitor and siRNA were used to inhibit Sirt1 expression. Mrc1 and ATG5 expression were sharply abrogated by EX527 in macrophages. Subsequently, Sirt1 siRNA reversed the AsC induced alterations in Mrc1, Arg1 and Cd86 expression and inhibited the AsC-induced increase in LC3-II/LC3-I, BECN1 and ATG5 expression, and the reduction in p62 expression. Consistent with previous studies, the present results showed that the upregulation of Sirt1 by AsC promoted cell autophagy [28] and polarization [42]. Along with the in vitro, in vivo and molecular docking results, these data led us to hypothesize that AsC modulates autophagy and polarization in macrophages by targeting Sirt1 and upregulating its expression.
In conclusion, our findings in this paper provide novel insights into the molecular mechanisms of AsC against atherosclerosis (Figure 8). AsC should be developed as an efficient candidate, and macrophage polarization should also be considered as a drug target in the clinic. However, in the present work, we selected Sirt1 as the only target gene for the study. However, the antiatherosclerotic effects of AsC are intricate, and one gene cannot fully explain the actions of the compound. Multiple targets, multiple signaling pathways, and multiple biological processes should be explored in future studies to fully elucidate the molecular mechanisms of AsC against atherosclerosis.
Materials and Methods
Reagents
Ox-LDL (by copper ion-induced LDL oxidation, MDA=35 nM) was obtained from Union-Bio Technology (Beijing, China). AsC was isolated in our previous study (Wang et al., 2014) at the Institute of Medicinal Plant Development (Beijing, China). Dimethylsulfoxide (DMSO), 3-(4,5-dimethylthiazol-2yl-)-2, oil red O, DAPI and 3-methyladenine (3-MA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The DMEM Basal Medium and fetal calf serum (FBS) were obtained from HyClone (Logan, UT, USA). The CytoID Autophagy Detection Kit was obtained from Enzo Life Sciences (Farmingdale, NY, USA). Cd36 and Mrc1 flow cytometry antibodies were purchased from Biolegend (San Diego, CA, USA). EX527 was obtained from Target Molecule Corp. (Shanghai, China). Antibodies against P62, LC3II and pronase were obtained from Sigma-Aldrich (St. Louis, MO, USA). NP40 lysis buffer was obtained from Beyotime Biotechnology (Shanghai, China). Anti-mouse and anti-rat IgG (H+L) F(ab')2 fragment antibodies were acquired from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Sirt1, Cd86, Mrc1, Arg1, and BECN1 were purchased from Abcam (Cambridge, United Kingdom). All other antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. Six-week-old (17 ± 1 g) male ApoE-/- mice with a C57BL/6 N background were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and maintained in conventional cages in a temperature controlled facility (temperature: 22 ± 1°C; humidity: 60%) with a 14 h light/10 h dark cycle. A BECN1 knockdown mouse model was constructed by injecting a lentivirus (Lv) expressing RNA inference (RNAi) targeting BECN1, which was purchased from Shanghai GeneChem Co. Ltd. (Shanghai, China), into the tail vein of ApoE-/- mice, according to a previous study [44]. A Lv vector expressing green fluorescence protein only was used as the control. Mice were randomly divided into five experimental groups (n = 8/group): (I) the C57 mouse group; (II) ApoE-/- mouse group; (III) ApoE-/- mouse + AsC group; (IV) ApoE-/- mouse + BECN1 RNAi group; and (V) ApoE-/- mouse + AsC + BECN1 RNAi group. One month after the injection of Lv, the mice were subjected to either AsC (20 mg·kg-1·day-1, i.g.) or normal saline for 4 weeks as described above (Figure 2A). All mice were fed with a high fat diet (HFD, 0.3% cholesterol and 20% pork fat) for 4 weeks. AsC was dissolved in normal saline.
Serum lipids
At the end of the experiment, all mice were fasted overnight and their sera were acquired from the inner canthus. Then, the heart and aortas were separated immediately, and the aortas were stored at -80°C. Serum lipid levels were determined based on commercial kits using a Beckman AU480 biochemical autoanalyzer (Fullerton, CA, USA).
Histological and immunohistochemical assays
Serial cryo-sections (6 μm thick) of the aortic root were stained with oil red O. Sirt1, Cd86, Arg1 expression in the aortic root was detected by immunofluorescence. Briefly, the aortic sections were incubated with primary antibody (1:50) overnight at 4°C. After rinsing, the sections were incubated for 1 h with the secondary antibody at room temperature and then incubated with 0.5 g/L DAPI containing antifluorescence quenching agent for 5 min. Images were obtained with a the Tissue FAXS microscope (Tissue FAX plus; Tissue Gnostics, Vienna, Austria) and analyzed based on our previous method [22].
Cell culture and treatment
RAW264.7 macrophages were obtained from the National Infrastructure of Cell Line Resource (Beijing, China), and cultured in DMEM basic medium supplemented with 10% (v/v) FBS and 1% penicillin-streptomycin. Macrophages were maintained in a 5% CO2 incubator at 37°C. AsC was dissolved in DMSO to generate a solution and diluted with culture medium. Then, the cells were seeded in various plates, pretreated with AsC, and exposed to ox-LDL (80 μg·mL-1) for 24 h.
Oil red O staining
Macrophages were pretreated with AsC (20 μM) for 12 h and then incubated with ox-LDL for 24 h. The cells were washed three times with PBS and fixed with 4% (w/v) paraformaldehyde for 10 min at room temperature. After that, the cells were stained with filtered oil red O solution (30 min, 60°C) and observed under a microscope (Olympus, Tokyo, Japan). Then, the absorbance was measured at 358 nm by a Tecan Infinite M1000 Microplate Reader (Tecan, Männedorf, Switzerland).
Transmission electron microscopy (TEM)
After all treatments, the cells were collected and fixed in 2.5% glutaraldehyde (TAAB, Berkshire, England) in 0.1 M sodium phosphate buffer (pH 7.4) overnight. The cells were washed in the same buffer 3 times and postfixed in 1% osmic acid at 4°C for 2 h and then dehydrated and embedded in epoxypropane according to a standard procedure. Ultrathin sections were stained with uranyl acetate and lead citrate and observed under a JEOL JEM1230 (JEOL Ltd., Tokyo, Japan).
Flow cytometry
Autophagosome formation in macrophages was investigated using a CytoID Autophagy Detection Kit (Enzo Life Sciences, NY, USA) according to the manufacturer’s instructions. The CytoID fluorescent reagents specifically evaluated autophagic vacuoles formed during autophagy. Briefly, macrophages were obtained and washed twice in PBS. The cells were resuspended in 0.5 mL of freshly diluted CytoID reagents and incubated at 37 °C for 30 min. The cells were washed and the CytoID fluorescence of the cells was immediately analyzed by flow cytometry (BD Biosciences, NJ, USA). The percentage of cells with CytoID staining was used to represent the formation of autophagosomes.
Macrophages were surface stained with anti-Cd36 and anti-Mrc1 antibodies and then analyzed by flow cytometry. Briefly, macrophages were collected and stained with anti-Cd36 and anti-Mrc1 antibodies for 20 min at 37°C, washed with PBS and analyzed using flow cytometry. The fluorescence intensity was statistically compared with model group.
Arginase assay
Arginase activity was measured based on a previous study [49]. Briefly, macrophages were collected and lysed with 0.1% Triton X-100 (containing protease and phosphatase inhibitors). Then, the cell lysate was incubated with arginase activation solution (5 mM MnCl2 in 25 mM Tris HCl [pH 7.4]) for 10 min at 56°C. Subsequently, the mixture was added to the arginase substrate solution (0.5 M L-arginine in water [pH 9.7]) and incubated at 37°C for 1 h. The reaction was terminated by the addition of an acid mixture (H2SO4, H3PO4, and water at a ratio of 1:3:7), followed by the addition of a-isonitrosopropiophenone (9% w/v), which was heated to 100°C for 45 min. The OD value was measured at 540 nm on a microplate reader (Tecan, Switzerland).
Immunofluorescence
Cell immunofluorescence staining was performed using rat anti-LC3II, anti-Mrc1, anti-ATG5 antibodies as well as rat IgG (H+L) F(ab')2 fragment. DAPI was used to visualize the nuclei. The cells were observed by the ImageXpress® Micro system (Molecular Device, CA, USA) and analyzed by MetaXpress Software according to our previous study [36].
Quantitative real-time PCR
The RAW264.7 cells were pretreated with AsC (20 μM) 12 h before exposure to ox-LDL for 24 h. Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, United States). The isolated RNA was reverse transcribed into cDNA using the GoScriptTM Reverse Transcription System (Promega). Then, amplification was carried out using real-time RT-PCR with the Power SYBR Premix Ex TaqTM II (TaKaRa Biotechnology, Dalian, China) in an iQ5 Real-time PCR detection system with analysis software (Bio-Rad, Santa Rosa, CA, United States). Primers (Table 1) were designed using premier primer Software 6.0 (Canadian Premier Life Insurance Company, ON, Canada). The 2-ΔCT method was used to analyze the results according to our previous research [24].
Table 1. Primers used for quantitative real-time PCR.
| Gene | Primer sequence (5' to 3') | Product (bp) |
| GAPDH | F: CTGCGGCATCCACGAAACT | 126 |
| R: AGGGCCGTGATCTCCTTCTG | ||
| IL-1β | F: TGCCACCTTTTGACAGTGATGA | 135 |
| R: TGTGCTGCTGCGAGATTTGA | ||
| iNOS | F: CTGCAGCACTTGGATCAGGAACCTG | 311 |
| R: GGAGTAGCCTGTGTGCACCTGGAA | ||
| CD206 | F: TGCTACTGAACCTCCTCAACTGC | 121 |
| R: AGCCTGACCCCAACTTCTCGT | ||
| Arg-1 | F: TGCATATCTGCCAAAGACATCG | 137 |
| R: TCCATCACCTTGCCAATCCC |
Cellular thermal shift assay (CETSA)
CETSA was performed according to a previous study [26]. Briefly, the cells were collected and heated individually at different temperatures (42, 46, 50, 54, 58, and 62°C) for 3 min and then cooled for 3 min at room temperature. Then, the samples were centrifuged, and the obtained cells were analyzed by western blotting.
Drug affinity responsive target stability (DARTS) assay
Briefly, total cell protein was isolated using NP40 lysis buffer. The lysate was equally distributed into seven groups and treated with different concentrations of DMSO as a control or AsC (0, 5, 10, 20, 50 and 100 μM) separately for 1 h at room temperature. Then, pronase (25 μg·mL-1) was added to the lysates for a further an additional 30 min at 37°C. The reactions were stopped by adding SDS-PAGE loading buffer, and the samples were analyzed via western blotting.
Molecular docking
The binding poses of AsC in the active site of Sirt1 (PDB code: 4ZZJ) were analyzed using the docking program Lib Dock according to our previous method [21]. The binding affinities (LibDockScore) in Discovery Studio 4.5 were used to evaluate the interactions between AsC and Sirt1.
siRNA assay
siRNAs targeting BECN1 and Sirt1 were purchased from Santa Cruz Biotechnology along with control siRNA and siRNA transfection reagent, according to our previous report [36]. Briefly, 80% confluent cells were transiently transfected with 100 nM siRNA per dish for 7 h according to the manufacturer's method. Then, the cells were switched to DMEM complete medium and incubated for an additional 24 h. Where indicated, macrophages were treated with AsC (20 μM) for 12 h and then exposed to ox-LDL for another 24 h. The knockdown efficiency of the target proteins was measured by western blotting.
Western blot analysis
Macrophages were harvested and lysed with cell lysis buffer containing 0.1 mM dithiothreitol and proteinase inhibitor cocktail. Protein concentration was detected using a Bio-Rad DC protein determination kit. A western blot assay was then performed, and immunoblotting was developed using an ECL kit. Band intensities were analyzed using Gel Pro software (Media Cybernetics, Rockville, MD, United States).
Statistical analysis
All analyses were performed with GraphPad Prism 6.0 software (San Diego, CA, USA). The data are presented as the mean ± S.D. Multigroup comparisons were analyzed by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Comparisons between two groups were performed by use of Student's unpaired t-test. Values of P < 0.05 were considered to indicate statistical significance. The data and statistical analysis complied with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Abbreviations
CVD: cardiovascular disease; Ox-LDL: oxidized low-density lipoprotein; AsC: Araloside C; TASAES: total saponins of Aralia elata (Miq.); FBS: fetal calf serum; HFD: high fat diet; 3-MA: 3-methyladenine; Sirt1: Silent information regulator 1; CETSA: cellular thermal shift assay; DARTS: drug affinity responsive target stability; TEM: transmission Electron Microscopy.
Author Contributions
Yun Luo performed the in vivo experiments and wrote the manuscript. Shan Lu performed the in vitro experiments. Ye Gao and Daoshun Wu participated in statistical analysis. Ke Yang performed the molecular docking. Xudong Xu isolated and identified AsC. All authors agreed on the final version. Xiaobo Sun and Guibo Sun applied for the projects and designed the experiments.
Conflicts of Interest
We declare all authors do not have any conflicts of interest for this paper.
Funding
This work was financially supported by CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-012); the Major Scientific and Technological Special Project “Drug Innovation Major Project” (No.2018ZX09711001-009); the National Natural Science Foundation of China (No. 81703745) and PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (No. 2017350016).
References
- 1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MS, et al, and American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019; 139:e56–528. https://doi.org/10.1161/CIR.0000000000000659 [PubMed]
- 2. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013; 13:709–21. https://doi.org/10.1038/nri3520 [PubMed]
- 3. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015; 12:10–17. https://doi.org/10.1038/nrcardio.2014.173 [PubMed]
- 4. Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V, Otsuka F, Gannon RL, Braumann RE, Dickinson MH, Gupta A, Jenkins AL, Lipinski MJ, et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest. 2018; 128:1106–24. https://doi.org/10.1172/JCI93025 [PubMed]
- 5. Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017; 79:541–66. https://doi.org/10.1146/annurev-physiol-022516-034339 [PubMed]
- 6. Aluganti Narasimhulu C, Fernandez-Ruiz I, Selvarajan K, Jiang X, Sengupta B, Riad A, Parthasarathy S. Atherosclerosis—do we know enough already to prevent it? Curr Opin Pharmacol. 2016; 27:92–102. https://doi.org/10.1016/j.coph.2016.02.006 [PubMed]
- 7. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013; 368:651–62. https://doi.org/10.1056/NEJMra1205406 [PubMed]
- 8. Vindis C. Autophagy: an emerging therapeutic target in vascular diseases. Br J Pharmacol. 2015; 172:2167–78. https://doi.org/10.1111/bph.13052 [PubMed]
- 9. Luo Y, Lu S, Zhou P, Ai QD, Sun GB, Sun XB. Autophagy: An Exposing Therapeutic Target in Atherosclerosis. J Cardiovasc Pharmacol. 2016; 67:266–74. https://doi.org/10.1097/FJC.0000000000000342 [PubMed]
- 10. Evans TD, Jeong SJ, Zhang X, Sergin I, Razani B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy. 2018; 14:724–26. https://doi.org/10.1080/15548627.2018.1434373 [PubMed]
- 11. Jeong SJ, Kim S, Park JG, Jung IH, Lee MN, Jeon S, Kweon HY, Yu DY, Lee SH, Jang Y, Kang SW, Han KH, Miller YI, et al. Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux. Autophagy. 2018; 14:120–33. https://doi.org/10.1080/15548627.2017.1327942 [PubMed]
- 12. Stein S, Lohmann C, Schäfer N, Hofmann J, Rohrer L, Besler C, Rothgiesser KM, Becher B, Hottiger MO, Borén J, McBurney MW, Landmesser U, Lüscher TF, Matter CM. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. 2010; 31:2301–09. https://doi.org/10.1093/eurheartj/ehq107 [PubMed]
- 13. Stein S, Schäfer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U, Tanner FC, Lüscher TF, Matter CM. SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging (Albany NY). 2010; 2:353–60. https://doi.org/10.18632/aging.100162 [PubMed]
- 14. Li L, Gao P, Zhang H, Chen H, Zheng W, Lv X, Xu T, Wei Y, Liu D, Liang C. SIRT1 inhibits angiotensin II-induced vascular smooth muscle cell hypertrophy. Acta Biochim Biophys Sin (Shanghai). 2011; 43:103–09. https://doi.org/10.1093/abbs/gmq104 [PubMed]
- 15. Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol. 2019; 16:155–65. https://doi.org/10.1038/s41569-018-0107-8 [PubMed]
- 16. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJ, Cornel JH, Pais P, et al, and CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017; 377:1119–31. https://doi.org/10.1056/NEJMoa1707914 [PubMed]
- 17. Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJ, de Lemos JA, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011; 305:2556–64. https://doi.org/10.1001/jama.2011.860 [PubMed]
- 18. Qiao L, Chen W. Atheroprotective effects and molecular targets of bioactive compounds from traditional Chinese medicine. Pharmacol Res. 2018; 135:212–29. https://doi.org/10.1016/j.phrs.2018.07.012 [PubMed]
- 19. Wang Z, Song S, Lu H, Chen G, Xu S, Sagara Y, Kitaoka N, Manabe M, Kodama H. Effect of three triterpenoid compounds isolated from root bark of Aralia elata on stimulus-induced superoxide generation and tyrosyl phosphorylation and translocation of p47(phox) and p67(phox) to cell membrane in human neutrophil. Clin Chim Acta. 2003; 336:65–72. https://doi.org/10.1016/S0009-8981(03)00326-7 [PubMed]
- 20. Du Y, Wang M, Liu X, Zhang J, Xu X, Xu H, Sun G, Sun X, Araloside C. Araloside C Prevents Hypoxia/Reoxygenation-Induced Endoplasmic Reticulum Stress via Increasing Heat Shock Protein 90 in H9c2 Cardiomyocytes. Front Pharmacol. 2018; 9:180. https://doi.org/10.3389/fphar.2018.00180 [PubMed]
- 21. Wang M, Tian Y, Du YY, Sun GB, Xu XD, Jiang H, Xu HB, Meng XB, Zhang JY, Ding SL, Zhang MD, Yang MH, Sun XB. Protective effects of Araloside C against myocardial ischaemia/reperfusion injury: potential involvement of heat shock protein 90. J Cell Mol Med. 2017; 21:1870–80. https://doi.org/10.1111/jcmm.13107 [PubMed]
- 22. Luo Y, Lu S, Ai Q, Zhou P, Qin M, Sun G, Sun X. SIRT1/AMPK and Akt/eNOS signaling pathways are involved in endothelial protection of total aralosides of Aralia elata (Miq) Seem against high-fat diet-induced atherosclerosis in ApoE-/- mice. Phytother Res. 2019; 33:768–78. https://doi.org/10.1002/ptr.6269 [PubMed]
- 23. Zhou P, Xie W, Luo Y, Lu S, Dai Z, Wang R, Sun G, Sun X. Protective Effects of Total Saponins of Aralia elata (Miq.) on Endothelial Cell Injury Induced by TNF-α via Modulation of the PI3K/Akt and NF-κB Signalling Pathways. Int J Mol Sci. 2018; 20:E36. https://doi.org/10.3390/ijms20010036 [PubMed]
- 24. Qin M, Luo Y, Lu S, Sun J, Yang K, Sun G, Sun X. Ginsenoside F1 Ameliorates Endothelial Cell Inflammatory Injury and Prevents Atherosclerosis in Mice through A20-Mediated Suppression of NF-kB Signaling. Front Pharmacol. 2017; 8:953. https://doi.org/10.3389/fphar.2017.00953 [PubMed]
- 25. Liu Y, Wang X, Pang J, Zhang H, Luo J, Qian X, Chen Q, Ling W. Attenuation of Atherosclerosis by Protocatechuic Acid via Inhibition of M1 and Promotion of M2 Macrophage Polarization. J Agric Food Chem. 2019; 67:807–18. https://doi.org/10.1021/acs.jafc.8b05719 [PubMed]
- 26. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016; 12:1–222. https://doi.org/10.1080/15548627.2015.1100356 [PubMed]
- 27. Ladoire S, Chaba K, Martins I, Sukkurwala AQ, Adjemian S, Michaud M, Poirier-Colame V, Andreiuolo F, Galluzzi L, White E, Rosenfeldt M, Ryan KM, Zitvogel L, Kroemer G. Immunohistochemical detection of cytoplasmic LC3 puncta in human cancer specimens. Autophagy. 2012; 8:1175–84. https://doi.org/10.4161/auto.20353 [PubMed]
- 28. Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, Xie Q, Zhou X, Chang H, Fu YJ, Zhu JD, Zhang QY, Mi MT. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013; 9:2033–45. https://doi.org/10.4161/auto.26336 [PubMed]
- 29. Liao LX, Song XM, Wang LC, Lv HN, Chen JF, Liu D, Fu G, Zhao MB, Jiang Y, Zeng KW, Tu PF. Highly selective inhibition of IMPDH2 provides the basis of antineuroinflammation therapy. Proc Natl Acad Sci U S A. 2017; 114:E5986–E5994. https://doi.org/10.1073/pnas.1706778114 [PubMed]
- 30. Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, Zhao H, Jiang Y, Sweitzer SM, Pietrak B, Schwartz B, Blum CA, Disch JS, et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun. 2015; 6:7645. https://doi.org/10.1038/ncomms8645 [PubMed]
- 31. Chang C, Su H, Zhang D, Wang Y, Shen Q, Liu B, Huang R, Zhou T, Peng C, Wong CC, Shen HM, Lippincott-Schwartz J, Liu W. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol Cell. 2015; 60:930–40. https://doi.org/10.1016/j.molcel.2015.10.037 [PubMed]
- 32. Song YM, Lee YH, Kim JW, Ham DS, Kang ES, Cha BS, Lee HC, Lee BW. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. 2015; 11:46–59. https://doi.org/10.4161/15548627.2014.984271 [PubMed]
- 33. Ward NC, Watts GF, Eckel RH. Statin Toxicity. Circ Res. 2019; 124:328–50. https://doi.org/10.1161/CIRCRESAHA.118.312782 [PubMed]
- 34. Zhang J, Wang H, Zheng Q. Cardioprotective effect of Aralia elata polysaccharide on myocardial ischemic reperfusion (IR) injury in rats. Int J Biol Macromol. 2013; 59:328–32. https://doi.org/10.1016/j.ijbiomac.2013.04.060 [PubMed]
- 35. Wang R, Yang M, Wang M, Liu X, Xu H, Xu X, Sun G, Sun X. Total Saponins of Aralia Elata (Miq) Seem Alleviate Calcium Homeostasis Imbalance and Endoplasmic Reticulum Stress-Related Apoptosis Induced by Myocardial Ischemia/Reperfusion Injury. Cell Physiol Biochem. 2018; 50:28–40. https://doi.org/10.1159/000493954 [PubMed]
- 36. Luo Y, Meng X, Zhou P, Lu S, Qin M, Xu X, Sun G, Sun X. Elatoside C protects against ox-LDL-induced HUVECs injury by FoxO1-mediated autophagy induction. Biochim Biophys Acta Mol Basis Dis. 2017; 1863:1654–65. https://doi.org/10.1016/j.bbadis.2017.01.017 [PubMed]
- 37. Bi Y, Chen J, Hu F, Liu J, Li M, Zhao L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019; 2019:6724903. https://doi.org/10.1155/2019/6724903 [PubMed]
- 38. Momtazi-Borojeni AA, Abdollahi E, Nikfar B, Chaichian S, Ekhlasi-Hundrieser M. Curcumin as a potential modulator of M1 and M2 macrophages: new insights in atherosclerosis therapy. Heart Fail Rev. 2019; 24:399–409. https://doi.org/10.1007/s10741-018-09764-z [PubMed]
- 39. Shen L, Sun Z, Nie P, Yuan R, Cai Z, Wu C, Hu L, Jin S, Zhou H, Zhang X, He B. Sulindac-derived retinoid X receptor-α modulator attenuates atherosclerotic plaque progression and destabilization in ApoE-/- mice. Br J Pharmacol. 2019; 176:2559–72. https://doi.org/10.1111/bph.14682 [PubMed]
- 40. Ma Y, Huang Z, Zhou Z, He X, Wang Y, Meng C, Huang G, Fang N. A novel antioxidant Mito-Tempol inhibits ox-LDL-induced foam cell formation through restoration of autophagy flux. Free Radic Biol Med. 2018; 129:463–72. https://doi.org/10.1016/j.freeradbiomed.2018.10.412 [PubMed]
- 41. Chen W, Li X, Guo S, Song N, Wang J, Jia L, Zhu A. Tanshinone IIA harmonizes the crosstalk of autophagy and polarization in macrophages via miR-375/KLF4 pathway to attenuate atherosclerosis. Int Immunopharmacol. 2019; 70:486–97. https://doi.org/10.1016/j.intimp.2019.02.054 [PubMed]
- 42. Zhang Q, Li Y, Miao C, Wang Y, Xu Y, Dong R, Zhang Z, Griffin BB, Yuan C, Yan S, Yang X, Liu Z, Kong B. Anti-angiogenesis effect of Neferine via regulating autophagy and polarization of tumor-associated macrophages in high-grade serous ovarian carcinoma. Cancer Lett. 2018; 432:144–55. https://doi.org/10.1016/j.canlet.2018.05.049 [PubMed]
- 43. Wang C, Wang Q, Lou Y, Xu J, Feng Z, Chen Y, Tang Q, Zheng G, Zhang Z, Wu Y, Tian N, Zhou Y, Xu H, Zhang X. Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J Cell Mol Med. 2018; 22:1148–66. https://doi.org/10.1111/jcmm.13368 [PubMed]
- 44. Zheng X, Xu F, Liang H, Cao H, Cai M, Xu W, Weng J. SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology. 2017; 66:809–24. https://doi.org/10.1002/hep.29238 [PubMed]
- 45. Miranda MX, van Tits LJ, Lohmann C, Arsiwala T, Winnik S, Tailleux A, Stein S, Gomes AP, Suri V, Ellis JL, Lutz TA, Hottiger MO, Sinclair DA, et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe-/- mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur Heart J. 2015; 36:51–59. https://doi.org/10.1093/eurheartj/ehu095 [PubMed]
- 46. Feng T, Liu P, Wang X, Luo J, Zuo X, Jiang X, Liu C, Li Y, Li N, Chen M, Zhu N, Han X, Liu C, et al. SIRT1 activator E1231 protects from experimental atherosclerosis and lowers plasma cholesterol and triglycerides by enhancing ABCA1 expression. Atherosclerosis. 2018; 274:172–81. https://doi.org/10.1016/j.atherosclerosis.2018.04.039 [PubMed]
- 47. Liu L, Zhu X, Zhao T, Yu Y, Xue Y, Zou H. Sirt1 ameliorates monosodium urate crystal-induced inflammation by altering macrophage polarization via the PI3K/Akt/STAT6 pathway. Rheumatology (Oxford). 2019; 58:1674–83. https://doi.org/10.1093/rheumatology/kez165 [PubMed]
- 48. Nakamura K, Kageyama S, Yue S, Huang J, Fujii T, Ke B, Sosa RA, Reed EF, Datta N, Zarrinpar A, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 regulates sirtuin-1-autophagy pathway in liver transplantation: from mouse to human. Am J Transplant. 2018; 18:1110–21. https://doi.org/10.1111/ajt.14586 [PubMed]
- 49. Wang XP, Zhang W, Liu XQ, Wang WK, Yan F, Dong WQ, Zhang Y, Zhang MX. Arginase I enhances atherosclerotic plaque stabilization by inhibiting inflammation and promoting smooth muscle cell proliferation. Eur Heart J. 2014; 35:911–19. https://doi.org/10.1093/eurheartj/eht329 [PubMed]