




Introduction
Bladder cancer is the second most common genitourinary malignancy in the world, with an estimated 549,393 new cases and 199,922 deaths in 2018 [1]. In the United States, an estimated 80,470 people will be diagnosed with this disease and 17,670 people are expected to have died from it in 2019 [2]. Statistically, 70% of patients are newly diagnosed with non-muscle invasive bladder cancers (NMIBC), which have a five-year survival rate of ~90%. However, NMIBCs have a high recurrence rate and a high probability of progressing toward muscle invasive bladder cancers (MIBCs) [3], with a dramatically reduced five-year survival rate once the disease becomes metastatic [4], since treatment for metastatic bladder cancer has seen little progress in decades [3]. For MIBC, the standard of care is radical cystectomy with platinum-based chemotherapy. The most active regimens are methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC), dose-dense MVAC, and gemcitabine plus cisplatin [5, 6]. Patients who received platinum-based chemotherapy have an overall survival rate of 9-15 months. Still, the median survival is reduced to 5 to 7 months in patients with resistance to platinum-based chemotherapy [7]. Immunotherapy by checkpoint inhibitors is the second-line of therapy for patients who fail to respond to first-line chemotherapy [8]. While erdafitinib, a pan-FGFR inhibitor, has been recently approved as a monotherapy option for patients with locally-advanced or metastatic urothelial cancer [9], caring for bladder cancer patients remains a huge social problem due to the high economic burden from end-of-life care, high recurrence rate of NMIBCs, and lack of effective treatments [10]. Accordingly, there is an unmet need to develop novel therapeutic agents for advanced bladder cancer patients.
CK1δ and CK1ε are two structure-related serine/threonine kinases with high homology in their kinase (98%) and C-terminal regulatory domains (53%) [11]. Several of their common substrates are involved in oncogenic signaling, such as Wnt (APC, β-catenin, NFATC3), p53 (TP53, MDM2), and death-receptor signaling (FADD, BID) [12], triggering gene transcription [13]. Due to the structural similarity and functional overlap, the contributions of CK1δ and CK1ε to the progression of human cancers remain elusive. Rosenberg et al. reported that CK1δ is widely overexpressed within a subset of breast tumors across all major classes, while CK1ε overexpression is restricted to the basal-like subclass by analyzing the transcription level of CK1 isoforms in datasets from the Cancer Genome Atlas (TCGA). Meanwhile, copy number gains of the CSNK1D locus were found in 36% of breast tumors, with higher frequencies in the basal-like and luminal B subtypes. The authors also revealed that CK1δ is a driver of Wnt/β-catenin activation, a molecular phenotype known to associate with poor prognosis in breast cancer patients [14, 15]. Importantly, either APC mutations or nuclear β-catenin accumulation are associated with poor outcome in patients with invasive bladder cancer [16]. Evidence from the microarray database of tumor cell lines and tissue samples indicated that CK1δ is overexpressed in many types of malignancy, including bladder cancer [12]. A TCGA dataset also showed that the copy number of CSNK1D, the gene that codes for CK1δ, is amplified in ~50% of bladder tumors, which correlated with enhanced CK1δ expression [14]. In addition, there was a large overlap between the CK1δ gene signature and Wnt signaling genes in bladder cancer [14, 15]. Together, the evidence suggests that CK1δ inhibition may be a promising strategy to treat human bladder cancer.
We previously identified 7H-pyrrolo-[2,3-d]pyrimidine derivatives as novel anticancer agents with potent anti-CK1δ activity [17]. Importantly, 4-((4-(4-ethylpiperazin-1-yl)phenyl)amino)-N-(3,4,5-trichlorophenyl)-7H-pyrrolo-[2, 3-d]pyrimidine-7-carboxamide hydrochloride (13i HCl) exhibits stronger anticancer activities than known CK1δ/ε inhibitors (PF-4800567, D4476, PF-670462) in human bladder and ovarian cancer cells. The inhibition of the CK1 δ/β-catenin pathway partly contributes to 13i HCl-mediated cell death. In the present study, we further elucidated the action mechanisms of 13i HCl in human bladder cancers. Our results here demonstrated that CSNK1D was upregulated in superficial and infiltrating bladder cancer patients from two independent datasets. Furthermore, compound 13i HCl suppresses proliferation and increases apoptosis in bladder cancer cells. For the first time, our data suggested that inhibition of CK1δ activates necroptosis in bladder cancer cells. Finally, 13i HCl inhibits migration of bladder cancer cells and reverses their mesenchymal characteristics. In conclusion, our findings describe the pharmacological mechanisms of compound 13i HCl in a preclinical setting, highlighting it as a potential therapeutic agent to treat bladder cancer.
Results
CK1δ is crucial to the growth of bladder cancer cells
To explore the relationship between CK1δ levels and bladder cancer progression in a clinical setting, we analyzed two independent microarray datasets of mRNA levels in normal tissues and patient samples. The results demonstrated that the gene expression of CSNK1D was upregulated in superficial and infiltrating bladder cancer patients (Figure 1A, 1B). We also examined CK1δ protein levels in different bladder cancer cell lines, and found that RT112 and T24 express the highest levels of CK1δ (Figure 1C). We therefore chose these two cell lines for subsequent experiments. To evaluate the contribution of CK1δ to cell growth, we stably knocked down CSNK1D by lentiviral transduction. The data suggested that CK1δ levels and those of its downstream target, β-catenin, were decreased in RT112 and T24 cells (Figure 1D). Meanwhile, viability decreased for RT112 and T24 cells at 72 h (Figure 1E, 1F). Together, the data suggest that CK1δ contributes to cell growth in bladder cancer cells.
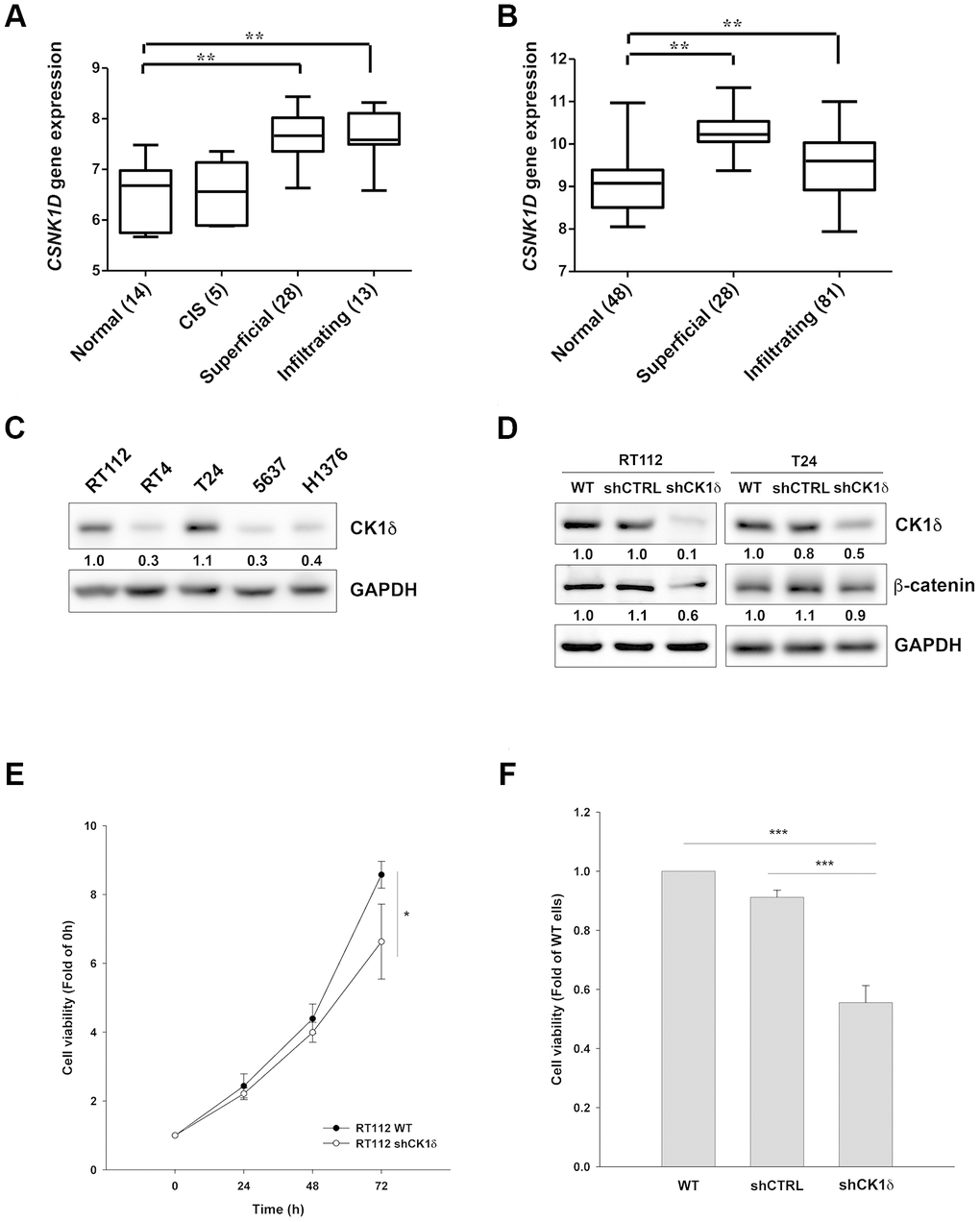
Figure 1. CK1δ promotes growth of bladder cancer cells. (A, B) Gene expression levels of CSNK1D in tissue samples of normal, carcinoma in situ (CIS), superficial and infiltrating bladder cancer patients obtained from Dyrskjot bladder dataset (A) or Sanchez-Carbayo bladder dataset (B). **P<0.01 compared to normal group. (C) Protein levels of CK1δ in different bladder cancer cell lines analyzed by Western blotting. (D) Control shRNA (shCTRL) or shCK1δ plasmids were transduced into RT112 and T24 cells by lentivirus and the cells were subjected to Western blotting with the indicated antibodies. (E, F) CK1δ knockdown decreases the viability of RT112 (E) and T24 (F) cells at 72 h by MTT assay. Date are represented as mean ± S.D. *P<0.05, ***P<0.001 compared to wild type (WT) cells (n=3).
Compound 13i HCl exhibits anti-proliferative activity in bladder cancer cells
We previously reported 7H-pyrrolo-[2,3-d]pyrimidine derivatives as novel anticancer agents with potent anti-CK1δ activity [17]. Among them, 13i HCl is the most potent against human RT-112 bladder cancer cells. In the current study, we evaluated the anti-proliferative activity of 13i HCl using MTT and SRB assay in two bladder cancer cell lines which express the highest levels of CK1δ. The data revealed that 13i HCl decreased the viability of RT112 and T24 cells in a concentration-dependent manner (Figure 2A) and inhibited their proliferation (Figure 2B). Notably, 13i HCl displayed weaker effects on the viability and proliferation of normal uroepithelial SV-HUC-1 cells (Figure 2A, 2B). As CK1δ and CK1ε are highly similar in their kinase domain, we further used autophosphorylation assay to examine the inhibitory activity of 13i HCl on CK1δ and CK1ε in RT112 cells [18]. CK1δ and CK1ε usually autophosphorylate their carboxyl-terminus regulatory domain, and the reaction is reversed by phosphatases which are sensitive to the inhibitor, okadaic acid, resulting in a rapid electrophoretic mobility shifts in western blots. Our data revealed that compound 13i HCl inhibited the autophosphorylation of CK1δ and CK1ε in a concentration-dependent manner, resembling the known dual CK1δ/ε inhibitor PF670462 (Figure 2C, 2D). In summary, 13i HCl exhibits potent anti-cancer activity in bladder cancer cell lines.
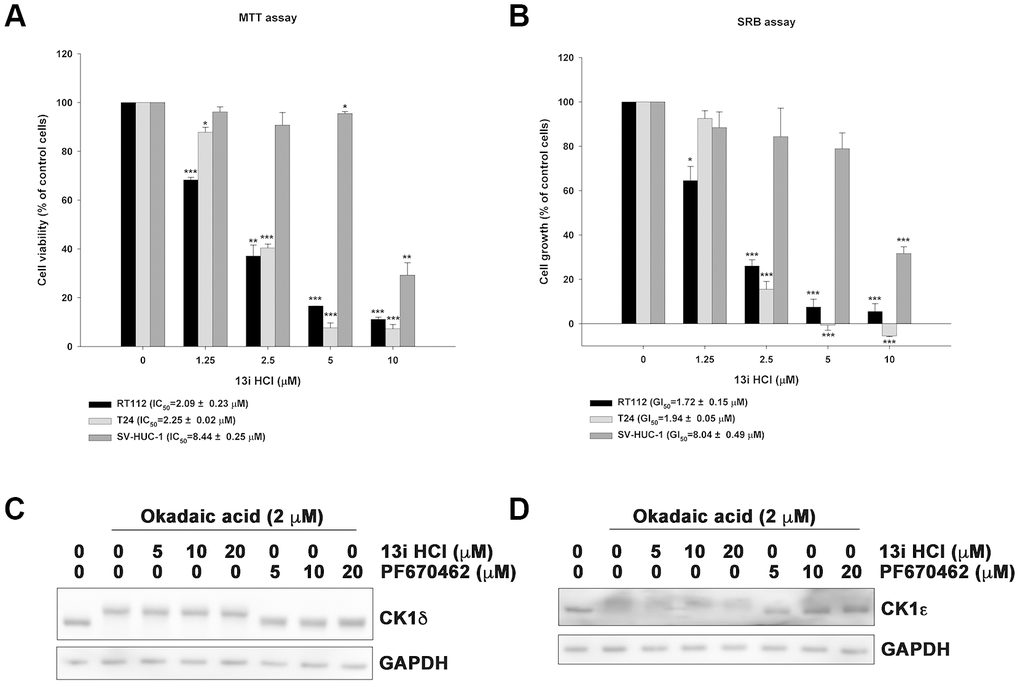
Figure 2. Compound 13i HCl exhibits anti-proliferative activity in bladder cancer cells. (A, B) RT112, T24 and SV-HUC-1 cells were exposed to the indicated concentrations of 13i HCl for 48 h and subjected to MTT assay (A) or SRB assay (B) to analyze cell viability and proliferation, respectively. Data are represented as mean ± S.D. (n=3) *P<0.05, **P<0.01, ***P<0.001 compared to control cells. (C, D) RT112 cells were treated with the indicated concentrations of 13i HCl and PF670462 in the presence of okadaic acid (2 μM) for 1 h and subjected to Western blotting by using CK1δ (C) and CK1ε (D) antibodies.
Effects of compound 13i HCl on cell cycle progression and apoptotic pathways in bladder cancer cells
To elucidate the mechanism underlying 13i HCl-induced cell death, we first examined 13i HCl’s effect on cell cycle progression by propidium iodide (PI) staining and flow cytometry. The data revealed that 13i HCl increased the population of sub-G1 cells in RT112 cells in a time- and concentration-dependent manner (Figure 3A, 3B). Accordingly, we further examined the regulatory proteins of apoptosis by western blotting. Compound 13i HCl activated the cleavage of caspase-3, -8, -9 as well as PARP in a time- and concentration-dependent manner in RT112 cells (Figure 3C, 3D). We also confirmed that 13i HCl increased the number of cells at the sub-G1 phase and apoptosis in T24 cells in a concentration-dependent manner (Supplementary Figure 1). Collectively, these data suggest that 13i HCl increases apoptosis in bladder cancer cells.
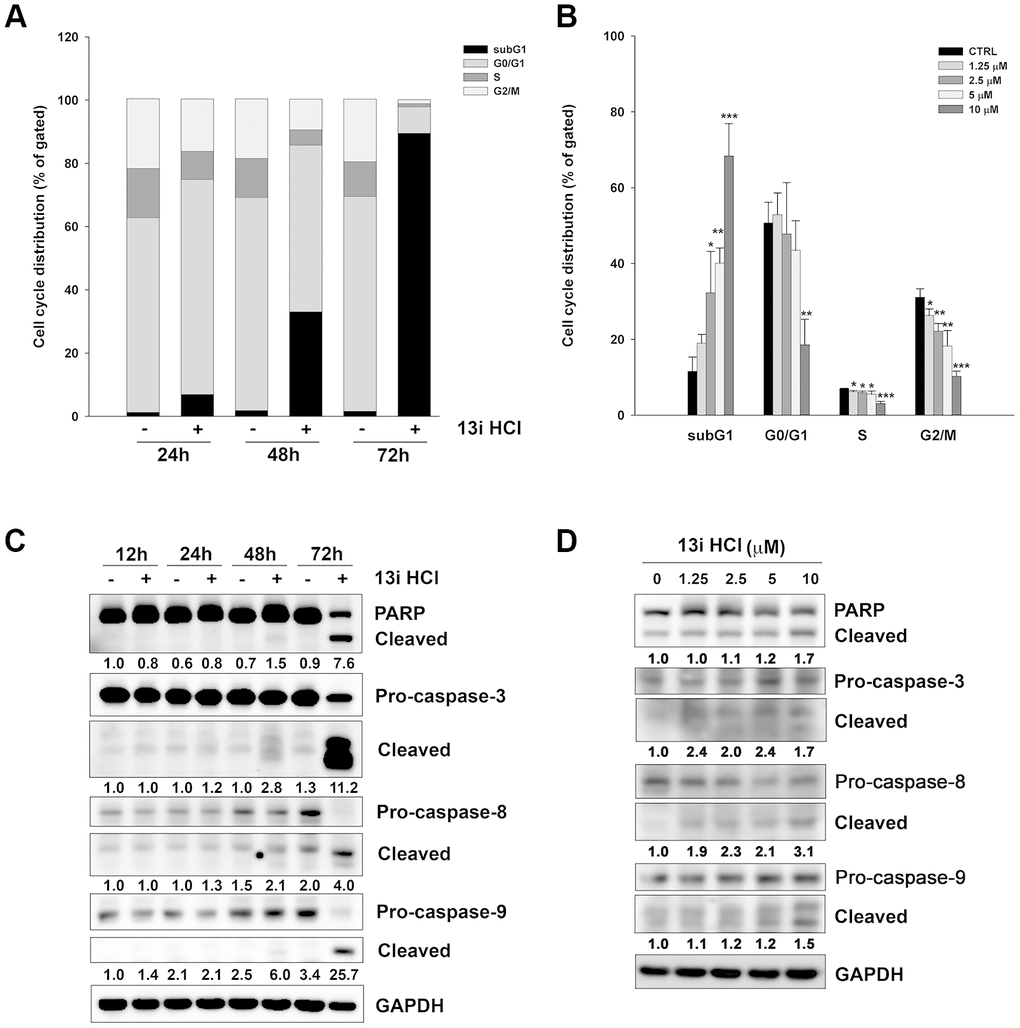
Figure 3. Compound 13i HCl induces apoptosis in RT112 cells. (A) RT112 cells were exposed to the indicated concentrations of 13i HCl for 72 h and subjected to cell cycle analysis. Data are represented as mean ± S.D. *P<0.05, **P<0.01, ***P<0.001 compared to control cells (n=3). (B) RT112 cells were treated with 13i HCl (10 μM) for the indicated times and cell cycle distribution was measured by PI staining and flow cytometry (n=2). (C, D) RT112 cells were exposed to the indicated concentrations of 13i HCl for 72 h (C) or 13i HCl (10 μM) for different times (D) and subjected to Western blotting with the indicated antibodies.
Inhibition of CK1δ activates necroptosis in bladder cancer cells
From the above findings, we observed that the protein levels of GAPDH were decreased under high concentrations of 13i HCl (Figure 3C, 3D). We hypothesized that compound 13i HCl increased membrane permeability in bladder cancer cells. Both apoptosis and necroptosis are classified as programmed cell death under drug-induced stress [19, 20]. We therefore examined the effect of apoptosis- and necroptosis-inhibitors on 13i HCl-induced cell death. The results showed that a pan-caspase inhibitor, Z-VAD-FMK, rescued cell death in the presence of 5 μM 13i HCl. Necrosulfonamide (NSA), a necroptosis inhibitor, reversed the cell death induced by 2.5 and 5 μM 13i HCl (Figure 4A). However, neither of these drugs reversed cell death at 10 μM. Because inhibition of caspases by Z-VAD-FMK might further increase the number of cells undergoing necroptosis, we therefore combined Z-VAD-FMK and NSA to examine their compounded effect on 13i HCl-induced cell death. The data showed that Z-VAD-FMK together with NSA reversed cell death induced by 2.5 to 10 μM of 13i HCl (Figure 4B). To further confirm the contribution of necroptosis in 13i HCl-induced cell death, we stably knocked down mixed lineage kinase domain-like protein (MLKL), the key signaling molecule in necroptosis. Surprisingly, MLKL-knockdown rescued 13i HCl-induced cell death (Figure 4C). Meanwhile, cell death induced by PF-670462, a specific CK1δ/ε inhibitor, was also rescued by MLKL-knockdown (Figure 4D). The knockdown efficiency was confirmed by western blotting (Figure 4E). Together, the data suggest that inhibition of CK1δ triggers not only apoptosis, but also necroptosis in bladder cancer cells.
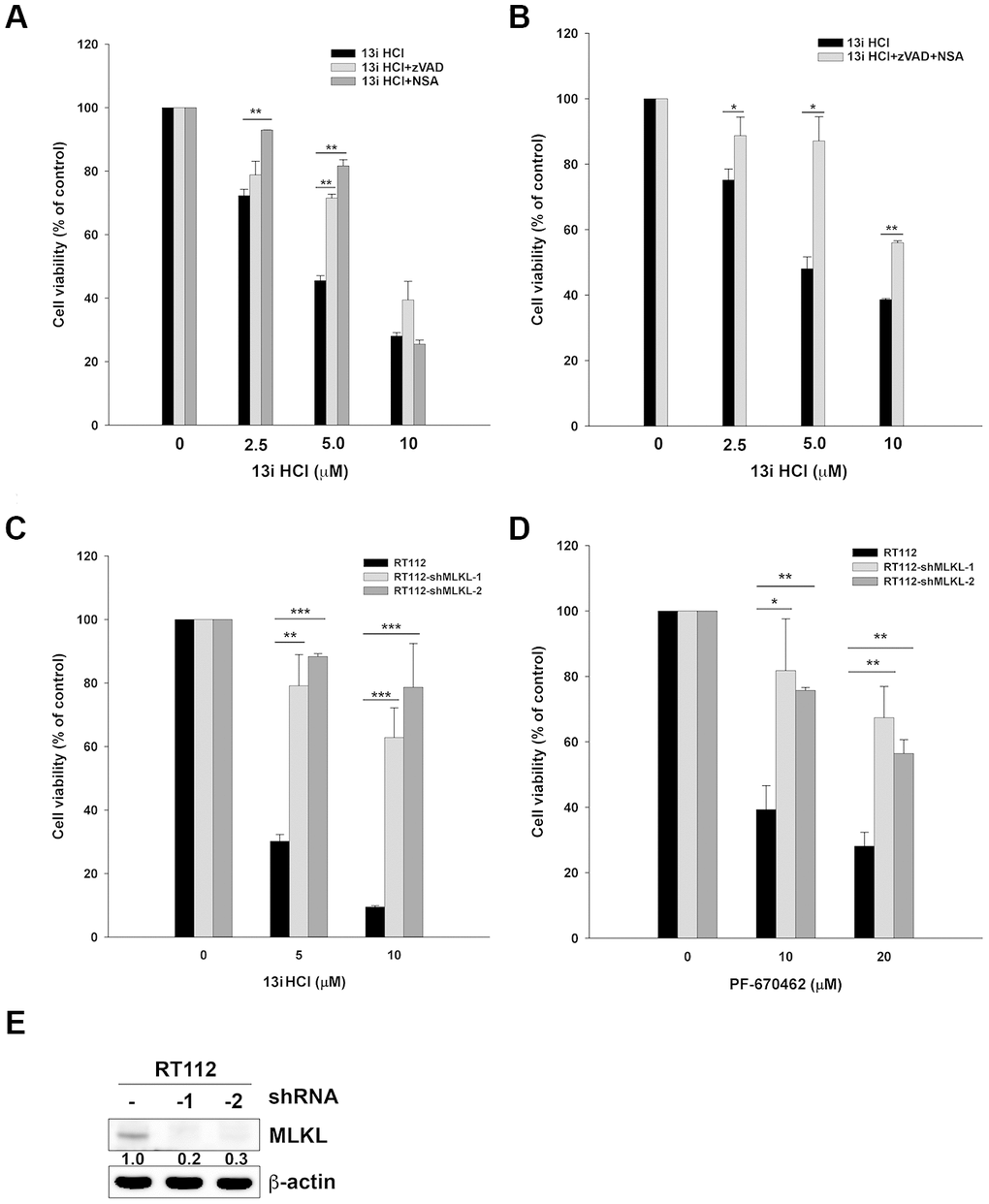
Figure 4. Inhibition of CK1δ activates necroptosis in bladder cancer cells. (A) RT112 cells were treated with the indicated concentrations of 13i HCl in the presence or absence of zVAD (20 μM) or NSA (10 μM) for 48 h and subjected to MTT assay. Data are represented as mean ± S.D. **P<0.01 compared to the group of 13i HCl alone (n=3). (B) RT112 cells were treated with the indicated concentrations of 13i HCl in the presence or absence of zVAD (20 μM) plus NSA (10 μM) for 48 h and subjected to MTT assay. Data are represented as mean ± S.D. *P<0.05, **P<0.01 compared to the group of 13i HCl alone (n=3). (C, D) RT112 and MLKL stable knocked-down clone (shMLKL-1, shMLKL-2) cells were treated with the indicated concentrations of 13i HCl (C) or PF-670462 (D) for 72 h and subjected to MTT assay. Data are represented as mean ± S.D. *P<0.05, **P<0.01, ***P<0.001compared to the group of 13i HCl or PF-670462 alone (n=3). (E) The knockdown efficiency of MLKL in RT112 cells was examined by Western blotting.
Compound 13i HCl triggers the phosphorylation of MLKL and ROS production in bladder cancer cells
To further test whether compound 13i HCl increases necroptosis in bladder cancer cells, we evaluated the marker of necroptosis, phosphorylated MLKL (pMLKL) by western blotting. The data showed that 13i HCl increased the phosphorylation of MLKL in a concentration-dependent manner at 72 h (Figure 5A). The same phenomenon was observed in RT112 cells treated with a known CK1δ/ε inhibitor, PF-670462 (Figure 5B). Meanwhile, 13i HCl increased extracellular LDH levels in a concentration-dependent manner (Figure 5C). It is known that reactive oxygen species (ROS) contribute to triggering necroptosis and apoptosis [21, 22]. Mitochondrial ROS reportedly promote RIP1 autophosphorylation, which is crucial for the recruitment of RIP3 in the necroptosome [23]. We therefore examined intracellular and mitochondrial ROS levels by using H2DCFDA and MitoSOX staining, respectively. Here we show that 13i HCl increased intracellular and mitochondrial ROS levels in RT112 cells (Figure 5D, 5E). Together, the data suggested that 13i HCl triggers necroptosis in bladder cancer cells.
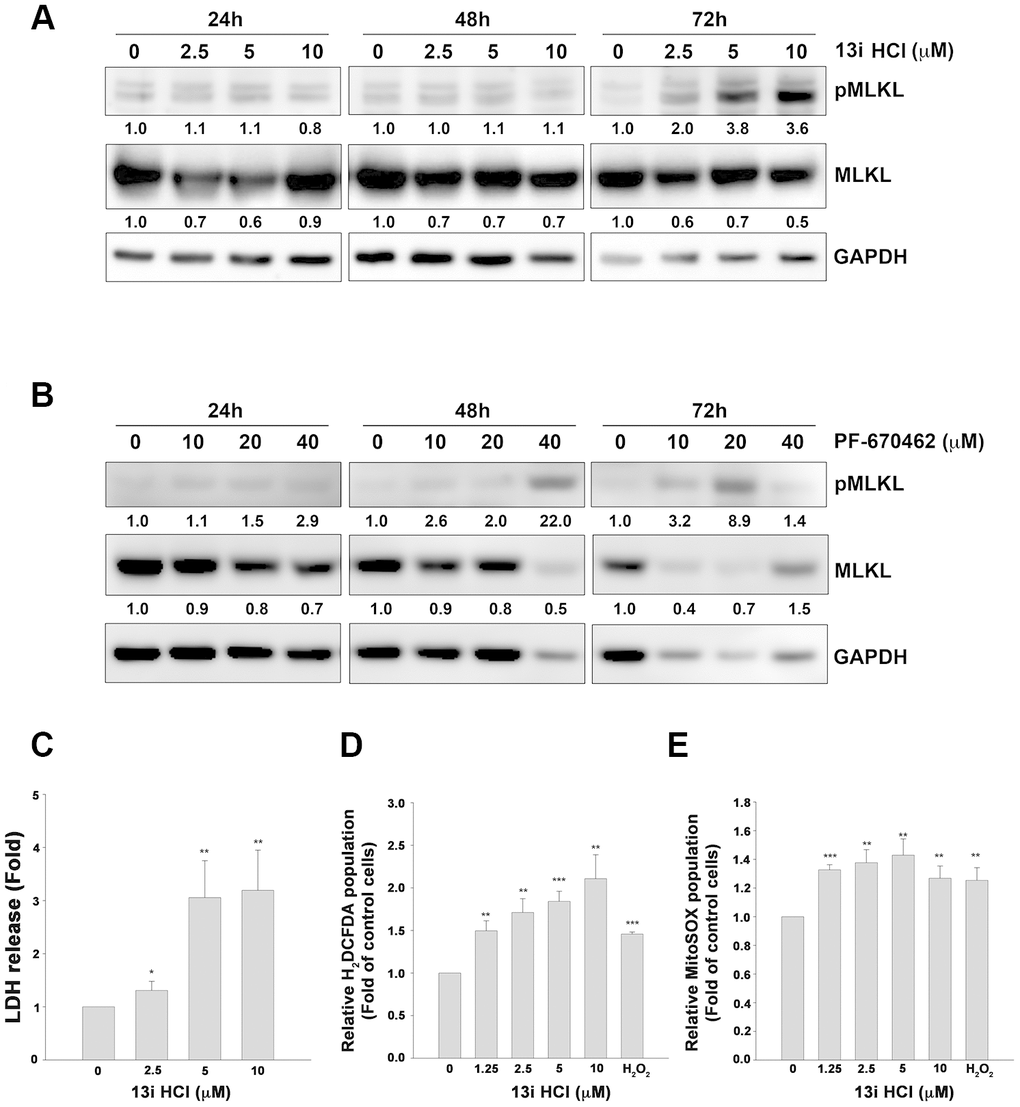
Figure 5. Compound 13i HCl increases MLKL phosphorylation and ROS production in bladder cancer cells. (A, B) RT112 cells were treated with the indicated concentrations of 13i HCl (A) or PF-670462 (B) for 24 h, 48 h and 72 h. The cells were subjected to Western blotting with the indicated antibodies. (C) RT112 cells were exposed to 13i HCl for 72 h and LDH levels in the supernatants were determined. (D, E) RT112 cells were exposed to the indicated concentrations of 13i HCl for 72 h or H2O2 (10 μM) for 1 h. The cells were stained with H2DCFDA (D) or MitoSOX (E) and subjected to flow cytometry. Data are represented as mean ± S.D. *P<0.05, **P<0.01, ***P<0.001 compared to control cells (n=3).
Effects of 13i HCl on migratory activity in bladder cancer cells
Our previous study demonstrated that compound 13i HCl displays inhibitory activity against CK1δ [17]. As CK1δ regulates several mediators of cancer metastasis, such as wnt/β-catenin and metastasis suppressor 1 (MTSS1) [12, 24], we examined the effects of 13i HCl on migration by wound-healing assay. Our data showed that 13i HCl inhibited the migratory activity of RT112 and T24 cells in a concentration-dependent manner (Figure 6A, 6B; Supplementary Figure 2A, 2B). Additionally, knocking-down CK1δ also decreased wound-healing in RT112 and T24 cells (Supplementary Figure 3A, 3B). Meanwhile, 13i HCl reversed the expression of epithelial-mesenchymal transition (EMT) markers, such as the increase in E-cadherin, and decrease in snail expression in RT112 cells (Figure 6C). Therefore, the data suggested that 13i HCl decreases migration and reversed EMT marker levels in bladder cancer cells.
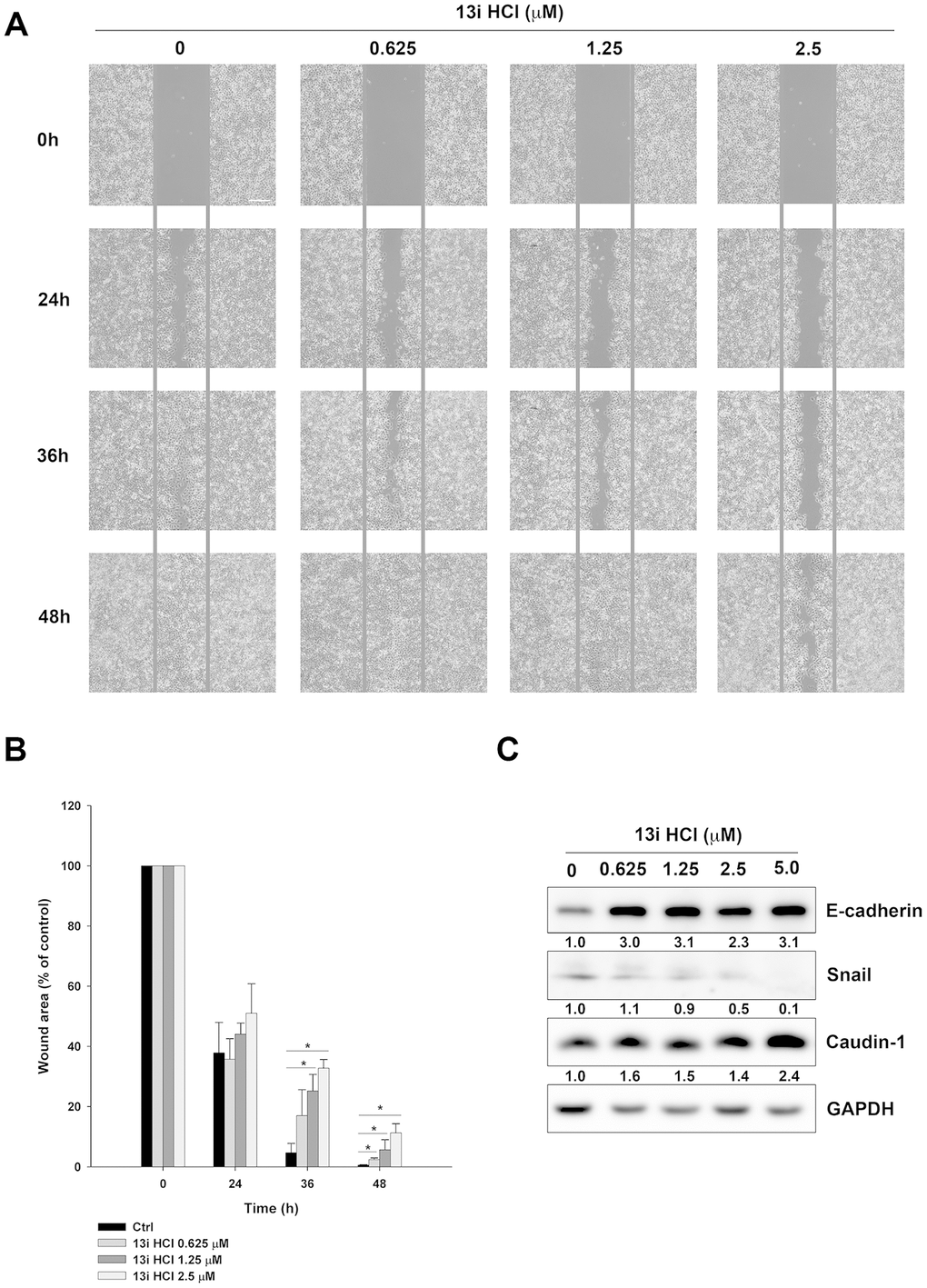
Figure 6. Effects of 13i HCl on migratory activity of bladder cancer cells. (A) RT112 cells were seeded into a 2-well insert on 6-well plate and allowed to attach overnight. The wound was created by removing the insert, and the cells were treated with or without 13i HCl and the images were captured at indicated times by EVOS XL Core Cell Imaging System (Thermo Scientific). Scale bar = 100 μm. (B) Quantification of wound healing assay. Data are represented as mean ± S.D. (n=2) *P<0.05 compared to control cells. (C) RT112 cells were exposed to indicated concentrations of 13i HCl for 72 h and subjected to Western blotting for the detection of EMT markers.
Discussion
Bladder cancer is the second most common genitourinary malignancy worldwide [1]. Approximately 30% of patients are first diagnosed as MIBC with a five-year survival rate of 50%, and that drops to only 6% once it progresses to the metastatic stage [4]. For decades, platinum-based chemotherapy has been the standard-of-care for advanced bladder cancer, but patients are often non-responsive or develop of resistance [3]. Hence, there is an unmet need to develop novel therapeutic agents to treat bladder cancer. CK1δ is a serine/threonine-protein kinase that regulates many cellular processes implicated in cancer, including Wnt/β-catenin signaling, apoptosis, DNA damage response and circadian rhythms [12]. Copy number amplification was observed in 50% of bladder tumors, which correlated with CK1δ overexpression [12, 14]. However, the anticancer activity and pharmacological mechanisms of CK1δ inhibitors on bladder cancer cells remain elusive. In the current study, we reported that mRNA levels of CSNK1D, the coding gene of CK1δ, are upregulated in both of superficial and invasive bladder cancers by analyzing two independent datasets (Figure 1A, 1B). Meanwhile, CK1δ is crucial to the growth and migration of bladder cancer cells (Figure 1D, 1F; Figure 6B). We also demonstrated that compound 13i HCl, a derivative of 7H-pyrrolo-[2,3-d]pyrimidines with CK1δ-inhibitory activity, inhibited proliferation and migration, and induced apoptosis in bladder cancer cells. Importantly, we describe for the first time that inhibition of CK1δ by 13i HCl or PF-670462 triggers necroptosis in bladder cancer cells. These results suggest that CK1δ could be a valuable therapeutic target to treat advanced bladder cancer, and that further development of compound 13i HCl is warranted to improve patient outcomes.
Several small molecules have shown Ck1δ-inhibitory activity, such as CKI-7, D4476, IC261, ®-DRF053, Bischof-524, and PF-670462 [25], but all present various problems that limit their application to bladder cancer therapy [26–31]. Recently, a series of purine scaffold inhibitors were discovered with IC50 values in the low nanomolar range, such as SR-1277, SR-2890, and SR-3029 [31]. Rosenberg et al. successfully demonstrated that the CK1δ/ε dual inhibitor SR-3029 inhibits growth in CK1δ-high breast cancer cells and several tumor xenografts in mice [14]. We also observed that compound 13i HCl inhibits both CK1δ and CK1ε isoforms as evidenced by autophosphorylation assays (Figure 2C, 2D). Currently, the challenge of using CK1 inhibitors to treat bladder cancer is the lack of selectivity between CK1δ and CK1ε, which may result from the high similarity in their kinase domain. Hence, drug design should focus on their highly variable C-terminal domains or the inhibition of auto-phosphorylation. The discovery of selective CK1δ- or CK1ε-specific inhibitors will provide new therapeutic possibilities for personalized medicine.
Although CK1δ inhibitors have shown promising anticancer activity in several studies, the molecular mechanisms underlying drug-mediated cell death are still unclear. In the current study, we observed that inhibition of CK1δ activity by compound 13i HCl triggers apoptotic and necroptotic cell death in bladder cancer cells. We also provided evidence that 13i HCl treatment or CK1δ knockdown inhibits RT112 cell migration (Figure 6A, 6B). CK1δ reportedly activates Wnt/β-catenin in breast cancer [14, 15] and CK1δ silencing reduces the migration and invasion of triple-negative breast cancer cells [32]. Importantly, either APC mutations or nuclear β-catenin accumulation are associated with poor outcome in patients with invasive bladder cancer [16]. In a previous study, we found that forced expression of β-catenin rescued cytotoxicity induced by 13i HCl in RT112 cells [17]. Therefore, targeting the CK1δ/β-catenin pathway is an attractive strategy to treat Wnt-driven bladder cancer. On the other hand, CK1δ has emerged as a potential target for other diseases. CK1δ inhibitors have been proposed to inhibit the phosphorylation of TDP-43, a pathological hallmark of central nervous system (CNS) diseases, such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia [33, 34]. CK1δ is also a potential target for Parkinson’s disease and pulmonary fibrosis [35, 36]. These observations further support the development of compound 13i HCl as a potential therapeutic target for other diseases.
Apoptosis, necroptosis and autophagic cell death are three main types of programmed cell death (PCD) induced by anticancer agents [19]. Apoptosis is the most studied PCD, characterized by cytoplasmic shrinkage, chromatin condensation, nuclear condensation, and membrane blebbing. The activation of caspases is a feature of apoptosis, accompanied by DNA/protein breakdown, and mitochondrial outer membrane permeabilization [37]. Necroptosis is a caspase-independent PCD, a form of necrotic death executed by receptor-interacting protein 1 (RIP1), RIP3, and Mixed Lineage Kinase Domain-Like (MLKL) protein [38]. Since resistance to apoptosis is one of the hallmarks of cancer, necroptosis-based cancer therapy has been proposed as a novel strategy for antitumor treatment [39]. Necroptosis is a unique cell-killing mechanism in response to severe stress or impaired apoptosis, which can be triggered by inflammatory cytokines, chemotherapeutic agents, and natural compounds [40]. Meanwhile, necroptotic tumor cells initiate adaptive immune responses by releasing damage-associated molecular patterns (DAMPs) into the microenvironment, activating dendritic and cytotoxic T cells that may suppress tumor progression [41]. For the first time, we observed that inhibition of CK1δ activity by compound 13i HCl or PF-670462 triggers necroptosis in bladder cancer cells. Knocking down of MLKL rescued cytotoxicity induced by 13i HCl or PF-670462 in RT112 cells (Figure 4C, 4D). However, the mechanisms by which CK1δ regulates necroptosis remain elusive. From the literature, CK1δ is able to phosphorylate T362 in the catalytic domain of protein phosphatase 5 (PP5), enhancing its phosphatase activity [42]. A crucial target of PP5 is Cdc37, a cochaperone of the Hsp90 complex, regulating the activation of protein kinase clients by Hsp90-Cdc37 [43, 44]. Interestingly, RIPK3 and MLKL, two important regulators in necrosomes, are clients of Hsp90-Cdc37 [45, 46]. Therefore, it would be useful to elucidate any potential linkages between CK1δ inhibition and of necroptosis induction in future studies.
Materials and Methods
Cell culture, antibodies, and reagents
RT112, RT4, 5637 cells were cultured in RPMI 1640; T24 cells were cultured in McCoy's 5a, H1376 cells were cultured in EMEM, and SV-HUC-1 were cultured in F-12K, supplemented with 10% (v/v) FBS and 1% (v/v) antibiotic-antimycotic solution (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in a humidified incubator containing 5% CO2. 4-((4-(4-ethylpiperazin-1-yl)phenyl)amino)-N-(3,4,5-trichlorophenyl)-7H-pyrrolo-[2, 3-d]pyrimidine-7-carboxamide hydrochloride (13i HCl) was synthesized by Dr. Jing-Ping Liou, as previously described [17]. PF-670462, okadaic acid, necrosulfonamide (NSA), and z-VAD-FMK were purchased from Cayman Chemical (Ann Arbor, MI, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Sulforhodamine B (SRB) were obtained from Sigma Chemical Corp (St. Louis, MO, USA). Antibodies against various proteins were obtained from the following sources: caspase-3 from Novus biologicals (Littleton, CO, USA); CK1δ, pMLKL from Abcam (Cambridge, MA, USA); β-catenin from Santa Cruz Biotechnology (Santa Cruz, CA, USA); PARP, caspase-8, caspase-9 from Cell Signaling Technology (Danvers, MA, USA), and β-actin, MLKL, GAPDH from Genetex (Irvine, CA, USA).
Cell viability, proliferation and lactate dehydrogenase (LDH) assays
Cells were seeded in 96-well plates and exposed to indicated compounds for 48 or 72 h. Cell viability was examined by MTT assay as described previously [47]. Cell proliferation was measured with the SRB assay as described previously [48]. For LDH assay, cells were seeded in 96-well plates and treated with drugs at the indicated concentrations for 72 h. LDH levels in culture supernatants were analyzed by using the CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (Promega; Madison, WI, USA) according to the manufacturer's protocol.
Cell cycle analysis
Cells were seeded in 6-well plates, and exposed to indicated compounds for 24 to 72 h. The cells were collected by trypsinization, washed one time by PBS, and fixed with ethanol (70%) at -20 °C overnight. The cells were pelleted by centrifugation, and incubated in 0.1 mL of phosphate-citric acid buffer (0.2 M NaHPO4, 0.1 M citric acid, and pH 7.8) for 15 min at room temperature, and then resuspended in propidium iodide staining buffer containing Triton X-100 (0.1%, v/v), RNase A (100 μg/mL), and propidium iodide (80 μg/mL) for 30 min in the dark. Cell cycle distribution was analyzed by flow cytometry with CellQuest software (Becton Dickinson, Mountain View, CA, USA) following previously published methods [17].
Intracellular and mitochondrial ROS analysis
Cells were seeded in 6-well plates, exposed to DMSO or indicated compounds for 72 h, and harvested by trypsinization. For total cell ROS analysis, cells were stained with 0.1 μM H2DCFDA (Biotium, Fremont, CA, USA) at 37 °C for 20 min. For mitochondrial ROS analysis, cells were stained with 5 μM MitoSOX Red (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for 10 min. After washing with PBS three times, cells were subjected to ROS detection via flow cytometry with CellQuest software according to the manufacturer's instructions (Becton Dickinson, Mountain View, CA, USA), as previously described [47].
Cell migration assay
Cells were seeded into a 2-well insert (Ibidi, Munich, Germany) on a 6-well plate and allowed to attach overnight. A wound was created by removing the insert, and the cell-free gap was around 500 μm. Cells were allowed to migrate in the presence or absence of drugs, and images were captured at the indicated times by EVOS XL Core Cell Imaging System (Thermo Scientific).
Microarray datasets analysis
Dyrskjot et al. published a dataset in which 14 normal bladder, 5 carcinoma in situ (CIS), 28 superficial bladder cancer, and 13 invasive bladder cancer samples were analyzed using Affymetrix U133A microarrays [49]. Array data were obtained from the NCBI Gene expression omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database with the accession number GSE3167. RMA log expression units were calculated using ‘affy’ package for the R statistical programming language. The default RMA settings were used to background correct, normalize and summarize all expression values. Second dataset was published by Sanchez-Carbayo et al., in which 81 infiltrating bladder urothelial carcinoma, 28 superficial bladder cancer, and 48 normal bladder samples were analyzed on Affymetrix U133A microarrays [50]. The gene expression level of CSNK1D was obtained from this study, and log2 expression level was used for statistical analysis. A 2-tailed Student’s t-test was then applied for the calculation of the p value between two different groups.
Statistical analysis
Each experiment was performed independently with at least two biological replicates. Data in the bar graphs are presented as means ± S.D and analyzed by using the Student’s t-test with p values < 0.05 considered significant.
Supplementary Materials
Abbreviations
BC: bladder cancer; EMT: epithelial-mesenchymal transition; MIBC: muscle invasive bladder cancer; MTSS1: metastasis suppressor 1; NMIBC: non-muscle invasive bladder cancer; PCD: programmed cell death; ROS: reactive oxygen species.
Author Contributions
Conceptualization, CHC.; investigation, YCL and MCC; data curation, YC. and CHC; Analysis, MCC and THH; resources, JPL; writing original draft, YCL and CHC; review and editing of manuscript, MCC and CHC; funding acquisition: CHC; supervision, CHC.
Conflicts of Interest
The authors declare that they have no competing interests.
Funding
This research was funded by the Ministry of Science and Technology of the Republic of China, grant No. MOST-107-2320-B-038-039. This work was also financially supported by the “TMU Research Center of Cancer Translational Medicine” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492 [PubMed]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019; 69:7–34. https://doi.org/10.3322/caac.21551 [PubMed]
- 3. Berdik C. Unlocking bladder cancer. Nature. 2017; 551:S34–35. https://doi.org/10.1038/551S34a [PubMed]
- 4. Funt SA, Rosenberg JE. Systemic, perioperative management of muscle-invasive bladder cancer and future horizons. Nat Rev Clin Oncol. 2017; 14:221–34. https://doi.org/10.1038/nrclinonc.2016.188 [PubMed]
- 5. Ghosh M, Brancato SJ, Agarwal PK, Apolo AB. Targeted therapies in urothelial carcinoma. Curr Opin Oncol. 2014; 26:305–20. https://doi.org/10.1097/CCO.0000000000000064 [PubMed]
- 6. Bukhari N, Al-Shamsi HO, Azam F. Update on the Treatment of Metastatic Urothelial Carcinoma. ScientificWorldJournal. 2018; 2018:5682078. https://doi.org/10.1155/2018/5682078 [PubMed]
- 7. Flaig TW, Spiess PE, Agarwal N, Bangs R, Boorjian SA, Buyyounouski MK, Downs TM, Efstathiou JA, Friedlander T, Greenberg RE, Guru KA, Hahn N, Herr HW, et al. NCCN Guidelines Insights: Bladder Cancer, Version 5.2018. J Natl Compr Canc Netw. 2018; 16:1041–53. https://doi.org/10.6004/jnccn.2018.0072 [PubMed]
- 8. Ghasemzadeh A, Bivalacqua TJ, Hahn NM, Drake CG. New Strategies in Bladder Cancer: A Second Coming for Immunotherapy. Clin Cancer Res. 2016; 22:793–801. https://doi.org/10.1158/1078-0432.CCR-15-1135 [PubMed]
- 9. Nadal R, Bellmunt J. Management of metastatic bladder cancer. Cancer Treat Rev. 2019; 76:10–21. https://doi.org/10.1016/j.ctrv.2019.04.002 [PubMed]
- 10. Svatek RS, Hollenbeck BK, Holmäng S, Lee R, Kim SP, Stenzl A, Lotan Y. The economics of bladder cancer: costs and considerations of caring for this disease. Eur Urol. 2014; 66:253–62. https://doi.org/10.1016/j.eururo.2014.01.006 [PubMed]
- 11. Cheong JK, Virshup DM. Casein kinase 1: complexity in the family. Int J Biochem Cell Biol. 2011; 43:465–69. https://doi.org/10.1016/j.biocel.2010.12.004 [PubMed]
- 12. Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014; 13:231. https://doi.org/10.1186/1476-4598-13-231 [PubMed]
- 13. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004; 20:781–810. https://doi.org/10.1146/annurev.cellbio.20.010403.113126 [PubMed]
- 14. Rosenberg LH, Lafitte M, Quereda V, Grant W, Chen W, Bibian M, Noguchi Y, Fallahi M, Yang C, Chang JC, Roush WR, Cleveland JL, Duckett DR. Therapeutic targeting of casein kinase 1δ in breast cancer. Sci Transl Med. 2015; 7:318ra202. https://doi.org/10.1126/scitranslmed.aac8773 [PubMed]
- 15. Cheong JK, Virshup DM. CK1δ: a pharmacologically tractable Achilles’ heel of Wnt-driven cancers? Ann Transl Med. 2016; 4:433. https://doi.org/10.21037/atm.2016.11.07 [PubMed]
- 16. Kastritis E, Murray S, Kyriakou F, Horti M, Tamvakis N, Kavantzas N, Patsouris ES, Noni A, Legaki S, Dimopoulos MA, Bamias A. Somatic mutations of adenomatous polyposis coli gene and nuclear b-catenin accumulation have prognostic significance in invasive urothelial carcinomas: evidence for Wnt pathway implication. Int J Cancer. 2009; 124:103–08. https://doi.org/10.1002/ijc.23917 [PubMed]
- 17. Liu YM, Chen CH, Yeh TK, Liou JP. Synthesis and evaluation of novel 7H-pyrrolo-[2,3-d]pyrimidine derivatives as potential anticancer agents. Future Med Chem. 2019; 11:959–74. https://doi.org/10.4155/fmc-2018-0564 [PubMed]
- 18. Rivers A, Gietzen KF, Vielhaber E, Virshup DM. Regulation of casein kinase I epsilon and casein kinase I delta by an in vivo futile phosphorylation cycle. J Biol Chem. 1998; 273:15980–84. https://doi.org/10.1074/jbc.273.26.15980 [PubMed]
- 19. Mishra AP, Salehi B, Sharifi-Rad M, Pezzani R, Kobarfard F, Sharifi-Rad J, Nigam M. Programmed Cell Death, from a Cancer Perspective: an Overview. Mol Diagn Ther. 2018; 22:281–95. https://doi.org/10.1007/s40291-018-0329-9 [PubMed]
- 20. Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014; 35:14–23. https://doi.org/10.1016/j.semcdb.2014.07.013 [PubMed]
- 21. Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992; 267:5317–23. [PubMed]
- 22. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016; 1863:2977–92. https://doi.org/10.1016/j.bbamcr.2016.09.012 [PubMed]
- 23. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH, Huang D, Wu R, Han J. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017; 8:14329. https://doi.org/10.1038/ncomms14329 [PubMed]
- 24. Zhong J, Shaik S, Wan L, Tron AE, Wang Z, Sun L, Inuzuka H, Wei W. SCF β-TRCP targets MTSS1 for ubiquitination-mediated destruction to regulate cancer cell proliferation and migration. Oncotarget. 2013; 4:2339–53. https://doi.org/10.18632/oncotarget.1446 [PubMed]
- 25. Knippschild U, Krüger M, Richter J, Xu P, García-Reyes B, Peifer C, Halekotte J, Bakulev V, Bischof J. The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front Oncol. 2014; 4:96. https://doi.org/10.3389/fonc.2014.00096 [PubMed]
- 26. Chijiwa T, Hagiwara M, Hidaka H. A newly synthesized selective casein kinase I inhibitor, N-(2-aminoethyl)-5-chloroisoquinoline-8-sulfonamide, and affinity purification of casein kinase I from bovine testis. J Biol Chem. 1989; 264:4924–27. [PubMed]
- 27. Badura L, Swanson T, Adamowicz W, Adams J, Cianfrogna J, Fisher K, Holland J, Kleiman R, Nelson F, Reynolds L, St Germain K, Schaeffer E, Tate B, Sprouse J. An inhibitor of casein kinase I epsilon induces phase delays in circadian rhythms under free-running and entrained conditions. J Pharmacol Exp Ther. 2007; 322:730–38. https://doi.org/10.1124/jpet.107.122846 [PubMed]
- 28. Cheong JK, Nguyen TH, Wang H, Tan P, Voorhoeve PM, Lee SH, Virshup DM. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1δ/ɛ and Wnt/β-catenin independent inhibition of mitotic spindle formation. Oncogene. 2011; 30:2558–69. https://doi.org/10.1038/onc.2010.627 [PubMed]
- 29. Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004; 5:60–65. https://doi.org/10.1038/sj.embor.7400048 [PubMed]
- 30. Oumata N, Bettayeb K, Ferandin Y, Demange L, Lopez-Giral A, Goddard ML, Myrianthopoulos V, Mikros E, Flajolet M, Greengard P, Meijer L, Galons H. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J Med Chem. 2008; 51:5229–42. https://doi.org/10.1021/jm800109e [PubMed]
- 31. Bibian M, Rahaim RJ, Choi JY, Noguchi Y, Schürer S, Chen W, Nakanishi S, Licht K, Rosenberg LH, Li L, Feng Y, Cameron MD, Duckett DR, et al. Development of highly selective casein kinase 1δ/1ε (CK1δ/ε) inhibitors with potent antiproliferative properties. Bioorg Med Chem Lett. 2013; 23:4374–80. https://doi.org/10.1016/j.bmcl.2013.05.075 [PubMed]
- 32. Bar I, Merhi A, Larbanoix L, Constant M, Haussy S, Laurent S, Canon JL, Delrée P. Silencing of casein kinase 1 delta reduces migration and metastasis of triple negative breast cancer cells. Oncotarget. 2018; 9:30821–36. https://doi.org/10.18632/oncotarget.25738 [PubMed]
- 33. Bissaro M, Federico S, Salmaso V, Sturlese M, Spalluto G, Moro S. Targeting Protein Kinase CK1δ with Riluzole: Could It Be One of the Possible Missing Bricks to Interpret Its Effect in the Treatment of ALS from a Molecular Point of View? ChemMedChem. 2018; 13:2601–05. https://doi.org/10.1002/cmdc.201800632 [PubMed]
- 34. Alquezar C, Salado IG, de la Encarnación A, Pérez DI, Moreno F, Gil C, de Munain AL, Martínez A, Martín-Requero Á. Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia. Mol Neurodegener. 2016; 11:36. https://doi.org/10.1186/s13024-016-0102-7 [PubMed]
- 35. Morales-Garcia JA, Salado IG, Sanz-San Cristobal M, Gil C, Pérez-Castillo A, Martínez A, Pérez DI. Biological and Pharmacological Characterization of Benzothiazole-Based CK-1δ Inhibitors in Models of Parkinson’s Disease. ACS Omega. 2017; 2:5215–20. https://doi.org/10.1021/acsomega.7b00869 [PubMed]
- 36. Keenan CR, Langenbach SY, Jativa F, Harris T, Li M, Chen Q, Xia Y, Gao B, Schuliga MJ, Jaffar J, Prodanovic D, Tu Y, Berhan A, et al. Casein Kinase 1δ/ε Inhibitor, PF670462 Attenuates the Fibrogenic Effects of Transforming Growth Factor-β in Pulmonary Fibrosis. Front Pharmacol. 2018; 9:738. https://doi.org/10.3389/fphar.2018.00738 [PubMed]
- 37. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014; 15:81–94. https://doi.org/10.1038/nrm3735 [PubMed]
- 38. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016; 23:748–56. https://doi.org/10.1038/cdd.2016.8 [PubMed]
- 39. Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017; 24:1184–95. https://doi.org/10.1038/cdd.2017.65 [PubMed]
- 40. Chen D, Yu J, Zhang L. Necroptosis: an alternative cell death program defending against cancer. Biochim Biophys Acta. 2016; 1865:228–36. https://doi.org/10.1016/j.bbcan.2016.03.003 [PubMed]
- 41. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, Jin K, Ni Q, Yu X, Liu C. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019; 18:100. https://doi.org/10.1186/s12943-019-1029-8 [PubMed]
- 42. Dushukyan N, Dunn DM, Sager RA, Woodford MR, Loiselle DR, Daneshvar M, Baker-Williams AJ, Chisholm JD, Truman AW, Vaughan CK, Haystead TA, Bratslavsky G, Bourboulia D, Mollapour M. Phosphorylation and Ubiquitination Regulate Protein Phosphatase 5 Activity and Its Prosurvival Role in Kidney Cancer. Cell Rep. 2017; 21:1883–95. https://doi.org/10.1016/j.celrep.2017.10.074 [PubMed]
- 43. Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, Panaretou B, Neckers L, Clarke PA, Workman P, Piper PW, Prodromou C, Pearl LH. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell. 2008; 31:886–95. https://doi.org/10.1016/j.molcel.2008.07.021 [PubMed]
- 44. Li T, Jiang HL, Tong YG, Lu JJ. Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery. J Hematol Oncol. 2018; 11:59. https://doi.org/10.1186/s13045-018-0602-8 [PubMed]
- 45. Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA. 2015; 112:5017–22. https://doi.org/10.1073/pnas.1505244112 [PubMed]
- 46. Zhao XM, Chen Z, Zhao JB, Zhang PP, Pu YF, Jiang SH, Hou JJ, Cui YM, Jia XL, Zhang SQ. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016; 7:e2089. https://doi.org/10.1038/cddis.2015.390 [PubMed]
- 47. Chen CH, Changou CA, Hsieh TH, Lee YC, Chu CY, Hsu KC, Wang HC, Lin YC, Lo YN, Liu YR, Liou JP, Yen Y. Dual Inhibition of PIK3C3 and FGFR as a New Therapeutic Approach to Treat Bladder Cancer. Clin Cancer Res. 2018; 24:1176–89. https://doi.org/10.1158/1078-0432.CCR-17-2066 [PubMed]
- 48. Liu YM, Lee HY, Chen CH, Lee CH, Wang LT, Pan SL, Lai MJ, Yeh TK, Liou JP. 1-Arylsulfonyl-5-(N-hydroxyacrylamide)tetrahydroquinolines as potent histone deacetylase inhibitors suppressing the growth of prostate cancer cells. Eur J Med Chem. 2015; 89:320–30. https://doi.org/10.1016/j.ejmech.2014.10.052 [PubMed]
- 49. Dyrskjøt L, Kruhøffer M, Thykjaer T, Marcussen N, Jensen JL, Møller K, Ørntoft TF. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004; 64:4040–48. https://doi.org/10.1158/0008-5472.CAN-03-3620 [PubMed]
- 50. Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006; 24:778–89. https://doi.org/10.1200/JCO.2005.03.2375 [PubMed]