



Introduction
Globally, melanoma is a serious malignant skin cancer, and is estimated to kill approximately 55,500 individuals annually [1]. Increasing melanoma incidences are reported in older Caucasians, with males experiencing higher rates than females [2, 3]. Traditional surgical and chemo-radiotherapy treatments often fail to prolong patient survival, especially in those with advanced disease. This is due to metastasis and chemotherapy resistance, primarily due to acquired somatic mutations, e.g., B-Raf oncogene serine/threonine-kinase (BRAF), NRAS proto-oncogene (NRAS) and neurofibromin 1 (NF1) [4]. Targeted therapies such as dabrafenib and vemurafenib are proven melanoma treatments, generating positive results in clinical settings [5–9]. However, drug resistance after a period of treatment, leads to poor responses or relapses in advanced melanoma patients [10, 11]. Recent studies have demonstrated that metabolic reprogramming plays important roles in melanoma tumorigenesis, and may be responsible for drug resistance and poor prognoses [12]. Therefore, the identification of novel therapeutic strategies targeting cancer metabolism, and the concomitant development of therapeutic drugs are essential for melanoma cancer therapy.
Cellular cholesterol levels are tightly regulated by an intricate network of transcriptional regulation of cholesterol biosynthesis, deposition, cellular uptake and efflux programs [13–15]. Cholesterol metabolism dysregulation is associated with cancers. Oncogenic processes permits cholesterol synthesis in cancer cells, which is further metabolized to support rapid proliferation [16]. Recent investigations have shown that cholesterol biosynthesis gene activation is associated with poor prognoses in leukemia, sarcoma and melanoma [17]. Indeed, perturbed expression of key cholesterol biosynthesis enzymes, such as HMGCR and SQLE, prevents carcinogenesis in colorectal cancer, breast cancer, prostate cancer and hepatocellular carcinoma associated with nonalcoholic fatty liver disease (NAFLD-HCC) [18–22]. However, the role of cholesterol and its metabolites in cancer is still controversial [17, 23–25]. In breast cancer, decreased CYP7B1 expression triggers 27-hydroxycholesterol (27HC) accumulation [26], permitting 27HC to act as an estrogen receptor agonist that induces tumor growth and metastasis [25]. Other reports have observed that the cholesterol metabolites, dendrogenin A [27], cholestane-3β, 5α and 6β-triol [28] exert anti-proliferative effects in human breast and prostate cancer cells [28, 29]. Of note, the cholesterol metabolite, 7-dehydrocholesterol (7-DHC) exhibits toxic and pro-apoptotic effects towards melanoma cells [30], thereby encouraging us to develop a therapeutic strategy incorporating cholesterol conversion to 7-DHC.
Although the cholesterol 7-desaturase conversion of cholesterol to 7-DHC has not yet been reported in mammalian cells, several evolutionary conserved isoenzymes exist in orthologous organisms, such as NVD-BM from Bombyx mori, DAF-36 from Caenorhabditis elegans, NVD-DR from Danio rerio, TTHERM_00310640 from Tetrahymena thermophila SB210 and MGC154819 from Xenopus laevis [31–33]. Natural products including 7-DHC have been proven in cancer therapy, however, dose administration and side effects often hinder their clinical applications [34]. Designing and synthesizing genetic regulatory circuits in living cells, to program new biological behaviors, is a promising synthetic biological approach for medicinal research and potential therapeutic applications [35, 36]. Reprogrammed human cells can circumvent the shortcomings of conventional therapies, such as frequent administration and self-sufficient doses, providing new strategies to solve the complexities of metabolic disorders [37]. In this study, we performed codon optimization on several cholesterol 7-desaturases to examine their roles in melanoma cell proliferation, migration and tumorigenicity, both in vitro and in vivo. Furthermore, we developed a smart genetic circuit to induce 7-DHC accumulation by sensing transcription factors in melanoma cells.
Results
Cholesterol 7-desaturase expression inhibits melanoma cell growth and migration
Since no cholesterol 7-desaturases exists in mammalian cells, we optimized codon usage and synthesized several cholesterol 7-desaturases from other species; DAF-36 from Caenorhabditis elegans, NVD-BM from Bombyx mori, NVD-DR from Danio rerio (zebrafish), TTHERM_00310640 from Tetrahymena thermophila SB210 and MGC154819 from Xenopus laevis (Table 1, Supplementary Table 1). This approach facilitated anti-cancer studies in melanoma cells. Using transient transfection, the five humanized cholesterol 7-desaturases were expressed in A375 and A2058 melanoma cells by qRT-PCR assay (Figure 1A, 1B). Cell proliferation assays showed that the expression of NVD-BM significantly slowed cell growth of melanoma cells (A375 and A2058), however, DAF-36 failed to suppress A2058 cell proliferation and other cholesterol 7-desaturases did not inhibit melanoma cell proliferation (Figure 1C, 1D). Furthermore, the proliferation effects exerted by NVD-BM expression were dose dependent in A375 cells (Figure 2A). We next investigated cell migration capabilities in cells expressing NVD-BM, using the real-time cell analysis (RTCA). These data showed that A375-NVD-BM and A2058-NVD-BM cell migration capabilities were lower than the empty vector negative control (NC) group at 36 h, suggesting that NVD-BM severely impaired melanoma cell migration (Figure 2B, 2C).
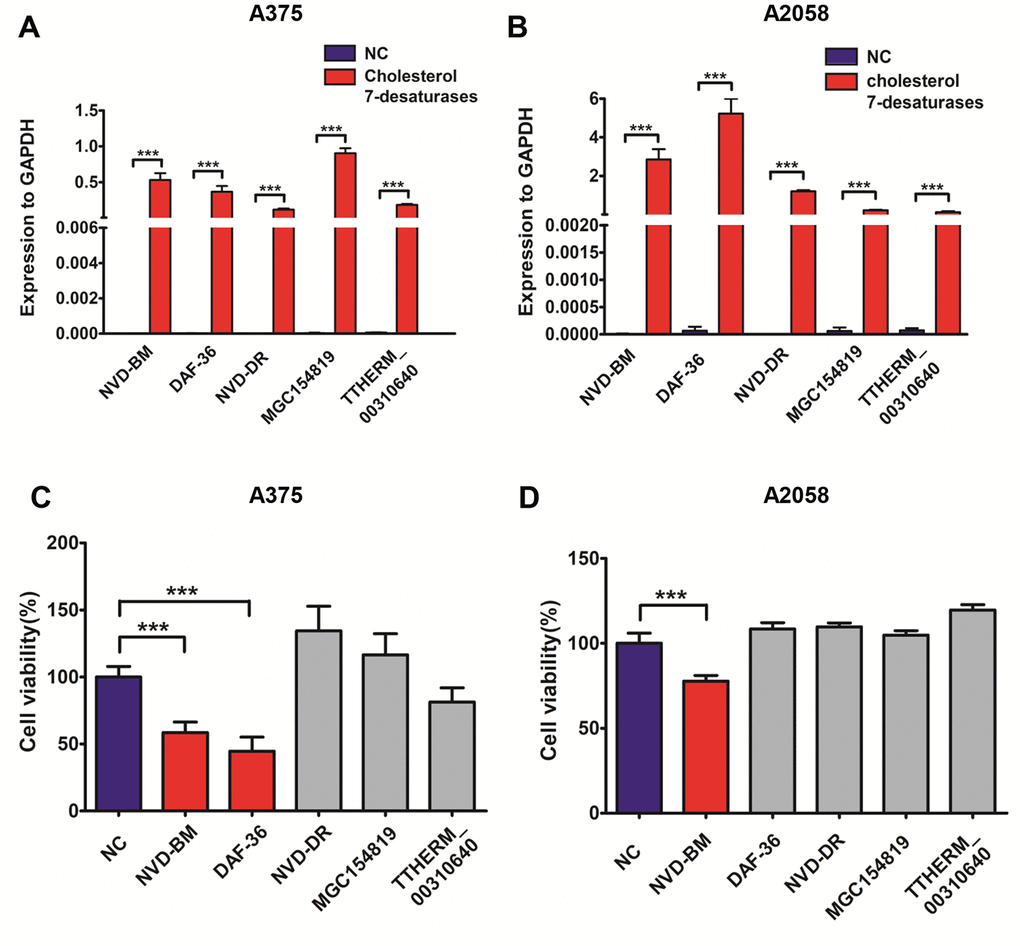
Figure 1. Heterogeneous cholesterol 7-desaturases inhibits melanoma cell growth. (A, B) Quantification of GAPDH, NVD-BM, DAF-36, NVD-DR, MGC154819 and TTHERM_00310640 gene expression using RT-qPCR in A375 and A2058 cells. (C, D) A375 and A2058 cell proliferation transfected with genes expressing different cholesterol 7-desaturases at 24 h. (***p < 0.001)
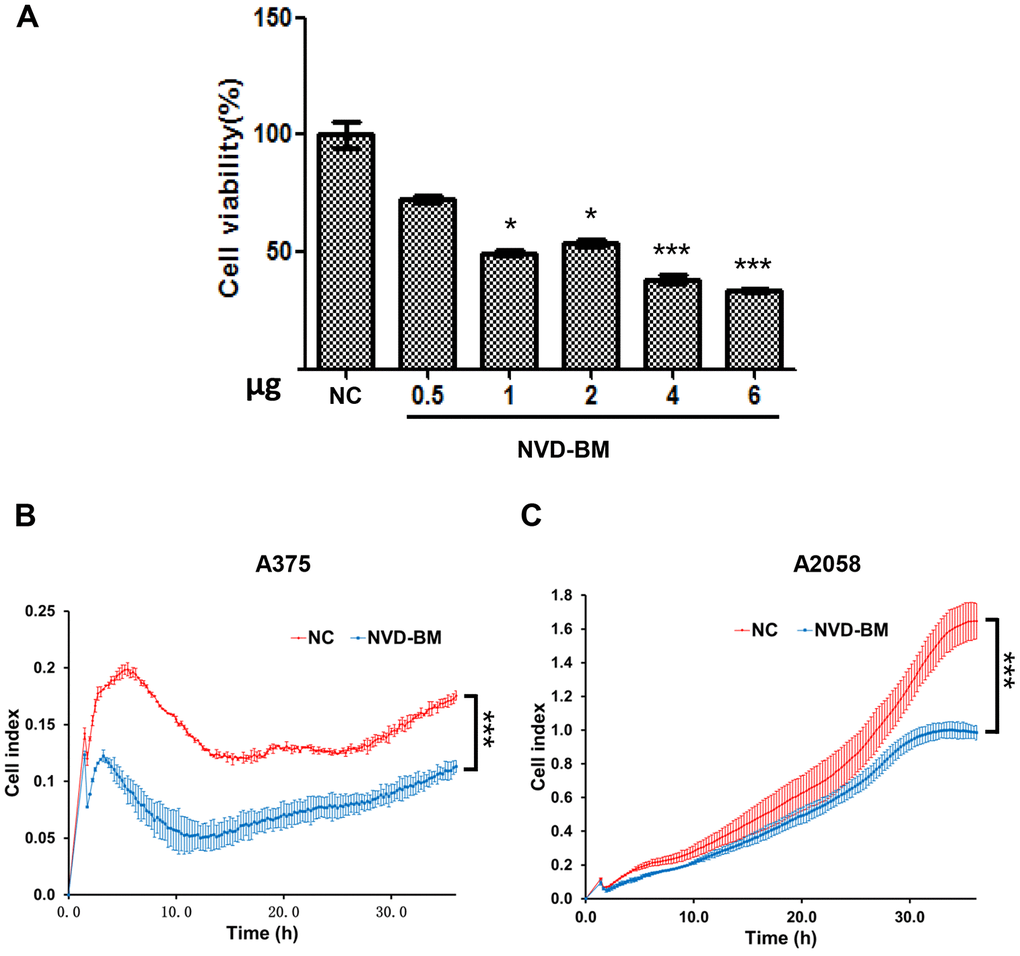
Figure 2. NVD-BM inhibits melanoma cell proliferation and migration. (A) A375 cell proliferation when transfected with different NVD-BM concentrations at 24 h. (B, C) The migration rates of 5×103 A375 and A2058 cells, transfected with pCDH and NVD-BM plasmid were assessed over 36 h by RTCA assay. (*p < 0.05, **p < 0.01, ***p < 0.001)
Table 1. Heterogeneous cholesterol 7-desaturase.
| Gene Symbol | Annotation | Species | Gene ID |
| DAF-36 | Cholesterol 7-desaturase | Caenorhabditis elegans | 179422 |
| NVD-BM | Rieske-domain protein Neverland | Bombyx mori | 733067 |
| NVD-DR | Neverland | Danio rerio | 436885 |
| TTHERM_00310640 | Rieske-like [2Fe-2S] domain protein | Tetrahymena thermophila SB210 | 7840976 |
| MGC154819 | MGC154819 protein | Xenopus laevis | 779348 |
NVD-BM inhibits melanoma cell proliferation in xenograft mice
The proliferation and migration inhibitory capabilities of NVD-BM in melanoma cells suggested anti-tumor effects in vitro, therefore we sought to recapitulate these effects in vivo. To confirm the anti-tumor effects of NVD-BM in vivo, A375 cells expressing NVD-BM and the empty vector NC, were subcutaneously injected into BALB/c-nu/nu mice (n = 4). Mice were sacrificed at the end of the fourth week, with tumors excised, measured and stained with hematoxylin-eosin (HE). Our data demonstrated that tumor diameter from A375 cells expressing NVD-BM, were approximately four folds smaller significantly than the NC group (p = 0.00063) (Figure 3A). Moreover, overall tumor structures were almost normal, and displayed no obvious necrosis (Figure 3B). In contrast, the NC group displayed large areas of necrosis, nuclear degradation and cytoplasmic protein degradation. These data demonstrated that NVD-BM served as an effective tumor suppressor in melanoma derived cells in vivo.
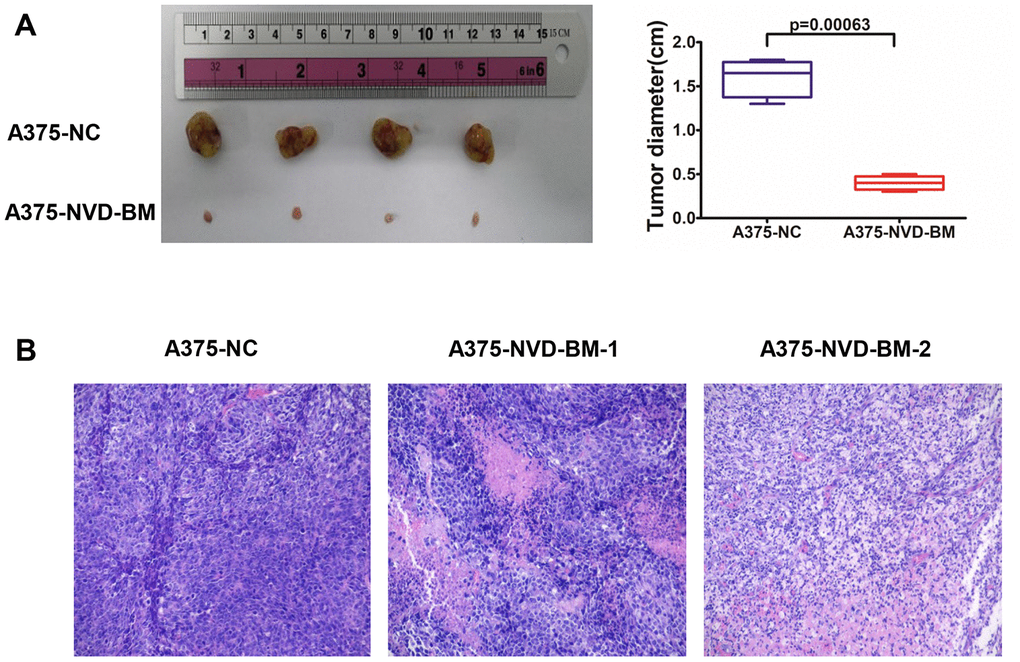
Figure 3. NVD-BM inhibits melanoma proliferation in xenograft mice. (A) Subcutaneous tumors generated in BALB/c-nu/nu mice, with NVD-BM transduced A375 cells. A375 cells transduced with blank lentiviral vector, and vectors expressing pCDH and NVD-BM were collected and subcutaneously injected into BALB/c-nu/nu mice, at a cell density of 1×107 to elicit tumorigenicity. Tumors were removed and diameters measured (n = 4/group) ± standard deviation (SD). (B) Hematoxylin eosin (HE) tumor staining in transduced A375 cells. Nuclei are blue. Cytoplasm is red. Magnification × 200.
NVD-BM catalyzes cholesterol to 7-DHC in melanoma cells
To test cholesterol 7-desaturase capability in converting cholesterol to 7-DHC, we extracted sterols from A375 cells with the expression of cholesterol 7-desaturases, and determined cholesterol and 7-DHC concentrations using GC-MS (Figure 4A). Our data revealed that NVD-BM converted cholesterol to 7-DHC, and was 2.1 folds compared with NC group (Figure 4B, 4C). DAF-36 failed to convert cholesterol to 7-DHC in A375 cells, suggesting that DAF-36 inhibited A375 cell proliferation via alternative, non-cholesterol mediated mechanisms. Therefore, NVD-BM appeared to effectively inhibit melanoma growth via cholesterol degradation, to increase 7-DHC levels in cells. This observation was consistent with the previous report showing that the proliferation of melanoma cells was inhibited by 7-DHC addition (Supplementary Figure 1).
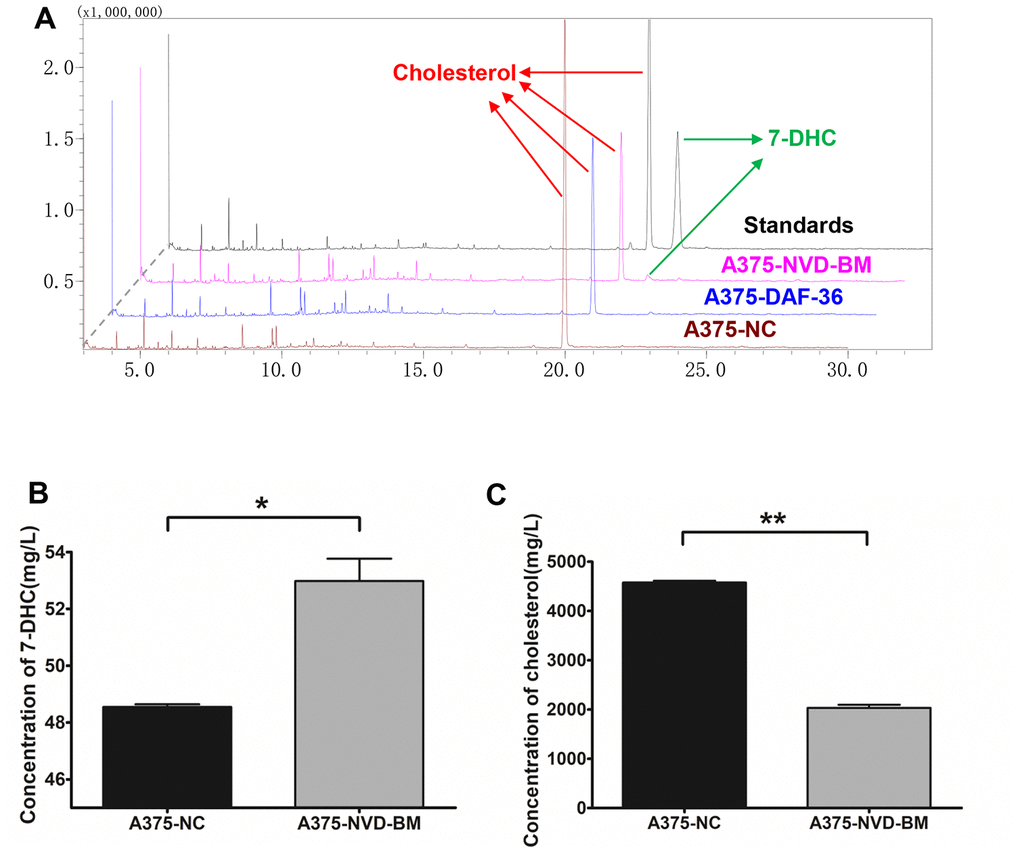
Figure 4. Heterogeneous cholesterol 7-desaturase NVD-BM converts cholesterol to 7-DHC. (A) GC/MS analysis of 7-DHC and cholesterol levels in A375 cells, transfected with NVD-BM and DAF-36, when compared with the negative control pCDH (NC), using 7-DHC and cholesterol standard curves. (B) 7-DHC concentrations in A375 cells, transfected with NVD-BM versus NC. (C) Cholesterol concentrations in A375 cells, transfected with NVD-BM versus NC. (*p < 0.05, **p < 0.01).
The development of a dual-input signal genetic biosensor
To enhance the efficacy and specificity of NVD-BM, based on a genetic circuit, we were ought to scan highly expressed transcription factors as dual-input sensors. We compared gene expression profiling data from melanoma tissue with normal skin tissue, and identified several differentially expressed melanoma genes. The top 10 highly expressed transcription factors, RELA, STAT1, FOXD1, POU3F4, SOX5, ELF1, TBX2, LEF1, HOXD13 and POU3F2, were then evaluated for their ability to drive luciferase expression (Figure 5A). Five repetitive transcription factor binding motifs were separately cloned into firefly luciferase vectors, driven by mini-promoters, to determine firefly luciferase expression in A375 and A2058 cells (Supplementary Table 2, Figure 5B). As shown in Figure 5C, RELA, STAT1, POU3F2 and POU3F4 were the top four input signals in A375 and A2058 cells. Next, vectors with different input signals, were divided into 16 permutation methods. Heatmaps showed that the optimum match was RELA and STAT1 in A375 and A2058 cells, respectively (Figure 5D). Finally, RELA and STAT1 were selected as dual-input signals for a genetic biosensor in melanoma cells.
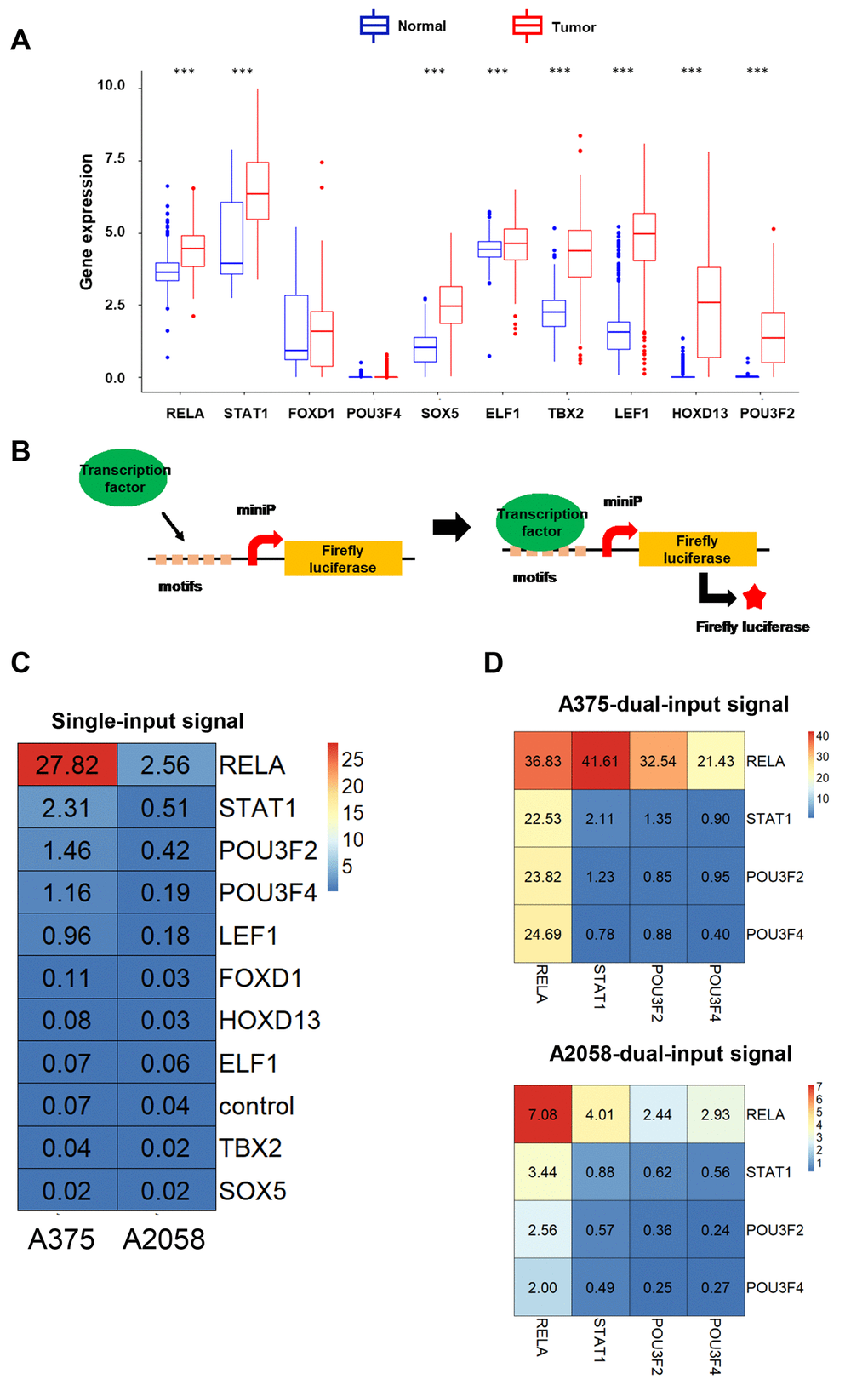
Figure 5. The development of dual-input biosensors. (A) Up-regulated transcription factors in melanoma tissues compared with normal skin tissues, were filtered from the TCGA database. (B) A schematic diagram of transcription factors as input signals, activating biosensors using the luciferase assay. (C) Heatmap of luciferase expression levels using a single-input promoter in A375 and A2058 cells. (D) Heatmap of luciferase expression levels using dual-input promoters in A375 and A2058 cells. (***p < 0.001).
The activated genetic biosensor inhibits melanoma cell growth and migration
We next developed a molecular switch combined with CRISPR-Cas9 and TET-ON, with four modules to activate NVD-BM expression and inhibit melanoma cell growth and decrease migration rates (Figure 6A). Five repetitive motifs from RELA (5’- GGGAATTTCC-3’), Cas9 and sgRNA, targeting the lacI gene were assembled into one construct, facilitating RELA binding to induce Cas9 expression. The Cas9 protein with sgRNA-lacI in the next module, inhibited lacI expression. Five repetitive motifs from STAT1 (5’- CATTTCCCGGAAACC-3’), lacO and rtTA were constructed in one vector, recruiting STAT1 binding to induce rtTA expression. In case of toxicity, NVD-BM was not expressed without doxycycline. When doxycycline was added to the culture medium, it entered cells and bound rtTA. Then, rtTA and doxycycline bound to the TRE gene to activate NVD-BM expression. Thus, when RELA and STAT1 were up-regulated in a tumor, parallel to doxycycline added in vitro, the genetic circuit was then switched on (Figure 6B).
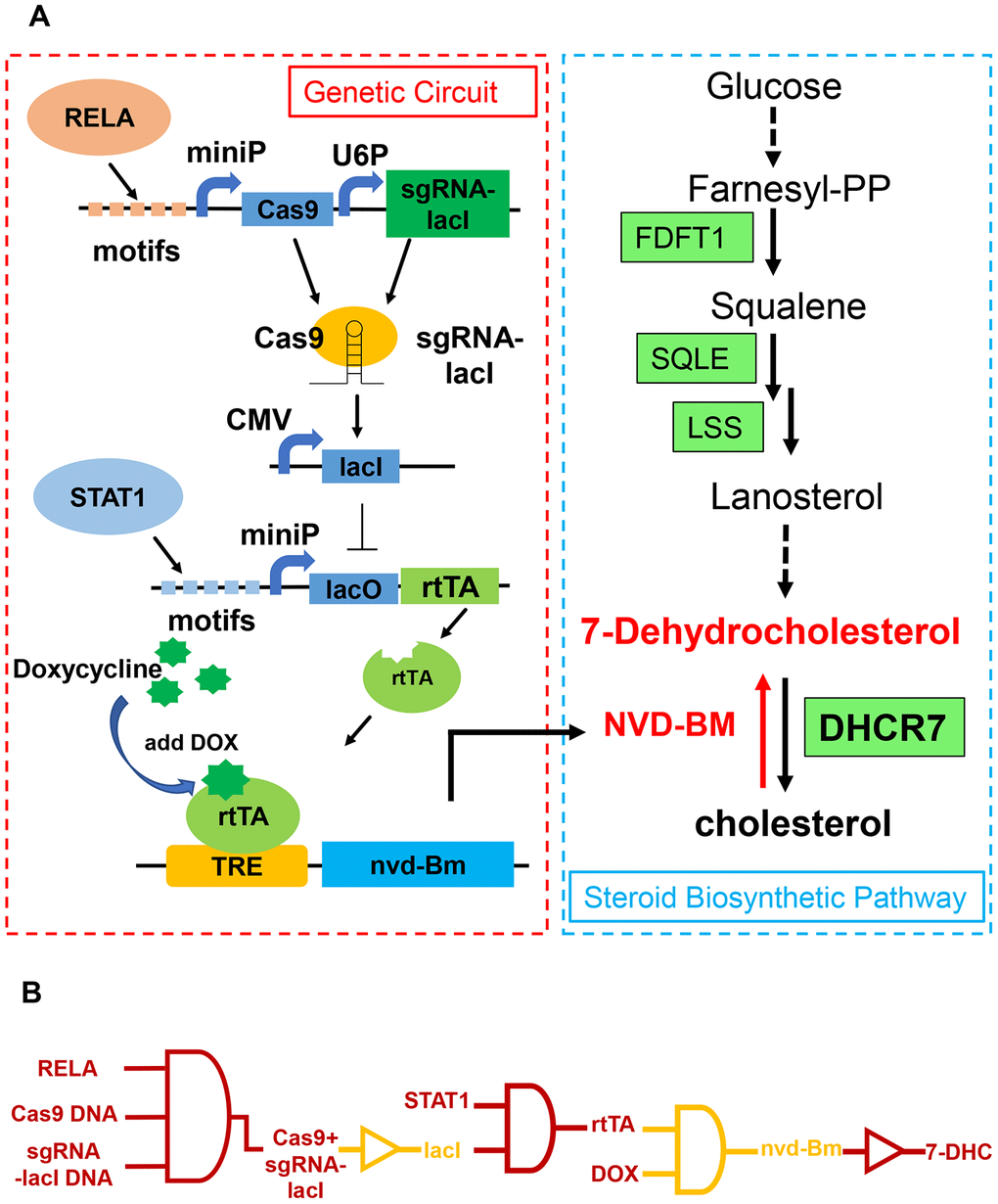
Figure 6. Schematic of a genetic circuit in melanoma. (A) A gene circuit, with four modules was activated by two up-regulated transcription factors (RELA and STAT1), to express cholesterol 7-desaturase NVD-BM, and directly catalyzing cholesterol to 7-DHC, to repress melanoma growth and migration. (B) Gene circuit diagram constructed in melanoma cells based on CRISPR/Cas9 and Tet-ON systems.
To test genetic circuit availability in melanoma cells, we co-transfected constructs into A375 and A2058 cells, and administered doxycycline at final concentration of 1 μM. Quantitative RT-PCR data showed that NVD-BM expression was activated by doxycycline (Figure 7A), and A375 and A2058 proliferation and migration rates were significantly impaired by NVD-BM activation (Figure 7B–7D). Thus, our genetic circuit effectively sensed the dual-input of RELA and STAT1 signals, and induced NVD-BM actuator expression to rebuild cholesterol metabolism by converting cholesterol to 7-DHC, promoting melanoma cell regression.
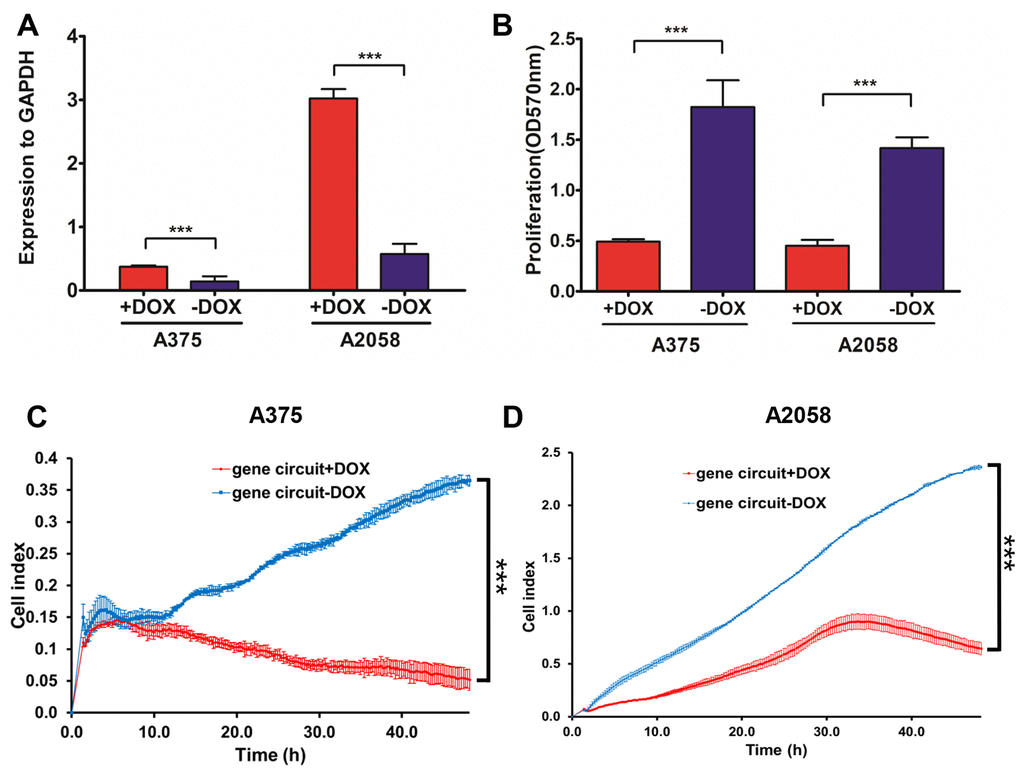
Figure 7. The activated genetic circuit inhibits melanoma cell growth and migration. (A) A375 and A2058 cells were co-transfected with genetic circuit plasmids. The relative expression of the reporter gene NVD-BM compared to GAPDH, was quantified by qRT-PCR. (B) A375 and A2058 cell proliferation was assessed after treatment with and without doxycycline for 48 h. (C, D) The migration rates of 6×104 A375 cells transfected with genetic circuit plasmids (± doxycycline) were assessed over 36 h by RTCA assay. (***p < 0.001).
NVD-BM functions as a tumor suppressor via Akt1/NF-κB signaling
Enhanced NF-κB activation, as a result of upstream signaling dysregulation, is a main molecular characteristic in melanoma [38]. Indeed, GSEA analysis of GSE3189 transcriptomic data between normal skin and melanoma samples, demonstrated that NF-κB signaling was up-regulated in melanoma patients (Figure 8A). Thus, we determined the expression of known NF-κB targets in melanoma, e.g., MMP9, CCL20 and IL1B [39, 40]. RT-PCR data demonstrated that MMP9, CCL20 and IL1B expression was significantly down-regulated by NVD-BM (Figure 8B). Similarly, by investigating the cellular localization of free RELA, which is an important transcription factor implicated in NF-κB signaling, we noted that RELA accumulated in the cytoplasm by western blot assay, in a dose dependent NVD-BM manner (Figure 8C), thus, suggesting that RELA translocation to the nucleus was inhibited under these conditions. Next, we determined the phosphorylation status of an upstream NF-κB kinase by western blot assay, and found that Akt1-ser473 phosphorylation was decreased by NVD-BM, in a dose dependent manner (Figure 8D). Collectively, these data indicated that NVD-BM promoted tumor regression by decreasing phosphorylated Akt1 levels, and inhibiting free RELA translocation into the nucleus in melanoma cells.
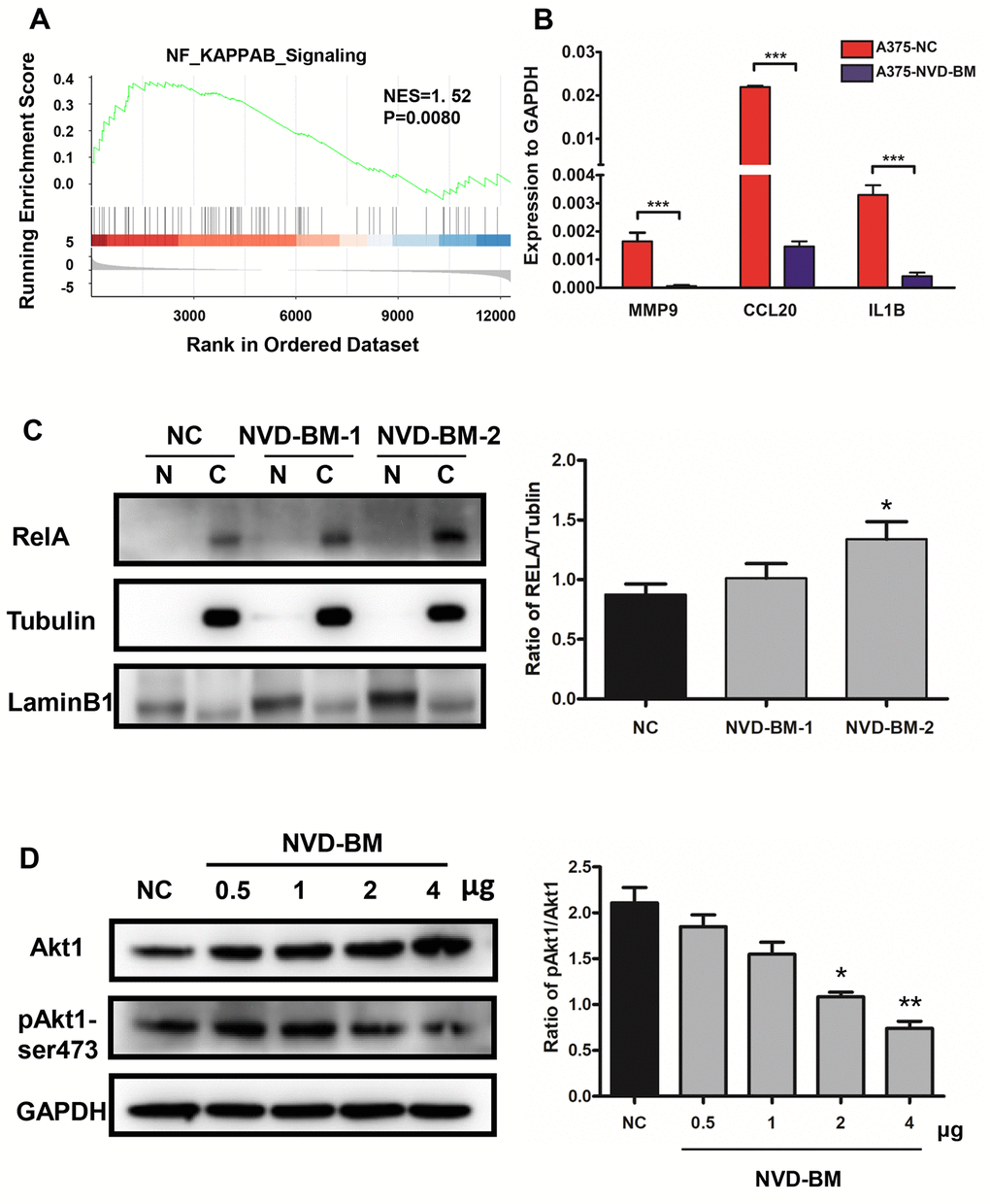
Figure 8. NVD-BM functions as a tumor suppressor via Akt1/NF-κB signaling. (A) GSEA revealed up-regulated genes associated with NF-κB signaling in melanoma (GSE3189 dataset). (B) Gene expression of the NF-κB target genes, MMP9, CCL20 and IL1B relative to GAPDH, in A375 cells transfected with NVD-BM, when compared with A375 cells transfected with blank vector, pCDH. (C) Western blot of A375 cells transfected with two different plasmid NVD-BM concentrations, when compared with pCDH blank vectors, analyzing the entry of RELA into the nucleus. (D) Western blot of A375 cells transfected with two different plasmid NVD-BM concentrations, when compared to the pCDH blank vector analyzed phosphorylated and unphosphorylated Akt1-SER473 with normalization to GAPDH. (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
Metabolic reprogramming has received considerable attention due to its ability to support growth and anti-apoptosis mechanisms in cancers [23]. Disrupting the expression of key enzymes in cholesterol biosynthesis, such as HMGCR and SQLE, appears to prevent carcinogenesis mechanisms in colorectal, breast, prostate and NAFLD-HCC cancers [18–22]. Though comprehensive studies showed that high cellular cholesterol contributes to tumor growth and metastasis, it is still not clear in melanoma yet. Indeed, transcriptome data shows that cholesterol biosynthesis gene activation is associated with poor prognoses in melanoma patients [17], however, the expression of cholesterol biosynthesis limiting enzymes HMGCR was down-regulated in melanoma patients according to analysis of TCGA-SKCM and GSE3189 transcriptome data (Supplementary Figure 2). In present study, administration of 7-DHC in vitro suppressed melanoma cell growth [38], suggesting that the strategic manipulation of cholesterol metabolism may be a potential strategy for melanoma treatment. Because no cholesterol 7-desaturases exist in mammalian cells, we screened and cloned evolutionary homologous cholesterol 7-desaturases from several species and tested them against several melanoma cell pathological characteristics, such as cell proliferation and migration. When compared with DAF-36, NVD-BM from Bombyx mori exhibited a stronger ability to inhibit growth in in vitro melanoma cells, and growth in an in vivo xenograft mouse model. Consistent with GC/MS data, DAF-36 failed to convert cholesterol to 7-dehydrocholesterol. Thus, we believed that NVD-BM is a novel tumor repressor in melanoma.
Synthetic cell-based therapies are ideal approaches for metabolic disorders, as they circumvent the shortcomings of conventional therapies, such as frequent administration and self-sufficient doses. Genetic circuits are typically composed of a sensor, processor and actuator [35]. From our results, NVD-BM was an ideal actuator in promoting 7-DHC accumulation in skin cancer. To ensure specificity, safety and efficacy of the genetic circuit, the sensor and processor were composed of four independent modules, which sensed highly expressed transcription factors in tumors, and activated downstream step by step. The CRISPR-Cas9 system and the Tet-on switch were central to our genetic circuit, guide RNA and Cas9 device were detached on separated module and dimerized once sensing up-regulated transcription factors. RELA and STAT1 binding motifs were blocked, and the switch was activated by doxycycline. Our results showed that RELA and STAT1 activated NVD-BM expression in the presence of doxycycline, and slowed melanoma cell growth and migration. However, endogenous transcription factors usually function as “master regulators”, exerting control over processes that specify cell types and developmental patterning in normal cells. Hence, preventing signal leakage and accurately setting specific threshold values that distinguish tumor and normal cells to trigger circuit activation, should be investigated in the future.
Taken together, we identified NVD-BM from Bombyx mori as a novel melanoma tumor suppressor. It decreased Akt1 phosphorylation levels and inhibited RELA translocation into nuclei. We also developed an effective genetic circuit, based on an “AND” gate approach, to activate NVD-BM expression by sensing RELA and STAT1. Our results provide insights to against cancer with cholesterol accumulation by depletion of cholesterol. More importantly, our genetic circuit dual-inputs and actuators are replaceable depend on cancer types, thus providing a general metabolic reprogramming strategy for cancer treatment.
Materials and Methods
Cell lines and cell culture
A375 malignant melanoma (A375) and 293T embryonic kidney (HEK293T) cells were purchased from Cell Resource Center of Peking Union Medical College (IBMS, CAMS/PUMC). The A2058 melanoma cell line was kindly provided by Dr. Fang from the Beijing Institute of Genomics. All cells were cultured in high-glucose DMEM complete medium (Gibco), with 1% penicillin-streptomycin (Solarbio), and 10% fetal bovine serum (FBS) (Gibco) in a 5% CO2, 37°C environment.
Subcutaneous tumor formation
BALB/c-nu/nu mice, weighing on average 10 g, were purchased from Beijing Huafukang Bioscience Co. Inc. Lentiviral vectors for NVD-BM and pCDH were transfected into A375 cells (n = 4/group), and selected using 1 μg/ml puromycin for two weeks. We then collected 1×107 cells for subcutaneous injection into mice. After four weeks, we measured mouse tumors, and tumor tissues were fixed in 10% neutral buffered formalin for hematoxylin-eosin (HE) staining. The animal protocol was reviewed and approved by the Animal Ethical and Welfare Committee (AEWC) (approval No. IRM-DWLL-2019102).
Plasmid construction
Codon optimization for cholesterol 7-desaturases from Caenorhabditis elegans, Bombyx mori, Danio rerio (Zebrafish), Tetrahymena thermophila SB210 and Xenopus laevis were performed by the OptimumGeneTM algorithm of GenScript, and then cloned into pCHD-CMV-MCS-EF1-CopGFP-T2A-puro (pCDH) via EcoRI/BamHI sites. To select input signals for genetic circuit construction, five repeated motifs from transcription factors were cloned into a firefly luciferase expressed plasmid. All motif sequences are listed (Supplementary Table 2). Furthermore, four genetic circuit plasmids, e.g. pCHD-RELA 5 motif (GGGAATTTCC)-miniP-Cas9-U6P-sgRNA-lacI, pCHD-CMV-lacI, pCHD-STAT1 5 motif (CATTTCCCGGAAACC)-miniP-lacO-rtTA and pCHD-TRE-NVD-BM, were also constructed based on the pCDH plasmid.
Cell transfection and infection
All cells were transfected with plasmids using Lipofectamine 3000 (Invitrogen) to facilitate DNA transient transfections. For cell infection, viral vectors were transfected into HEK293T cells, after which lentiviruses were collected and concentrated. A375 and A2058 cells were incubated in viral media and polybrene, and incubated in 1 μg/ml puromycin for two weeks.
Real-time quantitative PCR assay
Total RNA was isolated from cells using Trizol reagent (Invitrogen), and cDNA was generated using the reverse transcription kit (Genecopoeia). Real-time PCR was performed using SYBR PCR mix (TransGen), and GAPDH was used as a reference housekeeping gene. We calculated expression fold changes using the 2−ΔΔct method. All primers for qRT-PCR were designed using Snapgene and Primer 5 software (Supplementary Table 3).
MTT assay
Cells transfected with different plasmids were cultured in 96-well plates at 5×103 cells/well. After 48 h at 37°C in 5% CO2, 10 μl MTT solution was added to each well, and incubated for another 4 h. After this, the medium was removed and 110 μl/well DMSO added. Absorbance was measured at 570 nm on a plate reader (Thermo).
Real-time cell analysis assay
A375 and A2058 cell migration was analyzed by real-time cell analysis (RTCA). Both cell types were seeded into the upper chamber of an individual CIM-Plate-16 (5×103 cells/well) in 130 μL medium, without FBS. The upper chamber was placed on a lower CIM-Plate-16 chamber, which contained growth medium supplemented with 10% FBS (attractant). Impedance changes resulting from cell migration onto lower membranes were recorded every 5 min, and monitored over 36 h.
Sterol extraction and analysis
Sterol extraction and analysis were performed as recently described, with some modifications [41]. Approximately 1×107 cells/sample were collected and washed twice in phosphate buffer saline (PBS). Cells were then centrifuged, resuspended in 3 M HCl, and boiled for 3 min. This was performed three times. Cell fragments were collected by centrifugation, and pellets washed in distilled water. Cell precipitates were resuspended in a 2 M NaOH-methanol solution, and incubated at 60°C overnight. After cooling to room temperature, n-hexane was added to extract sterols, after which the n-hexane phase was removed in a vacuum desiccator. Eventually, desiccated sterol products were derived by N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) at 30°C for 4 h to generate samples for GC-MS analysis.
Sterols were separated on an Agilent 6890 gas chromatograph (GC) (USA), coupled to a Waters time-of-flight mass spectrometer (TOF-MS) (USA). The GC was equipped with a DB-5 fused-silica capillary column (30 m × 0.25 mm internal diameter, film thickness of 0.25 μm (J&W Scientific, CA, USA). Ions were generated by a 70 eV electron beam in EI mode, at an ionization current of 40 μA. Mass spectra were acquired in the 50–800 m/z range. The ion source temperature was 250°C, and the injection site temperature was 260°C. The temperature was initially 70°C for 2 min, then increased by 30°C/min to 250°C, and finally increased to 280°C at 10°C/min. 280°C was maintained for 15 min, and finally increased to 290°C at 5°C/min. The final temperature was maintained for 5 min. Standards (cholesterol and 7-DHC) were purchased from Sigma-Aldrich (USA).
Western blotting
Protein lysates from cells were lysed in RIPA buffer, with a protease inhibitor cocktail. For western blotting, proteins (20 μg/sample) were separated on 12% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes (Millipore). Membranes were blocked in 5% non-fat milk in TBST, and incubated with primary antibodies against Akt1 (dilution, 1:1000; Cat. No. 33224) (Signalway Antibody), pAkt1-ser473 (dilution, 1:1000; Cat. No. 13357) (Signalway Antibody), pAkt1-thr308/309 (dilution, 1:1000; Cat. No. 13311) (Signalway Antibody), LaminB1 (dilution, 1:1000; Cat. No. 40413) (Signalway Antibody), RELA (dilution, 1:1000; Cat. No. ab19870) (Abcam), Tubulin (dilution, 1:5000; Cat. No. Z0305) (Ray Antibody), and GAPDH (dilution, 1:5000; Cat. No. ab181602) (Abcam). After washing, membranes were incubated with anti-mouse (dilution, 1:5000; Cat. No. ab97040) (Abcam), or anti-rabbit (dilution, 1:5000; Cat. No. ab7090) (Abcam) peroxidase-conjugated secondary antibodies. Protein bands were revealed by ECL reagent on Exposure meter.
Luciferase assay
Cells were co-transfected with firefly luciferase and Renilla luciferase plasmids for 48 h. Growth medium was then removed, and monolayers washed gently and thoroughly in PBS, to avoid disruption. PBS was complexly removed. Then 1×PLB (Promega) was added to each well, and plates rocked at room temperature for 15 min. 100 μl LAR II (Promega) was added to luminometer tubes, including 20 μl cell lysates. Absorbance was measured at 560 nm. 100 μl Stop & Glo reagents (Promega) were then added to each tube, and absorbance measured at 560 nm. Luciferase results were determined by dividing absorbance at 560 nm by absorbance at 470 nm.
Bioinformatics analysis
R software was used to analyze bioinformatics data from GEO (https://www.ncbi.nlm.nih.gov/gds/), TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-enomics/tcga) and GTEX (http://genome.ucsc.edu/gtex.html) databases. Firstly, we downloaded transcriptome data from melanoma patient samples from the TCGA database, and normal skin sample data from the GTEX database, which are skin cutaneous melanoma (813 normal tissues, 471 tumor tissues). Then we downloaded one GEO dataset, GSE3189, i.e. transcriptome melanoma data comprising 7 normal and 45 melanoma tissues. Secondly, to compare gene expression between normal and tumor samples, we normalized RNA-Seq data from these public databases, and used the limma R package to analyze gene expression patterns, to screen significantly differentially expressed genes (log Fold Change > 1, adj-p value < 0.05). The clusterProfiler R package was used for Gene Set Enrichment Analyses (GSEA). In addition, statistical significance parameters (p value) for all data were calculated by T tests.
Author Contributions
BL designed and conceived the study. JL performed experiments, and processed data with assistance from LC and JZQ. YLH, ZJS and TTC assisted and guided animal experiments. BL and JL wrote the manuscript, with editing assistance from YJY, CL, WHX, YW and MDY.
Acknowledgments
We would like to thank Dr. Yanming Li and Dr. Qiong Yang from the Fang lab, Beijing Institute of Genomics, Chinese Academy of Sciences, for the kind gift of the A2058 cell line. We thank Futian Li from the Institute of Radiation Medicine, Chinese Academy of Medical Science for animal experiment assistance. We also acknowledge Dr. Dong Wang from the 309th hospital of China people's liberation army, for study advice.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81700153) and Natural Science Foundation of Tianjin city (Grant No. 18JCQNJC13300).
References
- 1. Schadendorf D, van Akkooi AC, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, Ugurel S. Melanoma. Lancet. 2018; 392:971–84. https://doi.org/10.1016/S0140-6736(18)31559-9 [PubMed]
- 2. Olazagasti Lourido JM, Ma JE, Lohse CM, Brewer JD. Increasing incidence of melanoma in the elderly: an epidemiological study in olmsted county, minnesota. Mayo Clin Proc. 2016; 91:1555–62. https://doi.org/10.1016/j.mayocp.2016.06.028 [PubMed]
- 3. Crocetti E, Mallone S, Robsahm TE, Gavin A, Agius D, Ardanaz E, Lopez MC, Innos K, Minicozzi P, Borgognoni L, Pierannunzio D, Eisemann N, and EUROCARE-5 Working Group. Survival of patients with skin melanoma in europe increases further: results of the EUROCARE-5 study. Eur J Cancer. 2015; 51:2179–90. https://doi.org/10.1016/j.ejca.2015.07.039 [PubMed]
- 4. Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol. 2014; 15:e371–81. https://doi.org/10.1016/S1470-2045(14)70072-5 [PubMed]
- 5. Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV, Di Giacomo AM, Svane IM, Lotem M, Bar-Sela G, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med. 2019; 25:941–46. https://doi.org/10.1038/s41591-019-0448-9 [PubMed]
- 6. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH
Jr , Kaempgen E, Martín-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 380:358–65. https://doi.org/10.1016/S0140-6736(12)60868-X [PubMed] - 7. Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, Chiarion Sileni V, Schachter J, Garbe C, Bondarenko I, Gogas H, Mandalá M, Haanen JB, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N Engl J Med. 2019; 381:626–36. https://doi.org/10.1056/NEJMoa1904059 [PubMed]
- 8. Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016; 17:1248–60. https://doi.org/10.1016/S1470-2045(16)30122-X [PubMed]
- 9. Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, Lewis KD, Tawbi HA, Hernandez G, Wongchenko MJ, Chang Y, Roberts L, Ballinger M, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med. 2019; 25:929–35. https://doi.org/10.1038/s41591-019-0474-7 [PubMed]
- 10. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013; 494:251–55. https://doi.org/10.1038/nature11814 [PubMed]
- 11. Helmbach H, Rossmann E, Kern MA, Schadendorf D. Drug-resistance in human melanoma. Int J Cancer. 2001; 93:617–22. https://doi.org/10.1002/ijc.1378 [PubMed]
- 12. Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer. 2017; 1868:1–6. https://doi.org/10.1016/j.bbcan.2016.12.004 [PubMed]
- 13. Simons K, Ikonen E. How cells handle cholesterol. Science. 2000; 290:1721–26. https://doi.org/10.1126/science.290.5497.1721 [PubMed]
- 14. Tall AR, Costet P, Wang N. Regulation and mechanisms of macrophage cholesterol efflux. J Clin Invest. 2002; 110:899–904. https://doi.org/10.1172/JCI16391 [PubMed]
- 15. Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002; 110:905–11. https://doi.org/10.1172/JCI16452 [PubMed]
- 16. Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013; 4:119. https://doi.org/10.3389/fphar.2013.00119 [PubMed]
- 17. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016; 76:2063–70. https://doi.org/10.1158/0008-5472.CAN-15-2613 [PubMed]
- 18. Jacobs EJ, Rodriguez C, Brady KA, Connell CJ, Thun MJ, Calle EE. Cholesterol-lowering drugs and colorectal cancer incidence in a large united states cohort. J Natl Cancer Inst. 2006; 98:69–72. https://doi.org/10.1093/jnci/djj006 [PubMed]
- 19. Wang C, Li P, Xuan J, Zhu C, Liu J, Shan L, Du Q, Ren Y, Ye J. Cholesterol enhances colorectal cancer progression via ROS elevation and MAPK signaling pathway activation. Cell Physiol Biochem. 2017; 42:729–42. https://doi.org/10.1159/000477890 [PubMed]
- 20. Borgquist S, Bjarnadottir O, Kimbung S, Ahern TP. Statins: a role in breast cancer therapy? J Intern Med. 2018; 284:346–57. https://doi.org/10.1111/joim.12806 [PubMed]
- 21. Stopsack KH, Gerke TA, Sinnott JA, Penney KL, Tyekucheva S, Sesso HD, Andersson SO, Andrén O, Cerhan JR, Giovannucci EL, Mucci LA, Rider JR. Cholesterol metabolism and prostate cancer lethality. Cancer Res. 2016; 76:4785–90. https://doi.org/10.1158/0008-5472.CAN-16-0903 [PubMed]
- 22. Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, Zhou Y, Zhang Y, Xu W, Yang Y, Wu B, Cheng G, Lai PB, et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci Transl Med. 2018; 10:eaap9840. https://doi.org/10.1126/scitranslmed.aap9840 [PubMed]
- 23. Silvente-Poirot S, Poirot M. Cancer. Cholesterol and cancer, in the balance. Science. 2014; 343:1445–46. https://doi.org/10.1126/science.1252787 [PubMed]
- 24. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006; 9:358–65. https://doi.org/10.1097/01.mco.0000232894.28674.30 [PubMed]
- 25. McDonnell DP, Park S, Goulet MT, Jasper J, Wardell SE, Chang CY, Norris JD, Guyton JR, Nelson ER. Obesity, cholesterol metabolism, and breast cancer pathogenesis. Cancer Res. 2014; 74:4976–82. https://doi.org/10.1158/0008-5472.CAN-14-1756 [PubMed]
- 26. Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA, Umetani M, Euhus DM, Xie Y, Shaul PW. 27-hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013; 5:637–45. https://doi.org/10.1016/j.celrep.2013.10.006 [PubMed]
- 27. Bauriaud-Mallet M, Vija-Racaru L, Brillouet S, Mallinger A, de Medina P, Rives A, Payre B, Poirot M, Courbon F, Silvente-Poirot S. The cholesterol-derived metabolite dendrogenin A functionally reprograms breast adenocarcinoma and undifferentiated thyroid cancer cells. J Steroid Biochem Mol Biol. 2019; 192:105390. https://doi.org/10.1016/j.jsbmb.2019.105390 [PubMed]
- 28. Lin CY, Huo C, Kuo LK, Hiipakka RA, Jones RB, Lin HP, Hung Y, Su LC, Tseng JC, Kuo YY, Wang YL, Fukui Y, Kao YH, et al. Cholestane-3β, 5α, 6β-triol suppresses proliferation, migration, and invasion of human prostate cancer cells. PLoS One. 2013; 8:e65734. https://doi.org/10.1371/journal.pone.0065734 [PubMed]
- 29. de Medina P, Paillasse MR, Segala G, Voisin M, Mhamdi L, Dalenc F, Lacroix-Triki M, Filleron T, Pont F, Saati TA, Morisseau C, Hammock BD, Silvente-Poirot S, Poirot M. Dendrogenin a arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat Commun. 2013; 4:1840. https://doi.org/10.1038/ncomms2835 [PubMed]
- 30. Gelzo M, Granato G, Albano F, Arcucci A, Dello Russo A, De Vendittis E, Ruocco MR, Corso G. Evaluation of cytotoxic effects of 7-dehydrocholesterol on melanoma cells. Free Radic Biol Med. 2014; 70:129–40. https://doi.org/10.1016/j.freeradbiomed.2014.02.013 [PubMed]
- 31. Yoshiyama-Yanagawa T, Enya S, Shimada-Niwa Y, Yaguchi S, Haramoto Y, Matsuya T, Shiomi K, Sasakura Y, Takahashi S, Asashima M, Kataoka H, Niwa R. The conserved rieske oxygenase DAF-36/neverland is a novel cholesterol-metabolizing enzyme. J Biol Chem. 2011; 286:25756–62. https://doi.org/10.1074/jbc.M111.244384 [PubMed]
- 32. Yoshiyama T, Namiki T, Mita K, Kataoka H, Niwa R. Neverland is an evolutionally conserved rieske-domain protein that is essential for ecdysone synthesis and insect growth. Development. 2006; 133:2565–74. https://doi.org/10.1242/dev.02428 [PubMed]
- 33. Najle SR, Nusblat AD, Nudel CB, Uttaro AD. The sterol-C7 desaturase from the ciliate tetrahymena thermophila is a rieske oxygenase, which is highly conserved in animals. Mol Biol Evol. 2013; 30:1630–43. https://doi.org/10.1093/molbev/mst076 [PubMed]
- 34. Zhang T, Bao J, Zhang M, Ge Y, Wei J, Li Y, Wang W, Li M, Jin Y. Chemo-photodynamic therapy by pulmonary delivery of gefitinib nanoparticles and 5-aminolevulinic acid for treatment of primary lung cancer of rats. Photodiagnosis Photodyn Ther. 2020; 101807. https://doi.org/10.1016/j.pdpdt.2020.101807 [PubMed]
- 35. Brophy JA, Voigt CA. Principles of genetic circuit design. Nat Methods. 2014; 11:508–20. https://doi.org/10.1038/nmeth.2926 [PubMed]
- 36. Lienert F, Lohmueller JJ, Garg A, Silver PA. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat Rev Mol Cell Biol. 2014; 15:95–107. https://doi.org/10.1038/nrm3738 [PubMed]
- 37. Teixeira AP, Fussenegger M. Synthetic biology-inspired therapies for metabolic diseases. Curr Opin Biotechnol. 2017; 47:59–66. https://doi.org/10.1016/j.copbio.2017.06.004 [PubMed]
- 38. De Donatis GM, Le Pape E, Pierron A, Cheli Y, Hofman V, Hofman P, Allegra M, Zahaf K, Bahadoran P, Rocchi S, Bertolotto C, Ballotti R, Passeron T. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene. 2016; 35:2813. https://doi.org/10.1038/onc.2015.468 [PubMed]
- 39. Cheng Q, Wu J, Zhang Y, Liu X, Xu N, Zuo F, Xu J. SOX4 promotes melanoma cell migration and invasion though the activation of the NF-κB signaling pathway. Int J Mol Med. 2017; 40:447–53. https://doi.org/10.3892/ijmm.2017.3030 [PubMed]
- 40. Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, Jin L, Pupo G, Cullinane C, McArthur GA, Tummino PJ, Rizos H, Hersey P. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. 2014; 27:1126–37. https://doi.org/10.1111/pcmr.12282 [PubMed]
- 41. Guo XJ, Xiao WH, Wang Y, Yao MD, Zeng BX, Liu H, Zhao GR, Yuan YJ. Metabolic engineering of Saccharomyces cerevisiae for 7-dehydrocholesterol overproduction. Biotechnol Biofuels. 2018; 11:192. https://doi.org/10.1186/s13068-018-1194-9 [PubMed]