



Introduction
For decades, researchers have investigated the process of aging: a decline in health over time, which is generally considered to be due to an accumulation of cellular damage [1, 2]. This cellular damage has different causes, but genomic instability is considered one of the main factors that contribute to cellular aging [3]. Genetic damage such as point mutations, chromosomal rearrangements, aneuploidy and copy number variations accumulate during life and are caused by endogenous and exogenous sources, including UV radiation, ionizing radiation and reactive oxygen species (ROS) [3, 4]. While the accumulation of genomic instability over time can cause cellular senescence and apoptosis in the case of aging, in other cases it can lead to uncontrolled cellular proliferation, and is therefore associated with an increased cancer risk [3, 4].
Defects in DNA repair proteins lead to an accumulation of DNA damage and thus contribute to accelerated aging, as is the case for several human diseases including Cockayne, Bloom and Werner syndromes, trichothiodystrophy and ataxia telangiectasia [5, 6]. In addition, several mouse models have confirmed that mutations in DNA repair proteins lead to accelerated aging. These mice exhibit accelerated aging and aging-related phenotypes such as alopecia, grey hair, osteoporosis, cachexia, neurological abnormalities, retinal degeneration and a predisposition to a wide variety of cancers [5, 7–12]. Taken together, this indicates that an exacerbated accumulation of DNA damage leads to premature aging, albeit the actual contribution of DNA damage to normal aging remains to be elucidated. More importantly, which types of DNA damage play a central role in the context of aging is still unknown.
Besides strategies to accelerate aging, genetic manipulation studies in worms, flies and mice have also successfully lead to increases in lifespan, suggesting a direct implication of the manipulated genes in normal aging [1, 13]. For instance, mice overexpressing the spindle assembly checkpoint protein BubR1 show a reduction in age-dependent aneuploidy, reduced incidence of cancer, and an increased healthy lifespan compared to WT mice [14]. Another relevant mouse model with delayed aging is a mouse model with extra copies of tumor suppressor genes Trp53 and its positive regulator Arf, which in addition to having an increased median lifespan has a reduced tumor incidence [15]. The median survival is increased further in mice with constitutive telomerase reverse transcriptase (TERT) overexpression in addition to extra copies of Trp53 and Arf [16].
In recent years, replication stress (RS) has been acknowledged as an important source of endogenous DNA damage [17]. RS is a type of DNA damage that occurs when obstacles to replication lead to an accumulation of single stranded DNA (ssDNA) at stalled replication forks, which is recognized by ssDNA binding protein RPA. This initiates a signaling cascade involving Ataxia Telangiectasia and Rad3-related (ATR) kinase and CHK1 which promotes DNA repair, cell cycle arrest, and apoptosis [18–20]. Similar to other types of DNA damage, RS has been linked to aging. For instance, aged hematopoietic stem cells (HSCs) exhibit increased levels of RS compared to young HSCs [21]. In addition, mutations in the ATR gene cause Seckel syndrome in humans, which is characterized by progeria, growth retardation, microcephaly, mental retardation and dwarfism [22] (OMIM210600). The involvement of RS in premature aging has also been shown experimentally with a mouse model for Seckel syndrome [12]. ATR-Seckel mice exhibit a phenotype similar to that of human patients, which is further aggravated in combination with several cancer-driving mutations such as the Myc oncogene or the absence of the tumor suppressor p53 [12, 23]. ATR-Seckel mice show high levels of RS during embryonic development, accelerated aging in adult life and early lethality [12]. Interestingly, mice harbouring extra alleles of Chk1 (Chk1Tg) or of the ribonucleotide reductase (RNR) regulatory subunit Rrm2 (Rrm2Tg), which is a limiting factor for dNTP production, improved the lifespan and alleviated the progeroid phenotype of ATR mutant mice [24, 25]. These Chk1 and Rrm2 transgenic mice carry bacterial artificial chromosome (BAC) alleles of the respective genes, including exons and introns, under their own endogenous promoters. This strategy provides supraphysiological levels of CHK1 and RRM2 while preventing overexpression in tissues where these genes are normally not expressed, and was proven successful with the Trp53 BAC-transgenic mouse model [26]. Collectively, these studies suggested that RS might have important implications in mammalian aging. However, the effect of Chk1 and Rrm2 expression levels on normal aging, in mice with physiological levels of ATR, remains to be elucidated.
In the current study, we investigated the effect of supraphysiological levels of CHK1 and RRM2, which confer extra protection against RS, on normal aging. We utilized cohorts of WT, Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg mice to asses tumor-free survival of these mice. We found no differences in survival between the genotypes and all mice exhibited similar signs of aging, although there was a higher tumor incidence in the transgenic mice compared to WT mice. Thus, supraphysiological levels of CHK1 and RRM2 do not affect normal aging in mice.
Results
Generation of mice with supraphysiological levels of CHK1 and RRM2
In order to investigate the effect of RS on lifespan, we used the Chk1Tg and Rrm2Tg mouse models previously generated in our laboratory [24, 25]. Chk1Tg and Rrm2Tg mice were crossed in order to obtain Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg mice. Transgenic mice were not phenotypically distinguishable from WT littermates and were born in accordance with Mendelian ratios (Chi-square p-value = 0.243) (Table 1).
Table 1. Offspring from Chk1Tg x Rrm2Tg and Chk1Tg;Rrm2Tg x WT crosses.
| WT | Chk1Tg | Rrm2Tg | Chk1Tg;Rrm2Tg | |
| Observed | 84 | 66 | 91 | 83 |
| Expected | 81 | 81 | 81 | 81 |
MEFs with increased levels of RRM2 and CHK1 show less damage after induction of RS
To confirm that supraphysiological levels of CHK1 and RRM2 could protect cells against RS, we generated mouse embryonic fibroblasts (MEFs) from crosses between Chk1Tg and Rrm2Tg mice. Chk1Tg and Rrm2Tg MEFs have been previously described to be resistant to RS induced by the ribonucleotide reductase inhibitor hydroxyurea (HU) [24, 25]. Genomic PCR genotyping confirmed the four different MEF genotypes: WT, Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg (Figure 1A). In addition, increased protein levels of RRM2 and CHK1 in MEFs carrying the Rrm2 or Chk1 transgene were confirmed by Western blotting (Figure 1B).
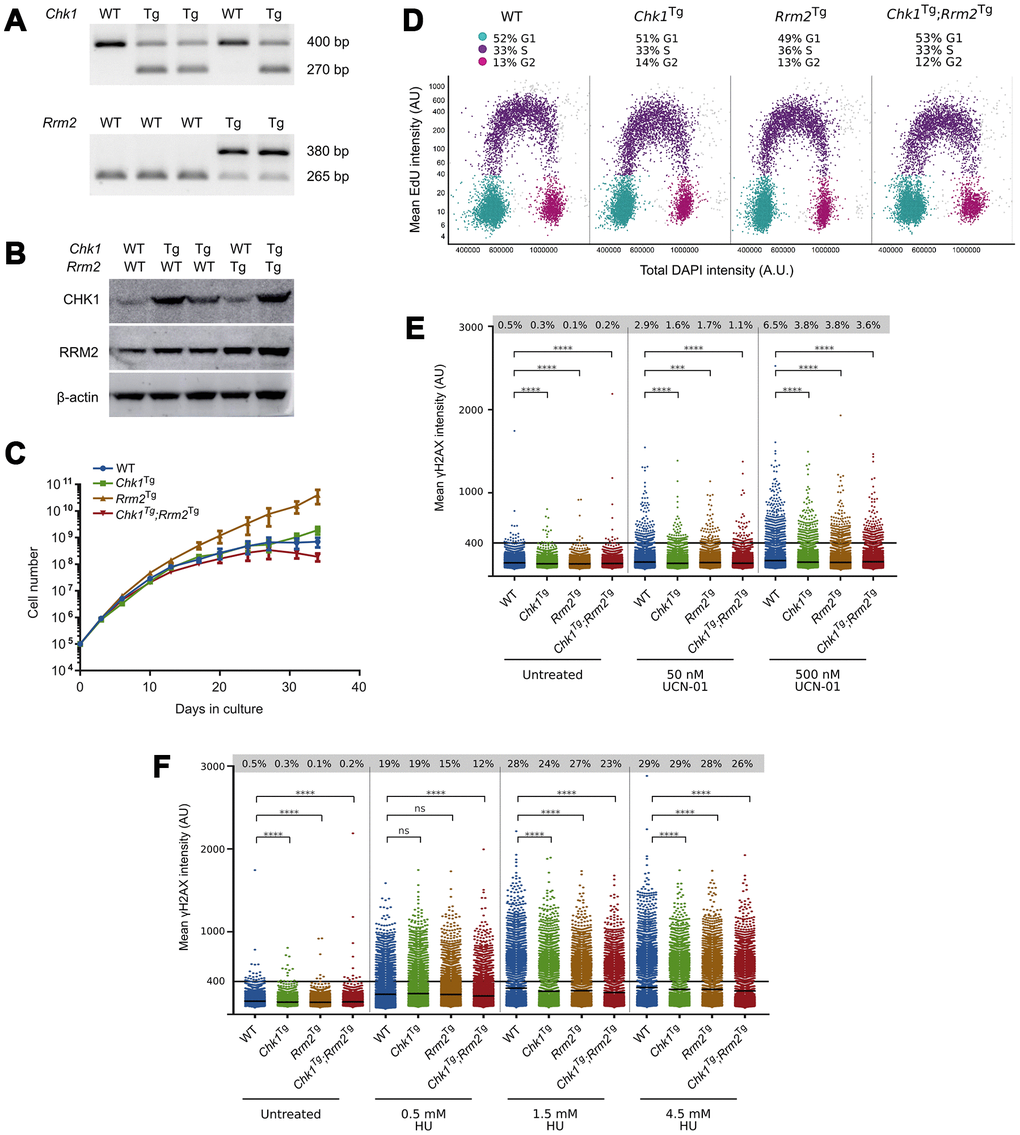
Figure 1. Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg MEFs are protected against replication stress compared to WT MEFs. (A) DNA genotyping results for Chk1 and Rrm2 alleles in MEFs; (B) Western blot showing CHK1 and RRM2 protein levels in MEFs; (C) Proliferation curves for Chk1 and Rrm2 transgenic MEFs. Cells were counted and replated every 3-4 days, in three technical replicates per genotype; (D) Cell cycle distribution of MEFs determined by EdU incorporation and DAPI profiles. At least 7000 cells were quantified per condition using high-content microscopy; (E, F) Quantification of γH2AX intensity in MEFs treated with UCN-01 (E) or HU (F) at indicated concentrations for four hours. At least 7000 cells obtained from two technical replicates were quantified per condition using high-content microscopy. Percentages indicate cells with γH2AX intensity above a threshold of 400 AU, and means are indicated by horizontal black lines for each condition. The control cells are the same for (E) and (F), as the results were obtained from the same experiment. **** = P ≤0.0001; *** = P ≤0.001; ns = P > 0.05. Statistical significance was calculated using the unpaired t-test.
We then assessed whether elevated levels of CHK1 and RRM2 would influence cell proliferation, and found that Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg MEFs proliferated similarly to WT MEFs (Figure 1C). Similarly, Edu incorporation analyses showed similar cell cycle distribution and percentage of replicating cells in all the genoytypes (Figure 1D and Supplementary Figure 1).
Next, we assessed whether MEFs carrying extra copies of Rrm2 and Chk1 were protected against RS by assessing γH2AX levels in these cells. Using high-content microscopy, we found that Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg MEFs have lower basal levels of γH2AX compared to WT MEFs (Figure 1E, 1F and Supplementary Figure 2A, 2B). In addition to assessing γH2AX levels in unstressed conditions, we induced RS by treating the cells with CHK1-inhibitor UCN-01 at different doses and found that the cells with higher CHK1 and/or RRM2 levels showed a decrease in γH2AX intensity compared to WT MEFs (Figure 1E and Supplementary Figure 2A). In addition, the MEFs were treated with HU (Figure 1F and Supplementary Figure 2B). In accordance with previously published data on Chk1 and Rrm2 transgenic MEFs [24, 25], we observed lower γH2AX intensity in these MEFs compared to WT. Importantly, these γH2AX analyses were performed in early passage MEFs (passage 3), when replication and cell proliferation were proficient and comparable among the different genotypes. Thus, the differences found in γH2AX cannot be explained by differences in replication or proliferation rates. These data confirm that cells from mice carrying extra copies of the Rrm2 or Chk1 genes show less DNA damage after induction of RS with HU and UCN-01 compared to cells from WT mice.
Supraphysiological levels of CHK1 and RRM2 do not influence lifespan in mice
Next, we aimed to investigate whether the protection against RS conferred by extra copies of Chk1 and Rrm2 would be reflected in the survival of mice, as they did in the context of reduced levels of ATR [24, 25]. To this end, we used mice containing the Chk1 and/or Rrm2 transgenes, and assessed tumor-free survival of these mice. Mice were euthanized when they had noticeable tumors or were visibly ill, as observed by rapid weight loss, hunched posture, rough hair coat, labored breathing, lethargy, impaired mobility or abdominal swelling. Only female mice were included in the cohorts, since they could be group-housed more easily than males.
Our results show that survival was not increased due to CHK1 or RRM2 levels: the survival of WT, Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg was not significantly different (log-rank p=0.3944) (Figure 2A). We collected tissues of two year-old mice (not included in survival curve). H&E staining of spleens, livers and kidneys of mice did not show obvious differences between the genotypes (Figure 2B). Furthermore, we observed no noticeable differences in appearance among the mice with different genotypes, as reflected by their weight at 1 year of age (Figure 2C). In addition, all mice displayed comparable signs of aging such as weight loss, grey hair and baldness (data not shown). In summary, unlike in an ATR-deficient background, extra copies of Chk1 and Rrm2 did not increase the overall survival of mice.
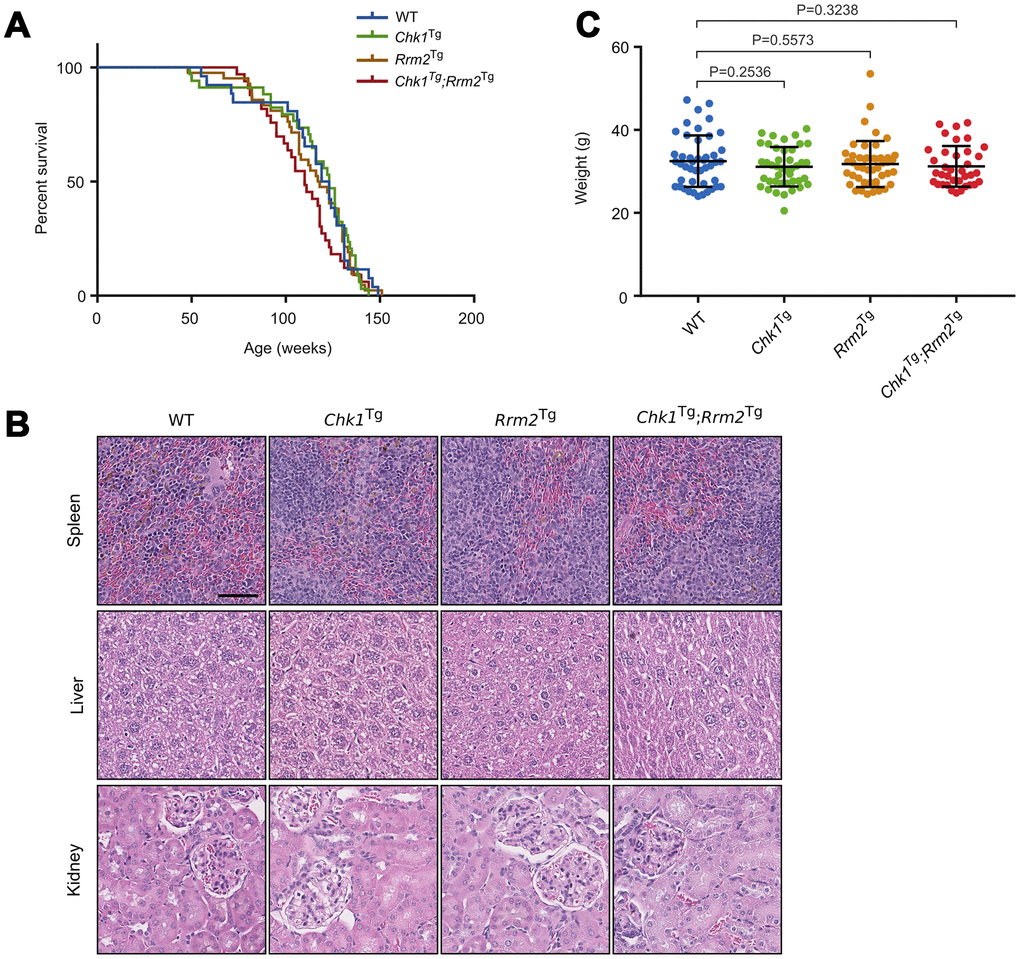
Figure 2. Chk1 and Rrm2 transgenes do not affect mouse lifespan. (A) Kaplan-Meier survival curves for WT (n=26), Chk1Tg (n=34), Rrm2Tg (n=42) and Chk1Tg;Rrm2Tg (n=33) mice. P-value = 0.3944 using the log-rank test; (B) Hematoxylin and eosin staining of mouse spleen, liver and kidney for mice with the indicated genotypes. Scale bar indicates 60 μm; (C) Total body weight of mice at one year of age. P-values were calculated using the unpaired t-test.
Extra copies of Chk1 and Rrm2 mildly increase the prevalence of spontaneous tumors
While none of the genetic combinations used in this study was able to extend mouse lifespan, the median survival of Chk1Tg;Rrm2Tg mice was the lowest with 100 weeks compared to 123 weeks for WT mice (not statistically significant). In addition, and despite no significant differences on overall survival, the percentage of mice that developed tumors was higher, although not statistically significant, for the mice containing extra copies of Chk1, Rrm2 or both transgenes (74%, 70% and 71%, respectively) compared to the percentage of WT mice with tumors (64%). This could be in line with previous reports indicating that overexpression of CHK1 or RRM2 favors tumorigenesis (Figure 3A) [25, 27]. The majority of the observed tumors were lymphomas, which was confirmed by an enlarged spleen or lymph nodes containing CD3e positive cells (Figure 3B). This is in agreement with our previous reports showing that increased CHK1 or RRM2 levels can reduce the DNA damage induced by oncogenes thereby facilitating oncogenic transformation or cell reprogramming [25, 28, 29].
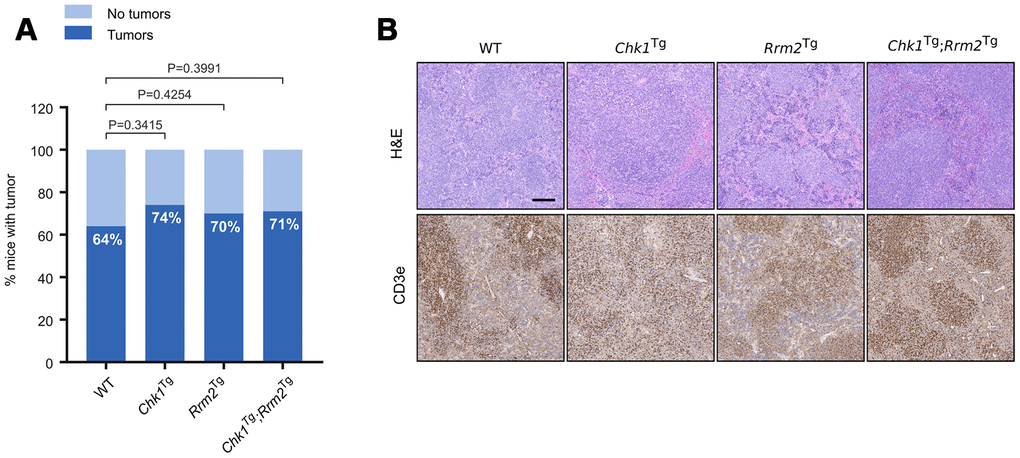
Figure 3. Increased incidence of spontaneous tumors in Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg mice compared to WT littermates. (A) Tumor incidence in WT (n=22), Chk1Tg (n=19), Rrm2Tg (n=27) and Chk1Tg;Rrm2Tg (n=24) mice subjected to necropsy. The differences observed in tumor incidence were not statistically significant according to the Chi-square test; (B) H&E and CD3e IHC staining of mouse spleens with tumor found with necropsy. Tumors are CD3e positive. Scale bar indicates 200 μm.
Discussion
In the presented study, we aimed to determine whether overexpression of Chk1 and/or Rrm2 could extend mammalian lifespan by utilizing transgenic mouse models carrying extra copies of the Chk1 and Rrm2 genes. These BAC-transgenic mouse models contain extra copies of the genes of interest, which keep the expression of the genes in a physiological range and maintain their endogenous regulation and has been a successful strategy in the past [15, 26]. Previous studies have shown that defects in RS-related proteins lead to increased RS levels and accelerated aging [12, 30, 31]. Lopez-Otin and colleagues described in 2013 the hallmarks of aging [3], one of them being DNA damage. However, a hallmark of aging should not only cause accelerated aging when aggravated, but also increase lifespan when attenuated [3].
Previous studies from our group showed that CHK1- and RRM2-overexpressing MEFs have lower levels of DNA damage in basal conditions and have greater protection against HU- and UCN-01-induced RS [24, 25], and we hypothesized that this protection against RS could lead to an increase in lifespan in vivo. However, we did not observe a difference in lifespan between Chk1Tg, Rrm2Tg and Chk1Tg;Rrm2Tg and WT mice, and all mice developed similar age-related symptoms. Based on these results we conclude that supraphysiological levels of CHK1 and RRM2 do not influence normal aging, and more generally, we propose that physiological RS might not be an important driver of aging. However, we cannot rule out that RS might influence aging in a less controlled environment where mice could be exposed to pathogens and other insults. Although Chk1Tg and Rrm2Tg mice were previously shown to extend the lifespan and alleviate the progeroid symptoms of ATR-deficient mice [24, 25], this is not a direct indication that CHK1 and RRM2 levels influence normal aging. CHK1 overexpression could compensate for ATR deficiency, as CHK1 is a downstream target of ATR. For RRM2 this connection to the ATR pathway is not as direct, although studies in yeast have shown that ATR ortholog Mec1 can activate the ribonucleotide reductase (RNR) complex [32, 33]. Therefore, extra levels of RRM2 could also partially compensate for ATR deficiency.
Interestingly, a higher fraction of the Chk1 and Rrm2 transgenic mice developed tumors, primarily lymphomas, compared to WT mice, although this difference was not statistically significant. The exact role of the RS response in regard to tumorigenesis is complex [34, 35]. On the one hand, CHK1 is known as a tumor suppressor, and its expression can induce senescence and apoptosis, thereby limiting tumorigenesis [36, 37]. On the other hand, experiments with Chk1Tg MEFs have shown that overexpression of CHK1 can promote oncogenic transformation, and elevated CHK1 levels are present in lymphomas, suggesting that CHK1 could also have a role in promoting tumorigenesis [25, 38]. In relation to RRM2, nucleoside supplementation has been shown to both limit and promote transformation [39, 40]. In addition, increased nucleotide levels can lead to errors during DNA replication and tumorigenesis [27, 41]. Our findings favor the concept that RS is not a requirement for transformation, but also that protection against RS does not prevent tumorigenesis.
Furthermore, the increased tumor incidence in Chk1 and Rrm2 transgenic mice could affect their survival, as is the case for TERT-overexpressing mice. Several Tert transgenic mouse models have an increased incidence of spontaneous tumors [42–45]. Thus, extension of lifespan by TERT-overexpression is only possible in the context of a cancer-resistant background [16]. Since we observed a slightly higher tumor incidence in Chk1 and Rrm2 transgenic mice, these mice could possibly survive longer in a cancer-resistant background. Also, the process of aging is complex and influenced by different factors. Conversely, overexpression of DNA damage proteins may improve several aspects of aging, but not the aging process as a whole, and therefore we cannot rule out the involvement of RS on aging. Nevertheless, our data indicate that RS might not be a relevant contributing factor to normal aging in mice.
Materials and Methods
Mouse husbandry
Chk1Tg and Rrm2Tg mouse models have been described previously [24, 25]. Both strains have a mixed C57BL/6-129/Sv background and carry a bacterial artificial chromosome (BAC) transgene. In case of Chk1Tg mice, there is one extra copy of the gene; Rrm2Tg mice carry multiple copies of Rrm2Tg in tandem. The mice in this study were housed at the University of Copenhagen, Department of Experiment Medicine and mouse work was monitored by the Institutional Animal Care and Use Committee and performed in compliance with Danish and European regulations.
Primers for PCR genotyping
Mice were genotyped using the following primers: gRrm2_Fw (TGTCCTGGAGAGCCAGTCTT), gRrm2_Rev (AAGGAGGGAGGGAGGCTATT) and gRrm2_Transgen (ACTGGCCGTCGTTTTACAAC) for the Rrm2 locus and gChk1_Fw (TGTCTTCCCTTCCCTGCTTA), gChk1_Rev (TCCCAAGGGTCAGAGATCAT) and gChk1_Transgen (GTAAGCCAGTATACACTCCGCTA) for the Chk1 locus. Expected band sizes are 265 bp for the Rrm2+, 380 bp for the Rrm2Tg, 400 bp for the Chk1+ and 270 bp for the Chk1Tg allele.
Cell culture
Mouse embryonic fibroblasts (MEFs) were generated from 13.5 dpc mouse embryos according to standard procedures. MEFs were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 15% heat-inactivated fetal bovine serum (FBS; ThermoFisher Scientific) and 1% penicillin-streptomycin (ThermoFisher Scientific). Three MEF lines were used for WT, Chk1Tg and Chk1Tg;Rrm2Tg, and two MEF lines for Rrm2Tg. The MEFs used in Figure 1 were derived from the same mating, and the MEFs used in Supplementary Figures 1 and 2 were derived from a second mating.
Proliferation assay
Early-passage MEFs were plated at a density of 100.000 cells per well of a 6-well plate. Every three to four days, cells were counted and re-seeded at a density of 100.000 cells per well. The experiment was carried out in three technical replicates.
Cell cycle analysis
10.000 cells were plated per well of a 96 well optical plate. After 24 hours, cells were incubated with 10 μM EdU (Life Technologies, A10044) for 30 minutes, before fixation in 4% PFA. Cells were permeabilized with 0.5% Triton X-100 in PBS for 15 min and washed three times with PBS. Click-iT reaction cocktail containing CuSO4, L-ascorbic acid and Alexa Fluor 647 fluorescent dye azide in PBS was added to each well and incubated according to the manufacturer’s protocol (Invitrogen C10269). Nuclei were counterstained with DAPI, and cell cycle profiles were made by image acquisition on an automated Olympus IX83 ScanR microscope and Olympus ScanR analysis software.
DNA damage experiments and drug treatments
MEFs at passage four were seeded in a 96 well optical plate at a density of 10.000 cells/well. After 24 hours, MEFs were incubated with hydroxyurea (Sigma-Aldrich H8627) or UCN-01 (Sigma-Aldrich U6508) at indicated concentrations for four hours, followed by fixation with 4% paraformaldehyde (PFA). For immunofluorescence experiments, cells were permeabilized with Triton X-100 (Sigma-Aldrich T8787), blocked in IF blocking buffer (3% BSA, 0.1% Tween in PBS), and incubated with γH2AX antibody (Merck Millipore 05-636) at 4°C overnight. Samples were incubated with goat anti-mouse IgG secondary antibody, Alexa Fluor 488 (Sigma-Aldrich A-11001) for 1 hour at room temperature, and nuclei were stained with DAPI. High-content microscopy was performed using an automated Olympus IX83 ScanR microscope.
Western blotting
Cells were lysed in RIPA buffer (Sigma-Aldrich R0278) supplemented with protease (Roche 4693132001) and phosphatase inhibitors (Sigma-Aldrich P0044). Western blot analysis was performed according to standard procedures. Primary antibodies used were anti-CHK1 (Novocastra), anti-RRM2 (Santa Cruz Biotechnology, SC-10844) and anti-β-actin (Sigma-Aldrich, A5441).
Histopathology
Mouse tissues were fixed in formalin and processed for immunohistochemistry (IHC) by HistoWiz Inc. (https://home.histowiz.com/; Figure2B) or the Histopathology unit at the Spanish National Cancer Research Center (CNIO; Figure 3B).
Statistical analysis
P-values were calculated in GraphPad Prism 7, unless otherwise indicated. For γH2AX analysis (Figure 1E, 1F and Supplementary Figure 2A, 2B), and for comparison of mouse weights (Figure 2C), an unpaired t-test was performed. The log-rank test was used for the mouse survival curve in Figure 2A. For Figure 3A, a Chi-square test was performed using GraphPad QuickCalcs (https://www.graphpad.com/quickcalcs/chisquared1.cfm).
Supplementary Materials
Author Contributions
A.J.L.-C. conceived and supervised the study. E.A. designed and performed most of the experiments. All the authors provided relevant intellectual input during this study. M.S. and A.A helped with the maintenance of the mouse colonies and generation of MEFs. The transgenic mouse models were generated in O.F. lab. A.J.L.-C. and E.A. wrote the manuscript. All of the authors read, edited and commented on the manuscript.
Acknowledgments
We thank the caretakers at Department of Experimental Medicine at the University of Copenhagen, and the Histopathology core unit at the Spanish National Cancer Research Center (CNIO, Madrid) for technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
Research in A.J.L.-C. lab was funded by Danish Council for Independent Research (Sapere Aude, DFF-Starting Grant 2014); Danish National Research Foundation (DNRF115); Danish Cancer Society (KBVU-2017_R167-A11063); European Research Council (ERC-2015-STG-679068); and Nordea-fonden (02-2017-1749). Research in O.F. lab was funded by grants from the Spanish Ministry of Science; Innovation and Universities (RTI2018-102204-B-I00, co-financed with European FEDER funds); and the European Research Council (ERC-617840).
References
- 1. Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008; 454:1065–71. https://doi.org/10.1038/nature07216 [PubMed]
- 2. Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005; 120:497–512. https://doi.org/10.1016/j.cell.2005.01.028 [PubMed]
- 3. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 4. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009; 361:1475–85. https://doi.org/10.1056/NEJMra0804615 [PubMed]
- 5. Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003; 299:1355–9. https://doi.org/10.1126/science.1079161 [PubMed]
- 6. Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015; 15:276–89. https://doi.org/10.1038/nrc3916 [PubMed]
- 7. Cogger VC, Svistounov D, Warren A, Zykova S, Melvin RG, Solon-Biet SM, O’Reilly JN, McMahon AC, Ballard JW, De Cabo R, Le Couteur DG, Lebel M. Liver aging and pseudocapillarization in a Werner syndrome mouse model. J Gerontol A Biol Sci Med Sci. 2014; 69:1076–86. https://doi.org/10.1093/gerona/glt169 [PubMed]
- 8. German J. Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine (Baltimore). 1993; 72:393–406. https://doi.org/10.1097/00005792-199311000-00003 [PubMed]
- 9. Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000; 26:424–29. https://doi.org/10.1038/82548 [PubMed]
- 10. van der Horst GT, van Steeg H, Berg RJ, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR, Bootsma D, Hoeijmakers JH. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997; 89:425–35. https://doi.org/10.1016/S0092-8674(00)80223-8 [PubMed]
- 11. Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006; 444:1038–43. https://doi.org/10.1038/nature05456 [PubMed]
- 12. Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009; 41:891–98. https://doi.org/10.1038/ng.420 [PubMed]
- 13. Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005; 120:449–60. https://doi.org/10.1016/j.cell.2005.02.002 [PubMed]
- 14. Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol. 2013; 15:96–102. https://doi.org/10.1038/ncb2643 [PubMed]
- 15. Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Viña J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007; 448:375–79. https://doi.org/10.1038/nature05949 [PubMed]
- 16. Tomás-Loba A, Flores I, Fernández-Marcos PJ, Cayuela ML, Maraver A, Tejera A, Borrás C, Matheu A, Klatt P, Flores JM, Viña J, Serrano M, Blasco MA. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell. 2008; 135:609–22. https://doi.org/10.1016/j.cell.2008.09.034 [PubMed]
- 17. Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 35:7545–56. https://doi.org/10.1093/nar/gkm1059 [PubMed]
- 18. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008; 9:616–27. https://doi.org/10.1038/nrm2450 [PubMed]
- 19. López-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair (Amst). 2010; 9:1249–55. https://doi.org/10.1016/j.dnarep.2010.09.012 [PubMed]
- 20. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003; 300:1542–48. https://doi.org/10.1126/science.1083430 [PubMed]
- 21. Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, Le Beau MM, Stohr BA, Méndez J, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014; 512:198–202. https://doi.org/10.1038/nature13619 [PubMed]
- 22. O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003; 33:497–501. https://doi.org/10.1038/ng1129 [PubMed]
- 23. Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, Artista L, Schleker T, Guerra C, Garcia E, Barbacid M, Hidalgo M, Amati B, Fernandez-Capetillo O. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011; 18:1331–35. https://doi.org/10.1038/nsmb.2189 [PubMed]
- 24. Lopez-Contreras AJ, Specks J, Barlow JH, Ambrogio C, Desler C, Vikingsson S, Rodrigo-Perez S, Green H, Rasmussen LJ, Murga M, Nussenzweig A, Fernandez-Capetillo O. Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev. 2015; 29:690–95. https://doi.org/10.1101/gad.256958.114 [PubMed]
- 25. López-Contreras AJ, Gutierrez-Martinez P, Specks J, Rodrigo-Perez S, Fernandez-Capetillo O. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J Exp Med. 2012; 209:455–61. https://doi.org/10.1084/jem.20112147 [PubMed]
- 26. García-Cao I, García-Cao M, Martín-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, Serrano M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002; 21:6225–35. https://doi.org/10.1093/emboj/cdf595 [PubMed]
- 27. Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, Lu Y, Bronson R, Alani E, Nikitin AY, Weiss RS. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008; 68:2652–60. https://doi.org/10.1158/0008-5472.CAN-07-5873 [PubMed]
- 28. Ruiz S, Lopez-Contreras AJ, Gabut M, Marion RM, Gutierrez-Martinez P, Bua S, Ramirez O, Olalde I, Rodrigo-Perez S, Li H, Marques-Bonet T, Serrano M, Blasco MA, et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat Commun. 2015; 6:8036. https://doi.org/10.1038/ncomms9036 [PubMed]
- 29. Schulze J, Lopez-Contreras AJ, Uluçkan Ö, Graña-Castro O, Fernandez-Capetillo O, Wagner EF. Fos-dependent induction of Chk1 protects osteoblasts from replication stress. Cell Cycle. 2014; 13:1980–86. https://doi.org/10.4161/cc.28923 [PubMed]
- 30. Weiss RS, Enoch T, Leder P. Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 2000; 14:1886–98. [PubMed]
- 31. Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun. 2014; 5:5744. https://doi.org/10.1038/ncomms6744 [PubMed]
- 32. Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998; 2:329–40. https://doi.org/10.1016/S1097-2765(00)80277-4 [PubMed]
- 33. Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998; 94:595–605. https://doi.org/10.1016/S0092-8674(00)81601-3 [PubMed]
- 34. Hsieh HJ, Peng G. Cellular responses to replication stress: Implications in cancer biology and therapy. DNA Repair (Amst). 2017; 49:9–20. https://doi.org/10.1016/j.dnarep.2016.11.002 [PubMed]
- 35. Lecona E, Fernández-Capetillo O. Replication stress and cancer: it takes two to tango. Exp Cell Res. 2014; 329:26–34. https://doi.org/10.1016/j.yexcr.2014.09.019 [PubMed]
- 36. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006; 444:633–37. https://doi.org/10.1038/nature05268 [PubMed]
- 37. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garrè M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006; 444:638–42. https://doi.org/10.1038/nature05327 [PubMed]
- 38. Höglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, Rudelius M, Nikolova V, Keller U, Nilsson JA. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res. 2011; 17:7067–79. https://doi.org/10.1158/1078-0432.CCR-11-1198 [PubMed]
- 39. Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, Zhang R. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013; 3:1252–65. https://doi.org/10.1016/j.celrep.2013.03.004 [PubMed]
- 40. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011; 145:435–46. https://doi.org/10.1016/j.cell.2011.03.044 [PubMed]
- 41. Martomo SA, Mathews CK. Effects of biological DNA precursor pool asymmetry upon accuracy of DNA replication in vitro. Mutat Res. 2002; 499:197–211. https://doi.org/10.1016/s0027-5107(01)00283-4 [PubMed]
- 42. González-Suárez E, Samper E, Ramírez A, Flores JM, Martín-Caballero J, Jorcano JL, Blasco MA. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001; 20:2619–30. https://doi.org/10.1093/emboj/20.11.2619 [PubMed]
- 43. González-Suárez E, Flores JM, Blasco MA. Cooperation between p53 mutation and high telomerase transgenic expression in spontaneous cancer development. Mol Cell Biol. 2002; 22:7291–301. https://doi.org/10.1128/MCB.22.20.7291-7301.2002 [PubMed]
- 44. Artandi SE, Alson S, Tietze MK, Sharpless NE, Ye S, Greenberg RA, Castrillon DH, Horner JW, Weiler SR, Carrasco RD, DePinho RA. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci USA. 2002; 99:8191–6. https://doi.org/10.1073/pnas.112515399 [PubMed]
- 45. Canela A, Martín-Caballero J, Flores JM, Blasco MA. Constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in Lck-Tert mice. Mol Cell Biol. 2004; 24:4275–93. https://doi.org/10.1128/MCB.24.10.4275-4293.2004 [PubMed]