



Introduction
The etiology of the human breast carcinoma (BC) is unknown, with the exception of hereditary tumors (HBC) caused by the hereditary transmission of mutated tumor suppressor genes, such as BRCA1 and BRCA2 [1], and of the rare radiation-induced tumors. Estrogens are well known to be relevant promotional factors for breast carcinoma, but their transforming role has never been definitively demonstrated [2]. Non-hereditary BCs, which represent approximately 90% of BC, are called sporadic (SBC), which means occasional, to indicate the current lack of specific causes.
Over the last almost 50 years the hypothesis of a viral origin of sporadic BC has received increasing attention, being defined as “highly likely” in a very recent review by Lawson and Glenn [3]. Such hypothesis is based on the high similarity of BC to the experimental model of mammary tumors induced in mice by the betaretrovirus Mouse Mammary Tumor Virus (MMTV) [4] transmitted from mother to newborn by lactation [5]. In fact, this model has been very useful to clarify several aspects of BC biology, such as the concept of cancer progression, the recognition of the so-called preinvasive lesions, and the promotional role of estrogens [6].
While the first investigations suggesting the existence of a human mammary tumor virus date back to the seventies [7], only in 1995 the advent of molecular biology allowed to detect MMTVenv-like sequences (MMTVels) in approximately 40% of infiltrating human breast carcinoma by Pogo and colleagues [8], who consequently introduced the term HMTV – human mammary tumor virus [9]. These data were later confirmed using laser capture microdissection and highly sensitive fluorescent PCR [10]. In contrast, it has been recently demonstrated that hereditary BC lacks MMTV sequences [1], as expected for the fact that HBCs, having a specific genetic etiology, do not need the action of a carcinogenetic viral agent. This result indirectly supports the reliability of the data in favor of the association between MMTV and sporadic breast carcinoma.
The hypothesis of a relationship between a human MMTV-like agent and BC raised the question of a potential zoonotic mouse-man transmission of MMTV. However, such a theory remained inconsistent with unconvincing sporadic reports of the possible transmission of MMTV from the mouse to humans directly or through animal vectors such as cat or dog [11–13]. Likewise, the possibility that human milk could be a mother-baby route of infection also appeared insufficiently supported [14, 15], as discussed later.
Subsequently, attention was focused on saliva – a common route of spreading of infectious diseases – and MMTVels were identified in human normal salivary glands and in saliva [16]. This finding, in the absence of sound data supporting mouse to man transmission of MMTV, suggested the existence of a human homologue of MMTV, possibly due to a MMTV mouse to man cross-species transmission that occurred in ancient times [17]. The demonstration of MMTVels in ancient individuals would strongly corroborate the hypothesis of a human MMTV-like betaretrovirus. To assess this possibility, we investigated the presence of MMTVels in the ancient dental calculus. The calculus, in fact, originates from saliva and is an excellent material for paleovirological studies.
Importantly, while no known human betaretroviruses are circulating in modern humans, the search for a human MMTV in human DNA can potentially be complicated by the presence of endogenous retroviruses (ERVs). ERVs are integrated sequences with retroviral origin that are present in our genome as well as in the genome of all vertebrates [18]. ERVs were acquired by a classical retroviral integration that occurred over millions of years ago within the genome of the host germ line cells, allowing their Mendelian inheritance through offspring. Human ERVs (HERVs), accounting for the 8% of the human genome, have accumulated mutations that generally compromised their coding potential, but several HERV groups have retained the capacity to be expressed and produce proteins. Some of the latter have been coopted during evolution for physiological functions, such as placenta development as well as the shaping of innate immunity pathways [19–21]. In addition, HERV expression is highly investigated for its possible contribution to human diseases, including autoimmunity and cancer [20, 22]. An updated general classification and characterization of approximately 3,200 HERV integrations in human genome reports the presence of 39 “canonical” HERV groups and 31 additional “non-canonical” groups of mosaic forms that arose from secondary integrations or recombination events [18]. Due to their similarity to exogenous retroviruses, HERV have also been broadly divided into three classes: Class I consists of gammaretrovirus-like and epsilon-like retroviruses, Class II of betaretrovirus-like retroviruses, and Class III of vaguely spumaretrovirus-like elements [18]. While no exogenous betaretroviruses are known to threaten humans nowadays, our genome harbors a betaretrovirus-like supergroup, namely HERV-K, as described in the early 80s when human sequences similar to the exogenous MMTV were found [23]. The HERV-K supergroup currently encompasses 10 distinct HERV groups, all sharing sequences similarities with MMTV. Consequently they were named HML (human MMTV-like) followed by a number from 1 to 10 [24].
The human genome harbors approximately 600 copies of HML sequences that share a significant level of intragroup identity [25]. Due to their abundant presence and to their remarkable similarity with MMTV, HML sequences can lead to misinterpretation when investigating the occurrence of exogenous betaretroviruses in humans, possibly leading to false positives.
This paper provides the first evidence that a MMTV-like betaretrovirus has been present in humans for thousands of years. The possibility of a contamination with murine material and endogenous betaretroviral sequences was carefully excluded.
Results
MMTVenv-like sequences are present in human remnants dated from the Copper Age to the 17th century
We had access to the skulls of 36 individuals dated from the Copper Age (details in Methods) to the 17th century, all from Italian regions, namely, Sardinia, Tuscany and Liguria (Figure 1 and Table 1). The ante Christum (AC) cases, all from Sardinia, were buried in typical collective tombs from the Neolithic to the Bronze Age, named Domus de Janas (house of fairies) and Tomb of Giants. From Sardinia, additional samples were collected from a Roman necropolis of the Imperial Roman Age (1st-3rd century), from a Medieval cemetery of the 14th-15th century, and from a late Renaissance plague mass burial (1582-1583). Other skulls were from the Italian peninsula: from an early Medieval cemetery (6th-8th century) in Liguria, a Medieval cemetery (12th-13th century) in Tuscany (Pisa), and from the Guinigi’s family tomb of the Modern Age (15th-17th century) again in Tuscany (Lucca). Dental calculus abundance (Figure 2) allowed the collection of specimens from all 36 cases and DNA extraction was successful in all cases. MMTVels were detected in six cases. Interestingly, the percentage of positivity (17%) was similar to that found in the saliva of healthy contemporary individuals [16].
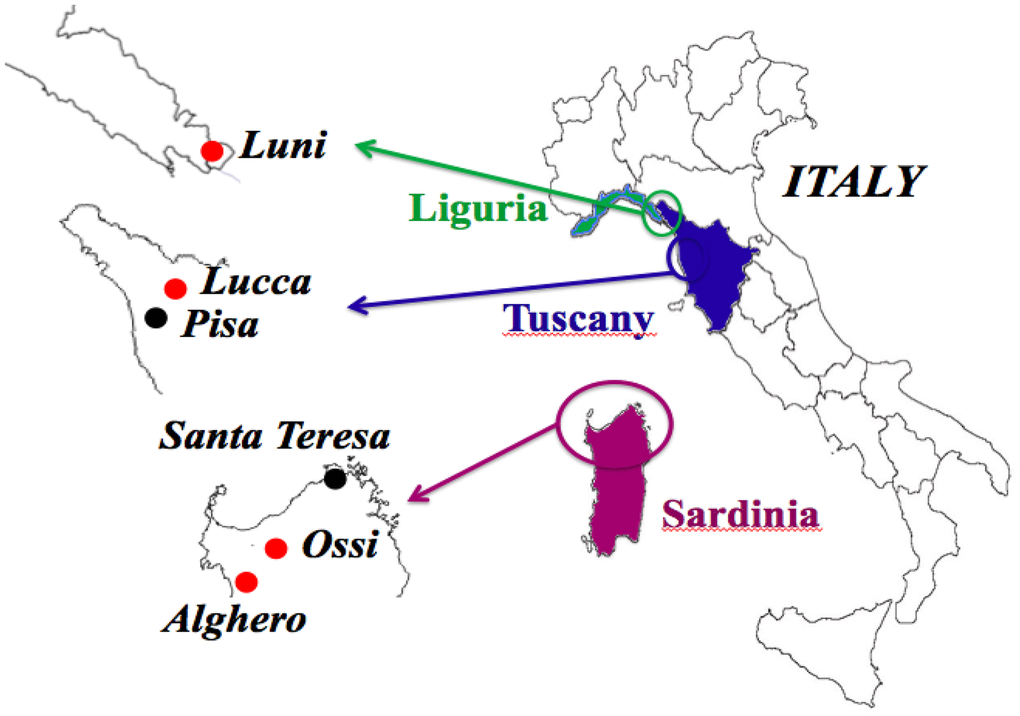
Figure 1. Geographic areas of origin of the 36 skulls objects of the study. Red dots indicate the four sites where the six cases positive for MMTVenv-like sequences were identified: two in Ossi, Sardinia; two in Alghero, Sardinia; one in Luni, Liguria; one in Lucca, Tuscany. In four cases, the sequencing of both ENV1 and ENV 2 amplicons was accomplished; one of the ENV1 amplicons showed a C > T polymorphism, that did not cause an amino acid substitution: Ossi (ENV2), Luni (ENV1-C), Alghero (ENV1-C, ENV1-T). The two cases that dated back to the Copper Age came from Ossi, Sardinia.
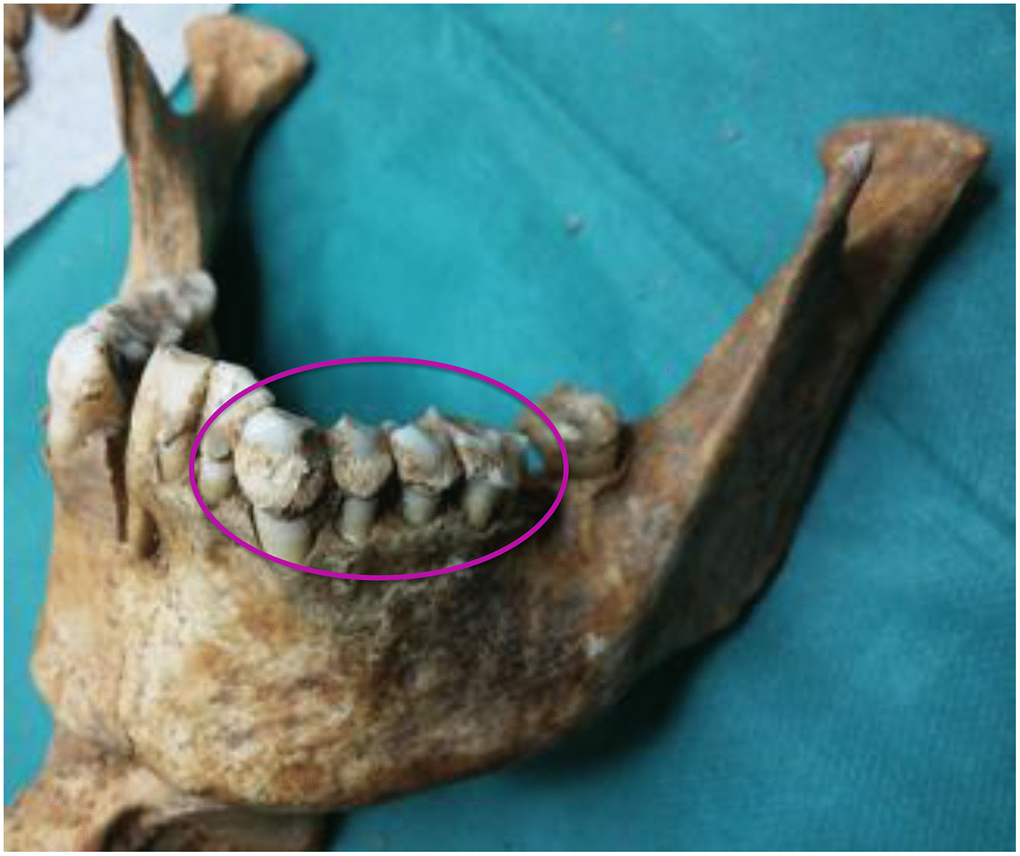
Figure 2. A mandible of one of the skulls examined. The circle indicates the abundance of calculus.
Table 1. Characteristics of the individuals and molecular results.
| Site | Type of burial | Date | Cases and sex | Age | MMTVels positive | ENV 1 | ENV 2 | Sequencing | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S’Adde’e Asile, Ossi, Sassari, Sardinia | Domus de Janas | 2712 ± 59 a.C. | 3 2♂, 1♀ | 16 - 22 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noeddale, Ossi, Sassari, Sardinia | Domus de Janas | 2560 ± 51 a.C. | 5 4♂, 1♀ | 20 - 29 | 2 ♂ | 2 ♂ | 1 ♂ | ENV2 ♂ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S’Isterridolzu, Ossi, Sassari, Sardinia | Domus de Janas | 2070 ± 35 a.C. | 4 1♂, 1♀, 2? | 20 - 28 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| La Testa, Santa Teresa, Sassari, Sardinia | Tomb of Giants | 1200 ± 132 a.C. | 3 3? | 25 - 35 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Monte Carru, Alghero, Sardinia | Roman necropolis | 1st-3rd century | 5 3♂, 2♀ | 24 - 34 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Amphitheatre, Luni, La Spezia, Liguria | early medieval cemetery | 6th-8th century | 1 1♂ | 20 - 30 | 1 ♂ | 1 ♂ | 1 ♂ | ENV1-C (♂) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sant’Alessandro, Vecchiano, Pisa, Tuscany | medieval cemetery | 12th-13th century | 5 1♂, 4♀ | 32 - 41 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| San Michele, Alghero, Sardinia | late medieval cemetery | 14th-15th century | 2 1♂, 1♀ | 32 - 43 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| San Michele, Alghero, Sardinia | plague mass burial | 1582-1583 | 6 3♂, 2♀, 1? | 35 - 44 | 2 ♂♀ | 2 ♂♀ | ENV1-C ♂ ENV1-T ♀ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Guinigi Chapel, Lucca, Tuscany | family tomb | 15th-17th century | 2 1♂, 1♀? | 20 - >50 | 1 ♀? | 1 ♀? | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| total | 36 17♂, 12♀, 7? | 6 4♂/1♀/1♀? | 6 4♂/1♀/1♀? | 2 ♂ | 4 3♂/1♀ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dental calculus was obtained from the skulls of 36 individuals dated from the Copper Age to the 17th century, all from Italian regions, namely Sardinia, Tuscany and Liguria. The a.C. cases were found in Domus de Janas and the Tomb of Giants, typical collective tombs from the Neolithic to the Bronze Age. Other Sardinian samples were from a Roman necropolis of the Imperial Roman Age, from a Medieval cemetery, and from a late Renaissance plague mass burial. Other skulls were from an early Medieval cemetery in Liguria, a Medieval cemetery in Tuscany and from the Guinigi’s family tomb of the Modern Age (15th-17th century) in Tuscany. Six cases were positive for MMTVels. ENV1 was detected in all of them, two of which were also positive for ENV2. In particular, two cases were from Ossi, Sardinia, two from Alghero, Sardinia, one from Luni, Liguria, and one from Lucca, Tuscany. Four of the six positive cases were sequenced, three for ENV1 and one for ENV2; one of the ENV1 cases showed a C > T polymorphism, that did not cause an amino acid substitution. One of these six cases was found in Ossi (ENV2), one in Luni (ENV1-C), and two in Alghero (ENV1-C, ENV1-T). Two cases were dated back to the Copper Age, both from Ossi, Sardinia. Four of the individuals who were positive for MMTVels were male, one female, and one probably female. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Two different ENV regions were amplified, named herein ENV1 and ENV2 (Supplementary Figure 1). Six of the 36 cases examined were positive for ENV1, and two of them were also positive for ENV2 (Table 1). Two of the six positive cases dated back to the Sardinian Copper Age, approximately 4,500 years ago. The other four cases were from the 6th-17th century. In four cases, both ENV1 and ENV2 were sequenced. One of the ENV1 amplicons had a C > T polymorphism (Figure 3), that has already been described and deposited in GenBank (accession number D16249.1), and does not cause an amino acid substitution. The ENV1 sequence with the T polymorphism was identified herein as ENV1-T, whereas the ENV1 wild-type sequence was identified as ENV1-C.
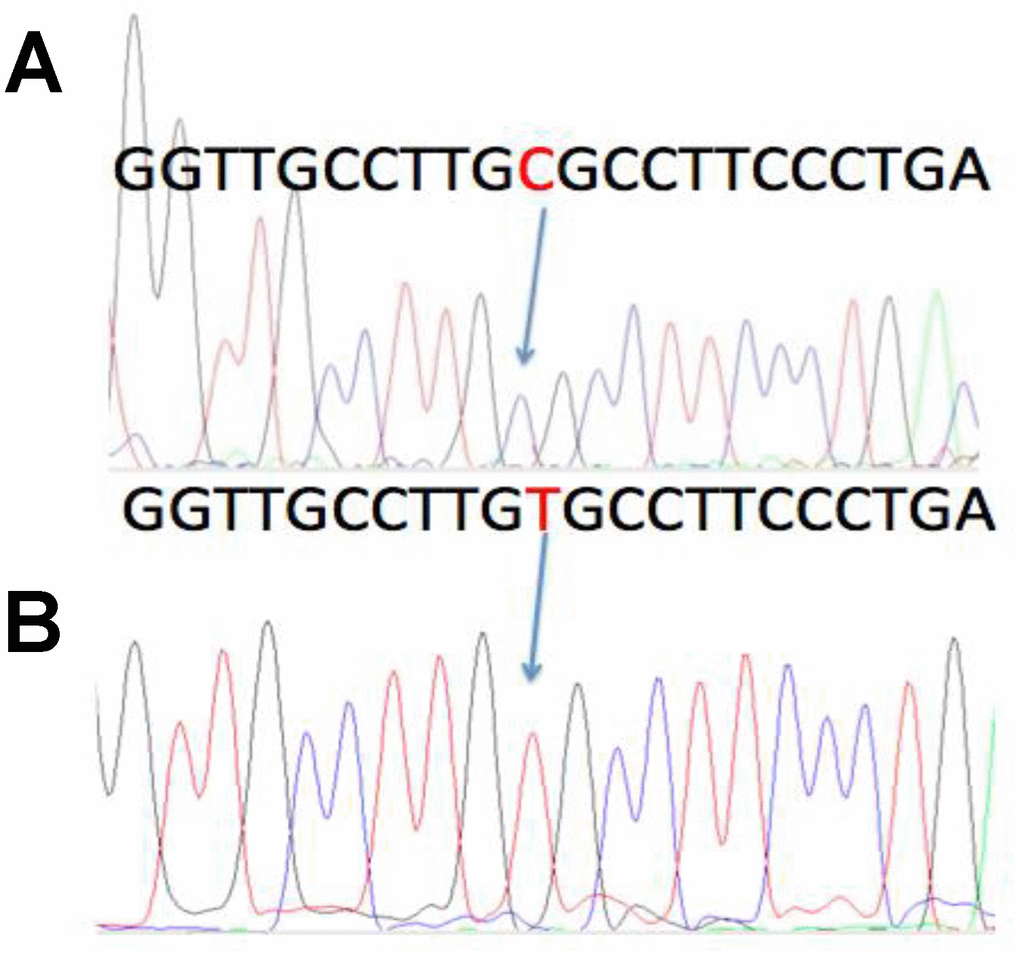
Figure 3. A previously described C > T polymorphism in the MMTV DNA sequence. In four of the MMTV positive cases, sequencing of both the ENV1 and ENV2 amplicons was accomplished. One of the ENV1 cases showed a previously described C > T polymorphism, that did not cause an amino acid substitution. The wild type sequence was named ENV1-C, whereas the polymorphic sequence was named ENV1-T. A: ENV1-C positive case from Luni, Liguria. B: The ENV1-T polymorphic case from Alghero, Sardinia.
The MMTVenv-like sequences in ancient individuals are not an artifact of contamination
Mice are regular residents of tombs, where they often choose skulls as a nest. Hence, the presence of murine mitochondrial and intracisternal A particle DNA as well as the murine and human housekeeping gene GAPDH was assessed [26]. The results (Figure 4) showed that all samples positive for MMTVels contained neither murine mitochondrial DNA (Figure 4B) nor intracisternal A particle DNA (Figure 4C). Furthermore, the murine housekeeping gene GAPDH was not detected (Figure 4D), whereas human GAPDH was amplified (Figure 4E). To determine whether any DNA was present in our samples, they were PCR-amplified with a universal bacterial primer pair targeting the 16S rRNA (Figure 4A).
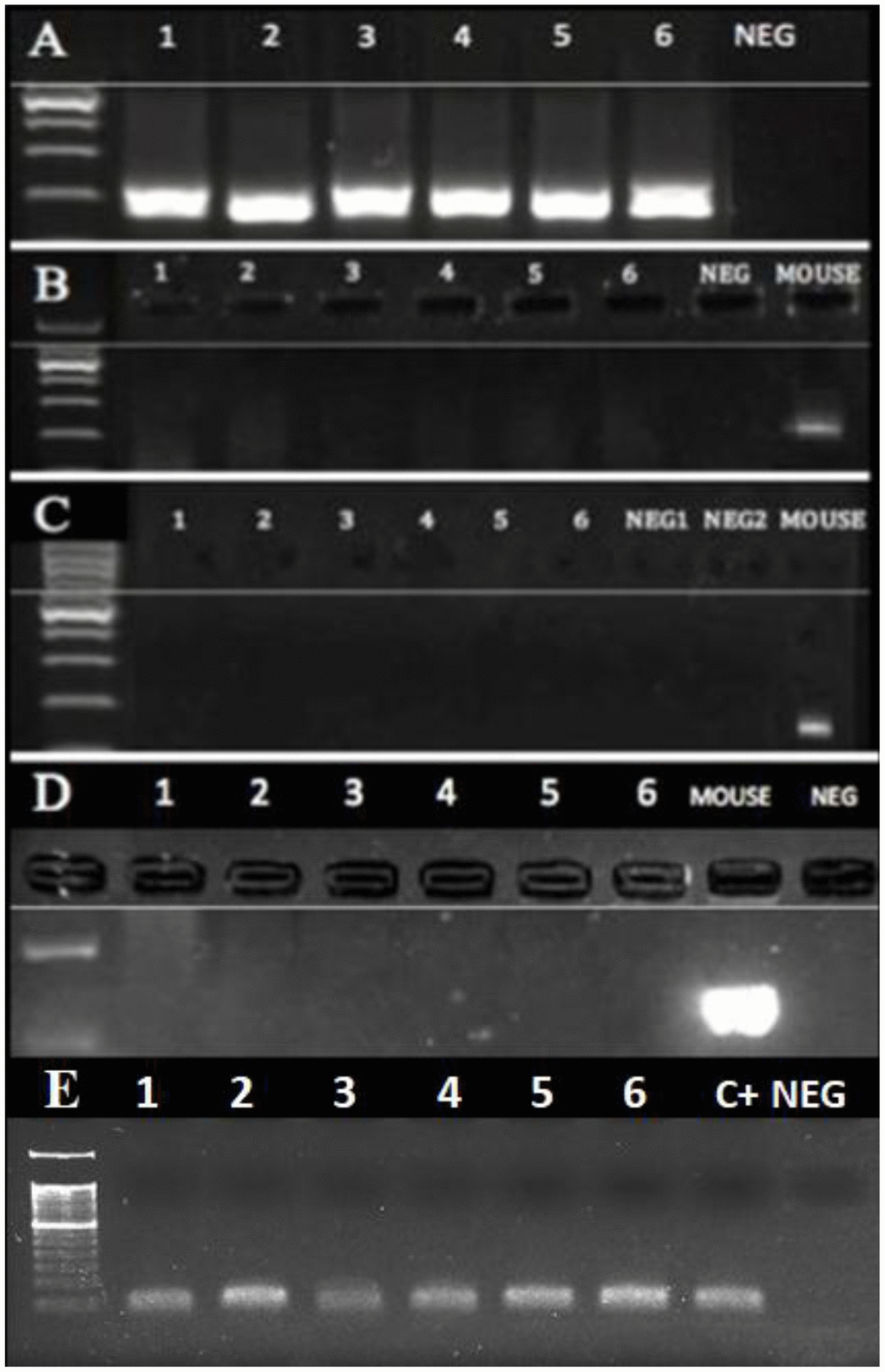
Figure 4. PCR of DNA extracted from the dental calculus. No mouse DNA was present in the six human cases (1-6) that were positive for MMTVels. (A) A 16S rRNA V3 region; all human cases were positive, whereas NEG was the negative control. (B) Intracisternal A Particle (IAP) LTRs; all the human cases were negative, as well as the negative control, whereas the mouse DNA was positive. (C) Murine mitochondrial DNA (mtDNA); all human cases were negative, whereas the mouse DNA was positive. (D) Murine GADPH; all the human cases were negative, whereas the mouse DNA was positive. (E) Human GADPH; all the human cases were positive; c+: a positive human control; neg: a negative control.
Amplified MMTVels regions have high identity with previously described HMTV and MMTV envs
To investigate the origin of DNA fragments obtained from dental calculus, the MMTVenv-like amplicons were aligned with respect to the HMTV and MMTV sequences retrieved from public repositories (Supplementary Figure 2; see the Methods for details and accession numbers). Both ENV1-C and ENV1-T (224 bp) mapped to HMTV sequence at nucleotides 1-224 and to MMTV sequence at nucleotides 6056-6279, whereas ENV2 (225 bp) mapped to HMTV sequence at nucleotides 984-1208 and to MMTV sequence at nucleotides 7039-7236 (Figure 5). Alignment analysis showed that both ENV1-C and ENV2 shared 100% similarity with respect to the corresponding portions of HMTV env, whereas ENV1-T differed only at the polymorphic site. Conversely, all three amplicons showed a few single nucleotide differences compared to the corresponding portions of MMTV. In fact, ENV1-C and ENV1-T had 2 and 3 nucleotides differences respectively, whereas ENV2 harbored 11 different nucleotides (Figure 5 and Table 2). To assess the presence of functional domains, the obtained env amplicons were analyzed with the GenBank BLASTx tool to detect conserved motifs [26]. The results showed that the ENV2 amplicon was localized within HMTV Heptad Repeat 2 (HR2) region that, together with Heptad Repeat 1 (HR1), forms the HIV-1-like HR1-HR2 ectodomain, constituting the Env transmembrane portion fusion core (Figure 5).

Figure 5. Graphical representation of MMTVenv-like amplicons compared to exogenous and endogenous betaretroviruses. The ENV1-C/ENV1-T (panel A) and ENV2 (panel B) amplicons were mapped to the HMTV env reference sequence and compared with the latter and to other reference sequences representative for exogenous and endogenous betaretrovirus env gene in a multiple nucleotide alignment. For each sequence, grey bases represent residues that were identical to the reference HMTV, while colored residues indicate single nucleotide changes to A (red), C (blue), G (yellow), and T (green). All the three amplicons presented very few nucleotide substitutions with respect to MMTV (1 discordant nucleotide for ENV1-T and 0 for both ENV1-C and ENV2) and HMTV (2 discordant nucleotides for ENV1-T, 3 for ENV1-C and 11 for ENV2). The presence of predicted functional domains in HMTV portions corresponding or near the env amplicons is also annotated: Heptad Repeats 1 and 2 (HR1 and HR2, respectively) and immunosuppressive domain (IS). N.B. in the ENV2 alignment, JSRV is not present due to the absence of any shared nucleotide sequence.
Table 2. Pairwise nucleotide identity of the MMTV env-like amplicons with respect to exogenous and endogenous betaretroviruses.
| ENV1-C | ENV1-T | ENV2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exogenous betaretroviruses | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HMTV AF243039 | 100·0% (0) | 99·6% (1) | 100·0% (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMTV NC 001503.1 | 99·1% (2) | 98·7% (3) | 95·1% (11) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMTV C3H AF228552.1 | 52·5% (125) | 52·5% (125) | 94·3% (13) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| JSRV (sheep) | 27·9% (150) | 27·9% (150) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MPMV (monkey) | 22·6% (181) | 22·6% (181) | 20·1% (191) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Endogenous betaretroviruses | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK14i HML1 | 29·2% (126) | 29·2% (126) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML1* | 28·9% (32) | 28·9% (32) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVKi HML2 | 28·9% (170) | 28·9% (170) | 33·8% (151) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML2* | 26·2% (200) | 26·2% (200) | 35·5% (147) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK9i HML3 | 33·6% (156) | 33·6% (156) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML3* | 33·9% (156) | 33·9% (156) | 32·5% (154) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK13i HML4 | 30·0% (161) | 30·0% (161) | 30·7% (158) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML4* | 36·1% (147) | 36·1% (147) | 34·2% (150) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK22i HML5 | 33·7% (156) | 33·7% (156) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML5* | 25·7% (168) | 25·7% (168) | 25·7% (171) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK3i HML6 | 27·5% (169) | 27·5% (169) | 30·0% (161) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML6* | 28·8% (166) | 28·8% (166) | 31·7% (157) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK11D HML7 | 31·3% (158) | 31·3% (158) | 35·5% (147) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML7* | 30·9% (159) | 30·9% (159) | 35·1% (148) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK11i HML8 | 30·0% (161) | 30·0% (161) | 35·5% (147) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML8* | 29·6% (162) | 29·6% (162) | 35·4% (153) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK14Ci HML9 | 31·7% (157) | 31·7% (157) | 32·5% (154) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML9* | 32·2% (156) | 32·2% (156) | 32·9% (153) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVKC4 HML10 | 21·4% (167) | 21·4% (167) | 32·9% (153) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HERVK HML10* | 27·4% (167) | 27·4% (167) | 33·3% (152) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The pairwise comparison between the identified MMTVels and other exogenous and endogenous betaretroviruses showed that all the amplicons shared a very high identity with MMTV and its putative human homologous HMTV, being instead highly divergent with respect to the other exogenous and endogenous betaretroviruses. Identity values are expressed as % of identical nucleotides. The number of divergent nucleotides is also indicated between brackets. Consensus sequences marked with * are from [18]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Amplified MMTVels do not derive from HERV-K HML amplification
Given the high degree of similarity between MMTV and HERV-K HML integrations, we examined whether the MMTVels fragments could have originated from the amplification of human endogenous sequences. The three MMTVenv-like amplified fragments were aligned with the consensus sequences of HERV-K HMLs 1 to 10, based on the Repbase Update repository [27] and the database published by Vargiu et al. [18] as well as with MMTV reference sequence, the MMTV strains C3H, JSRV, and MPMV, and the proposed HMTV reference sequence (see the Material and Methods for further details and accession numbers). Comparison of the ENV1-C and ENV1-T amplicons was possible with all the considered betaretroviruses, while the ENV2 amplicons showed no sequence correspondence in the HERV-K HML1, HML3, and HML5 Repbase consensuses or the JSRV reference sequence (Figure 6).
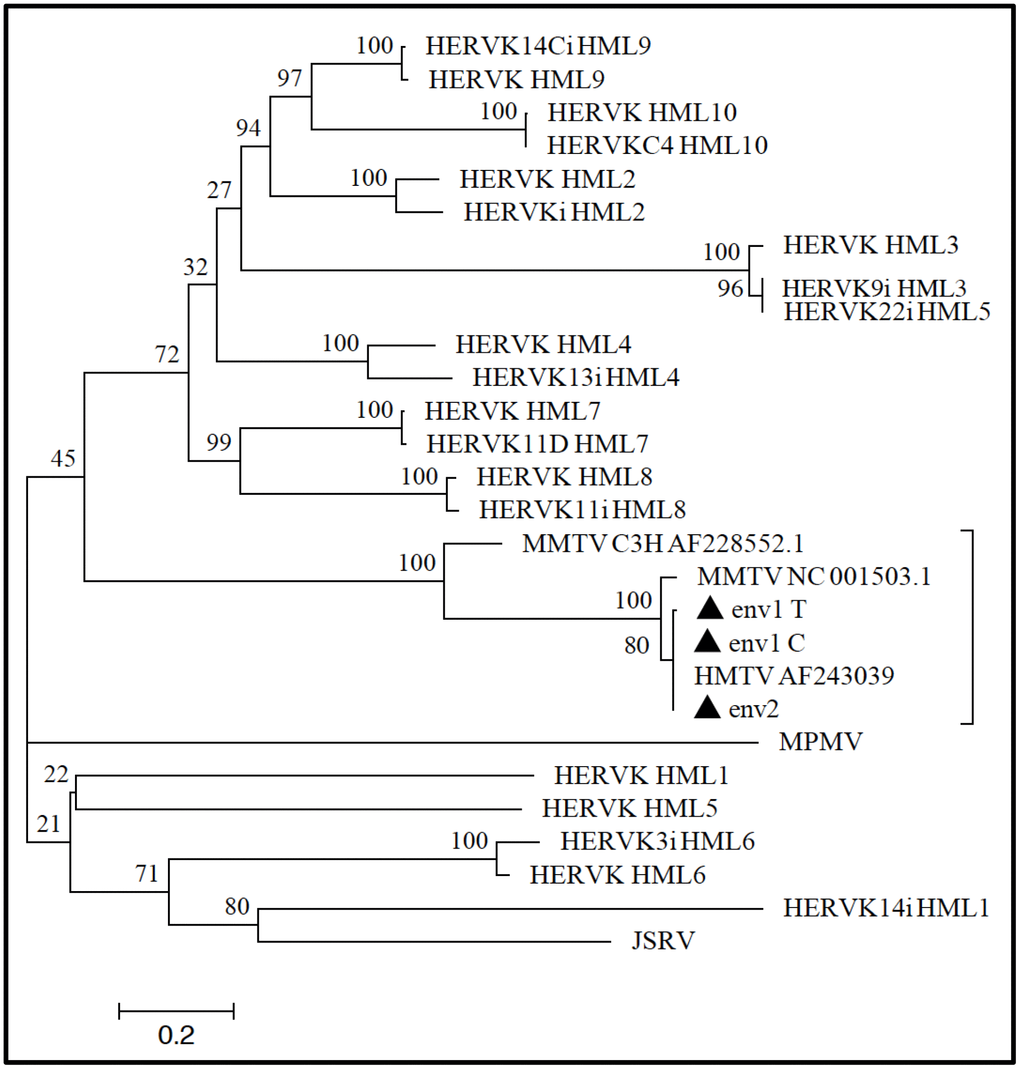
Figure 6. Phylogenetic analysis of MMTV env-like amplicons. ENV1-C, ENV1-T, and ENV2 amplicons (filled triangles) were analyzed to infer their phylogeny with respect to the other exogenous and endogenous betaretrovirus nucleotide sequences (see Materials and Methods for further details). The close relationship between the tree MMTVels amplicons and both MMTV and HMTV was confirmed at the phylogenetic level, given that all the sequences grouped together in the same clade that was statistically supported by the maximum bootstrap value. The amplicons were instead not related to any of the HERV-K HML groups’ sequences, which were clearly clustered in a different phylogenetic clade. Evolutionary relationships were inferred by using the maximum likelihood method and the Kimura-2-parameter model, and the resulting phylogeny was tested using the Bootstrap method with 100 replicates. The length of branches indicates the number of substitutions per site.
The results showed that all three MMTVenv-like amplified fragments shared overall very low identity (26% to 36%) with respect to all the HERV-K HML consensus sequences (Table 2). In contrast, the analysis demonstrated that all the amplicons shared remarkable sequence similarity with the HMTV and MMTV consensus sequences (Table 2). Particularly, both ENV1-C and ENV1-T showed high nucleotide identity with HMTV and MMTV (100% and 99.6%, respectively) and lower identity with the MMTV C3H strain (52.5%), while the ENV2 amplicon was highly identical to HMTV, MMTV, and the MMTV CH3 strain, with percentages of shared nucleotides of 100%, 95% and 94%, respectively (Table 2). In addition, all three MMTVenv-like amplicons showed high divergence compared to JSRV and MPMV exogenous betaretroviruses, with only 20% to 28% identity (the comparison between JSRV and the ENV2 amplicon was not possible due to the complete absence of a shared nucleotide sequence between the two sequences) (Table 2 and Figure 6). Overall, the above results exclude the possibility that the MMTVenv-like amplicons obtained from ancient dental calculus may have derived from betaretroviral HERV amplifications.
Phylogenetic analysis of the MMTVenv-like amplified regions
The alignment built to analyze the ENV amplicons nucleotide identity with other betaretroviruses was also used to infer their phylogeny with respect to the same exogenous and endogenous elements (Figure 6). Maximum likelihood analysis showed that all three MMTVenv-like amplicons grouped together with HMTV and MMTV, as supported by the highest bootstrap value (=100). This group was also significantly related to the MMTV C3H strain (bootstrap support=100), but it was clearly phylogenetically distinct from all the other exogenous and endogenous betaretroviruses (Figure 6), as suggested by the low nucleotide identity.
Discussion
Betaretroviruses are well-known pathogenic agents in animal models, such as Mason-Pfizer Monkey Virus (MPMV), Mouse Mammary Tumor Virus (MMTV), and Jaasiekte Sheep Retrovirus (JSRV), which cause immunodeficiency in macaque [28], mammary tumors in mouse [6] and pulmonary adenocarcinoma in sheep, respectively [29]. Contrarily, no human exogenous betaretrovirus is recognized as a causative agent in human diseases, given that, actually, no human exogenous betaretrovirus is known.
The strong similarity between human breast cancer and murine mammary tumors raised the question of the potential role of a betaretrovirus in breast carcinogenesis. In the 90s, molecular techniques allowed the detection of MMTVels in a high percentage of human infiltrating breast carcinomas [8, 9]. The authors suggested that the presence of such sequences was indicative of a human MMTV-like virus, named HMTV. Accordingly, a very recent review details all the data in favor of the viral etiology of human breast carcinoma [3]. Here we limit our discussion to the following: a) MMTV env sequences are absent in the human genome, whereas they are present in breast tumors and in normal breast tissues [30]; b) MMTVels have been identified in breast tissues prior to the development of MMTVels-positive breast cancer [31]; c) MMTVels have been detected in a high percentage of pre-invasive BC lesions, such as atypical epithelial hyperplasia and ductal carcinoma in situ (DCIS) [32]; d) the percentage of positivity of DCIS (80%) is double than that of the infiltrative tumors, which is confirmed to be between 30% and 40% [32]; and e) chromogenic in situ hybridization experiments have demonstrated the presence of viral hybridization signal in tumor nuclei, with a 50% reduction in infiltrating tumors compared to DCIS [32]. The strong reduction of positive cases moving from in situ to infiltrating tumors is of particular interest because it could indicate a possible relevance for the virus in cell transformation only and not for cancer progression. This phenomenon could be a consequence of DNA loss owing to the high level of chromosomal rearrangement characterizing breast tumors. Two findings are of special relevance and deserve a comment: the almost complete absence of MMTV sequences in hereditary breast carcinomas [1] and their presence in human salivary glands and saliva [16].
The difference in the results between sporadic tumors (with a high percentage of positive cases, 30.3%) and hereditary tumors (with a very high percentage of negative cases, almost 96%) is too distinct (p < 0.001) for it to be a mere coincidence or the consequence of a contamination with murine material, and does not allow to abandon the hypothesis of a viral etiology for human breast carcinoma. Moreover, the p14 protein, the signal peptide of the MMTV envelope precursor [33], is expressed only in cases that are positive for MMTVels [1].
The analysis of saliva was suggested by the fact that there are biological reasons that make milk improbable as a vehicle in MMTV-like virus transmission in humans [16] and that in humans a long duration of lactation is considered protective against breast cancer or ineffective [34]. The presence of MMTVels in saliva, suggesting the existence of a human MMTV-like betaretrovirus, inspired the present study.
We investigated the presence of MMTVels in ancient dental calculus, an important source for analyses of the paleomicrobiome and paleovirome. Saliva, in fact, provides many constituents of dental plaque, a bacterial biofilm covering the surface of teeth. Over time, plaque undergoes a process of calcification, giving origin to the dental calculus (tartar) through gradual calcium phosphate mineralization, in which all bacteria and viruses are entombed, providing excellent preservation of nucleic acids [35]. Dental calculus is commonly found in humans, both modern and ancient, including Neanderthals [36]. Paleomicrobiology has benefitted from the advances in high-throughput biomolecular sequencing, which can be easily applied to the analysis of calculus, which is considered an abundant and long-term reservoir of the ancient oral microbiome [35]. It must be stressed that the calculus is not easily colonized by environmental bacteria, does not have points of entry for exogenous bacteria, and lacks nutrient sources that are able to attract and support bacteria growth [37].
The study of dental calculus from 36 ancient skulls revealed MMTVels in 6 of them, of which two were from the Copper Age, i.e. 4,500 years ago, and 4 were from the 15th-17th century. The three identified sequences corresponded to the MMTV env gene and two of them differed only for an already described single nucleotide C>T polymorphism, being identified herein as ENV1-C, ENV1-T and ENV-2. We focused on the env gene because it is able to transform human epithelial mammary cells in culture and, for this reason, it has been object of all the previous studies concerning MMTV in human material [38].
The existence of a high number of Human MMTV-like (HML) sequences in the human genome over millions of years made it crucial to exclude the possibility that our env sequences derived from the amplification of HERVs. In fact, due to their abundance and remarkable similarity with MMTV, HML sequences can lead to misinterpretation when investigating the occurrence of exogenous betaretroviruses in humans, possibly leading to false positives.
The careful bioinformatics analysis conducted herein demonstrated that the MMTVels identified in the ancient calculus did not have a HERV origin. In fact, the amplified ENV sequences were aligned and compared with the corresponding sequences of the exogenous animal betaretroviruses and the HERV-K HML 1 to 10 group consensus sequences, showing very high identity to HMTV and MMTV and, in contrast, very low identity to the other animal exogenous betaretroviruses and to all HERV-K HMLs. In addition, the phylogenetic analysis demonstrated that the amplified env regions were significantly related to HMTV, MMTV and MMTV C3H, whereas they were quite distinct from all the other exogenous and endogenous betaretroviruses.
Overall, our study adds a piece to a puzzling scenario, which includes a number of scientists that are very skeptical about the hypothesis of a viral etiology of breast cancer and believe that all the positive results obtained over many decades are only a consequence of contamination with murine material or are due to the presence of endogenous betaretroviral sequences. This motif, even with dubious and inconclusive positions, has become recurrent, reducing the consideration of numerous biological and molecular data that tentatively link MMTV to human breast cancer, as summarized above and elsewhere [3]. However, the almost complete absence of MMTVels in hereditary breast carcinomas [1], the careful exclusion of possible contamination by murine material and bacteria [1, 26, 37], and the demonstration that the sequences identified in the present study do not have endogenous origin, do not allow to simply refuse such hypothesis, calling for further studies to finally demonstrate the possible existence of a modern human betaretrovirus.
Conclusions
This paper reveals that betaretroviral sequences sharing high identity with MMTV have been present in human species for at least 4,500 years.
This observation, together with all previous data concerning MMTV in humans, suggests the existence of a human exogenous betaretrovirus possibly derived from a cross-species transmission that occurred in prehistoric times, with consequent inter-human spread.
It is worth to note that mice became commensals of humans approximately 10,000 years ago, at the beginning of civilization, with the rise of agriculture in the area named the fertile crescent [39], which includes Western Asia, the Nile Valley, and the Nile Delta, guaranteeing mice an abundance of food, and giving origin to a period of strict cohabitation between the two species. In fact, humans also began to cohabit with other species, including those that they learned to domesticate. This situation played a relevant role in cross-species transmission – or species jump – with the evolution of animal microorganisms into human pathogens. An important example of cross-species transmission is represented, among others, by the human immunodeficiency virus (HIV), which has passed from chimpanzees to humans [40].
The data described herein strongly support the further investigation of the role of a MMTV-like element in human breast cancer. In this case, prophylactic strategies could be established based on the development of a specific vaccine or an immunotherapeutic approach. Interestingly, the immune-mediated targeting of MMTV-p14 protein has been recently proposed [41].
Materials and Methods
Study design
Dental calculus was collected from 36 skulls from the Copper Age to 17th century. Total DNA was extracted from calculus and whole genome amplification was performed. MMTVels were detected by fluorescence-nested PCR. Attention was focused on the env gene due to its ability to transform human mammary epithelial cells in culture [38]. Contamination by mouse DNA was excluded. The identified sequences were compared to endogenous betaretroviruses to assess their origin.
Characterization of the individuals
The dating of the a.C. individuals was performed by 14C and on the basis of archaeological and historical information for the A.D. cases. The two oldest cases (2560 ± 51 BC) belonged to the Copper Age (Chalcolithic, Eneolithic), which includes the period from approximately 3700/3400 BC to 2300 BC [42, 43]. The Copper Age was an archeological era characterized by the use of native copper for the production of metal tools. The more recent Bronze Age begins when humans discovered that adding tin to copper gave origin to the harder and stronger bronze.
Sex was determined according to Ferembach et al., whereas age at death was assessed by the sternal rib end modification and by dental wear [44, 45].
Collection of calculus
Operators wore a mask and sterilized gloves and used sterilized scalpels. New gloves and scalpels were used for each skull. Calculus was collected in sterilized tubes.
DNA extraction
Approximately 50 mg of dental calculus was crushed in a 2 ml tube and digested overnight at 56°C in 1 ml 0.45M EDTA with 10% proteinase K (Promega, Madison, WI, USA), followed by a 24 hours digestion at room temperature on an agitator. The entire volume was loaded in an automated system Maxwell 16 system (Promega, Madison, WI, USA) using the Maxwell® 16 LEV DNA Purification Kit.
Whole-genome amplification
Samples were subjected to whole-genome amplification using the Genomeplex Single Cell Whole Genome Amplification kit (Sigma-Aldrich, Saint Louis, MO, USA) starting from 7 μl of DNA and following the supplied protocol. The amplified DNA was measured with a Qubit 2.0 Fluorometer (Invitrogen, Life Technologies, Grand Island, NY) using the Qubit DNA assay kit to obtain a concentration ranging from 30 to 90 ng/μl per sample.
PCR housekeeping gene and murine contamination analysis
To determine whether any DNA was present in our samples, DNA extracts were PCR-amplified with a universal bacterial primer pair targeting the 16S rRNA V3 region: forward, 5’-ACTCCTACGGGAGGCAGCAGT-3’, reverse, 5’-GTATTACCGCGGCTGCTGGCAC-3’ [46].
The presence of contaminating mouse DNA was excluded by performing murine mitochondrial DNA and IAP LTRs PCR [25] and murine GAPDH PCR.
Detection of MMTVels: PCR and sequencing analysis
Fluorescence-nested PCR was used to detect the presence of DNA MMTVels. Two sets of primers were designed to amplify two different regions, ENV-1 and ENV-2 (Supplementary Figure 1), based on the sequence available in GenBank with accession number AF243039. ENV-1 was amplified by semi-nested 224-155 bp PCR using the following primers: ENV1Forward 5’-ccagatcgcctttaagaagga-3’, ENV1Reverse 3’-actccccctgtcaataaagagg-5’, and ENV1Reverse 3’-ccttccctcgcctagtgtag-5’. For amplification of ENV-2 (225 bp), we used a nested-PCR with the following primers: ENV2Forward 5’-ctgagagctgggaaagaacc-3’ and ENV2 Reverse 3’-ctgagagctgggaaagaacc-5’. Both ENV-1 and ENV-2 PCRs were carried out using a previously described protocol [32].
The products of PCR amplification were sequenced, after clean-up with the QIAquick PCR Purification Kit (Qiagen, Venlo, Netherlands), using Big Dye Terminator mix (Applied BioSystems, Warrington, UK). Sequencing reactions were run on an ABI3130 XL (Applied BioSystems, Warrington, UK).
Sequences considered in the study
To characterize their virological origin, the MMTVels amplicons were compared to the publicly available exogenous and endogenous betaretroviral reference/consensus sequences:
- Exogenous animal betaretroviruses: MMTV (NC001503), MMTV C3H strain (AF228552.1), JSRV (NC001494.1), MPMV (NC001550.1).
- HMTV (AF243039, env-LTR portion), the original MMTV identified in human breast tumors by Pogo et al. [8].
- Endogenous human betaretroviruses: HERV-K HML 1 to 10 based on the RepBase Update database and on the recently provided dataset of ~3200 HERV insertions [18, 27].
Bioinformatics analysis
The above nucleotide sequences and the corresponding amino acid translation have been aligned and compared regarding their identity and phylogenetic relationships as follows:
Sequences alignment: multiple alignments and their graphical depictions were generated using the Geneious bioinformatics software platform, version 8.1.4 with the MAFFT algorithms FFT-NS-i x1000 or G-INS-I with default parameters [47, 48]. All multiple alignments were visually inspected and, when necessary, manually optimized prior to subsequent analyses.
Phylogenetic analysis: the sequence phylogenetic analysis was performed by using the Maximum Likelihood (ML) method with the MEGA Software, version 6, based on the Kimura 2-parameter model [49]. Phylogenies were tested by the bootstrap method with 100 replicates. Pairwise deletion option was applied, allowing to compare only the nucleotide sequences of portions that were actually shared by the sequences. The tree was drawn to scale, with branch lengths measured as the number of substitutions per site.
Nucleotide divergence estimation: pairwise divergences between aligned nucleotide sequences were estimated using MEGA Software, version 6 [49], with a p-distance model and applying the pairwise deletion option.
Supplementary Materials
Author Contributions
GB had the idea for this study, designed and coordinated it. PB provided the skulls. AGN, GF, AF, VG, and CS performed the anthropological and paleopathological studies. FL, CMM, PC, and PA were involved in the molecular analysis. NG and ET performed the virological and bioinformatics study. All authors participated in interpretation of results and manuscript writing. GB, NG, and ET revised the manuscript. All authors approved the final manuscript.
Acknowledgments
The authors are grateful to the Archaeological Superintendence of Sassari and Nuoro, of Liguria and of Tuscany for their permission to perform the study on the ancient dental calculus.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
The study was partially funded by the project “On the antiquity of cancer: the contribution of paleopathology to the study of ancient tumors”, PRA 2018–19, University of Pisa.
References
- 1. Naccarato AG, Lessi F, Zavaglia K, Scatena C, Al Hamad MA, Aretini P, Menicagli M, Roncella M, Ghilli M, Caligo MA, Mazzanti CM, Bevilacqua G. Mouse Mammary Tumor Virus (MMTV) - like exogenous sequences are associated with sporadic but not hereditary human breast carcinoma. Aging (Albany NY). 2019; 11:7236–41. https://doi.org/10.18632/aging.102252 [PubMed]
- 2. Parl FF, Crooke PS, Plummer WD
Jr , Dupont WD. Genomic-epidemiologic evidence that estrogens promote breast cancer development. Cancer Epidemiol Biomarkers Prev. 2018; 27:899–907. https://doi.org/10.1158/1055-9965.EPI-17-1174 [PubMed] - 3. Lawson JS, Glenn WK. Evidence for a causal role by mouse mammary tumour-like virus in human breast cancer. NPJ Breast Cancer. 2019; 5:40. https://doi.org/10.1038/s41523-019-0136-4 [PubMed]
- 4. Bittner JJ. Some possible effects of nursing on the mammary gland tumor incidence in mice. Science. 1936; 84:162. https://doi.org/10.1126/science.84.2172.162 [PubMed]
- 5. Bevilacqua G, Marchetti A, Biondi R. Ultrastructural features of the intestinal absorption of mouse mammary tumor virus in newborn BALB/cfRIII mice. Gastroenterology. 1989; 96:139–45. https://doi.org/10.1016/0016-5085(89)90774-9 [PubMed]
- 6. Cardiff RD, Kenney N. Mouse mammary tumor biology: a short history. Adv Cancer Res. 2007; 98:53–116. https://doi.org/10.1016/S0065-230X(06)98003-8 [PubMed]
- 7. Moore DH. Evidence in favor of the existence of human breast cancer virus. Cancer Res. 1974; 34:2322–29. [PubMed]
- 8. Wang Y, Holland JF, Bleiweiss IJ, Melana S, Liu X, Pelisson I, Cantarella A, Stellrecht K, Mani S, Pogo BG. Detection of mammary tumor virus env gene-like sequences in human breast cancer. Cancer Res. 1995; 55:5173–79. [PubMed]
- 9. Liu B, Wang Y, Melana SM, Pelisson I, Najfeld V, Holland JF, Pogo BG. Identification of a proviral structure in human breast cancer. Cancer Res. 2001; 61:1754–59. [PubMed]
- 10. Zammarchi F, Pistello M, Piersigilli A, Murr R, Di Cristofano C, Naccarato AG, Bevilacqua G. MMTV-like sequences in human breast cancer: a fluorescent PCR/laser microdissection approach. J Pathol. 2006; 209:436–44. https://doi.org/10.1002/path.1997 [PubMed]
- 11. Stewart TH, Sage RD, Stewart AF, Cameron DW. Breast cancer incidence highest in the range of one species of house mouse, mus domesticus. Br J Cancer. 2000; 82:446–51. https://doi.org/10.1054/bjoc.1999.0941 [PubMed]
- 12. Szabo S, Haislip AM, Garry RF. Of mice, cats, and men: is human breast cancer a zoonosis? Microsc Res Tech. 2005; 68:197–208. https://doi.org/10.1002/jemt.20232 [PubMed]
- 13. Laumbacher B, Fellerhoff B, Herzberger B, Wank R. Do dogs harbour risk factors for human breast cancer? Med Hypotheses. 2006; 67:21–26. https://doi.org/10.1016/j.mehy.2006.01.016 [PubMed]
- 14. Johal H, Ford C, Glenn W, Heads J, Lawson J, Rawlinson W. Mouse mammary tumor like virus sequences in breast milk from healthy lactating women. Breast Cancer Res Treat. 2011; 129:149–55. https://doi.org/10.1007/s10549-011-1421-6 [PubMed]
- 15. Nartey T, Moran H, Marin T, Arcaro KF, Anderton DL, Etkind P, Holland JF, Melana SM, Pogo BG. Human Mammary Tumor Virus (HMTV) sequences in human milk. Infect Agent Cancer. 2014; 9:20. https://doi.org/10.1186/1750-9378-9-20 [PubMed]
- 16. Mazzanti CM, Lessi F, Armogida I, Zavaglia K, Franceschi S, Al Hamad M, Roncella M, Ghilli M, Boldrini A, Aretini P, Fanelli G, Marchetti I, Scatena C, et al. Human saliva as route of inter-human infection for mouse mammary tumor virus. Oncotarget. 2015; 6:18355–63. https://doi.org/10.18632/oncotarget.4567 [PubMed]
- 17. Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007; 447:279–83. https://doi.org/10.1038/nature05775 [PubMed]
- 18. Vargiu L, Rodriguez-Tomé P, Sperber GO, Cadeddu M, Grandi N, Blikstad V, Tramontano E, Blomberg J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology. 2016; 13:7. https://doi.org/10.1186/s12977-015-0232-y [PubMed]
- 19. Grandi N, Cadeddu M, Blomberg J, Tramontano E. Contribution of type W human endogenous retroviruses to the human genome: characterization of HERV-W proviral insertions and processed pseudogenes. Retrovirology. 2016; 13:67. https://doi.org/10.1186/s12977-016-0301-x [PubMed]
- 20. Grandi N, Tramontano E. HERV envelope proteins: physiological role and pathogenic potential in cancer and autoimmunity. Front Microbiol. 2018; 9:462. https://doi.org/10.3389/fmicb.2018.00462 [PubMed]
- 21. Grandi N, Tramontano E. Human endogenous retroviruses are ancient acquired elements still shaping innate immune responses. Front Immunol. 2018; 9:2039. https://doi.org/10.3389/fimmu.2018.02039 [PubMed]
- 22. Grandi N, Pisano MP, Tramontano E. The emerging field of human endogenous retroviruses: understanding their physiological role and contribution to diseases. Future Virol. 2019; 14:441–4. https://doi.org/10.2217/fvl-2019-0061
- 23. Callahan R, Drohan W, Tronick S, Schlom J. Detection and cloning of human DNA sequences related to the mouse mammary tumor virus genome. Proc Natl Acad Sci USA. 1982; 79:5503–07. https://doi.org/10.1073/pnas.79.18.5503 [PubMed]
- 24. Grandi N, Cadeddu M, Pisano MP, Esposito F, Blomberg J, Tramontano E. Identification of a novel HERV-K(HML10): comprehensive characterization and comparative analysis in non-human primates provide insights about HML10 proviruses structure and diffusion. Mob DNA. 2017; 8:15. https://doi.org/10.1186/s13100-017-0099-7 [PubMed]
- 25. Pisano MP, Grandi N, Cadeddu M, Blomberg J, Tramontano E. Comprehensive characterization of the human endogenous retrovirus HERV-K(HML-6) group: overview of structure, phylogeny, and contribution to the human genome. J Virol. 2019; 93:e00110–19. https://doi.org/10.1128/JVI.00110-19 [PubMed]
- 26. Robinson MJ, Erlwein OW, Kaye S, Weber J, Cingoz O, Patel A, Walker MM, Kim WJ, Uiprasertkul M, Coffin JM, McClure MO. Mouse DNA contamination in human tissue tested for XMRV. Retrovirology. 2010; 7:108. https://doi.org/10.1186/1742-4690-7-108 [PubMed]
- 27. Bao W, Kojima KK, Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015; 6:11. https://doi.org/10.1186/s13100-015-0041-9 [PubMed]
- 28. Bryant ML, Gardner MB, Marx PA, Maul DH, Lerche NW, Osborn KG, Lowenstine LJ, Bodgen A, Arthur LO, Hunter E. Immunodeficiency in rhesus monkeys associated with the original mason-pfizer monkey virus. J Natl Cancer Inst. 1986; 77:957–65. [PubMed]
- 29. Leroux C, Girard N, Cottin V, Greenland T, Mornex JF, Archer F. Jaagsiekte sheep retrovirus (JSRV): from virus to lung cancer in sheep. Vet Res. 2007; 38:211–28. https://doi.org/10.1051/vetres:2006060 [PubMed]
- 30. Lehrer S, Rheinstein PH. Mouse mammary tumor viral env sequences are not present in the human genome but are present in breast tumors and normal breast tissues. Virus Res. 2019; 266:43–47. https://doi.org/10.1016/j.virusres.2019.03.011 [PubMed]
- 31. Nartey T, Mazzanti CM, Melana S, Glenn WK, Bevilacqua G, Holland JF, Whitaker NJ, Lawson JS, Pogo BG. Mouse Mammary Tumor-Like virus (MMTV) is present in human breast tissue before development of virally associated breast cancer. Infect Agent Cancer. 2017; 12:1. https://doi.org/10.1186/s13027-016-0113-6 [PubMed]
- 32. Mazzanti CM, Al Hamad M, Fanelli G, Scatena C, Zammarchi F, Zavaglia K, Lessi F, Pistello M, Naccarato AG, Bevilacqua G. A mouse mammary tumor virus env-like exogenous sequence is strictly related to progression of human sporadic breast carcinoma. Am J Pathol. 2011; 179:2083–90. https://doi.org/10.1016/j.ajpath.2011.06.046 [PubMed]
- 33. Feldman D, Roniger M, Bar-Sinai A, Braitbard O, Natan C, Love DC, Hanover JA, Hochman J. The signal peptide of mouse mammary tumor virus-env: a phosphoprotein tumor modulator. Mol Cancer Res. 2012; 10:1077–86. https://doi.org/10.1158/1541-7786.MCR-11-0581 [PubMed]
- 34. Moore DH, Charney J, Kramarsky B, Lasfargues EY, Sarkar NH, Brennan MJ, Burrows JH, Sirsat SM, Paymaster JC, Vaidya AB. Search for a human breast cancer virus. Nature. 1971; 229:611–14. https://doi.org/10.1038/229611a0 [PubMed]
- 35. Weyrich LS, Dobney K, Cooper A. Ancient DNA analysis of dental calculus. J Hum Evol. 2015; 79:119–24. https://doi.org/10.1016/j.jhevol.2014.06.018 [PubMed]
- 36. Henry AG, Brooks AS, Piperno DR. Microfossils in calculus demonstrate consumption of plants and cooked foods in neanderthal diets (shanidar III, Iraq; spy I and II, Belgium). Proc Natl Acad Sci USA. 2011; 108:486–91. https://doi.org/10.1073/pnas.1016868108 [PubMed]
- 37. Warinner C, Speller C, Collins MJ, Lewis CM
Jr . Ancient human microbiomes. J Hum Evol. 2015; 79:125–36. https://doi.org/10.1016/j.jhevol.2014.10.016 [PubMed] - 38. Katz E, Lareef MH, Rassa JC, Grande SM, King LB, Russo J, Ross SR, Monroe JG. MMTV env encodes an ITAM responsible for transformation of mammary epithelial cells in three-dimensional culture. J Exp Med. 2005; 201:431–39. https://doi.org/10.1084/jem.20041471 [PubMed]
- 39. Brown TA, Jones MK, Powell W, Allaby RG. The complex origins of domesticated crops in the fertile crescent. Trends Ecol Evol. 2009; 24:103–09. https://doi.org/10.1016/j.tree.2008.09.008 [PubMed]
- 40. Pépin J. The origins of AIDS: from patient zero to ground zero. J Epidemiol Community Health. 2013; 67:473–75. https://doi.org/10.1136/jech-2012-201423 [PubMed]
- 41. Braitbard O, Roniger M, Bar-Sinai A, Rajchman D, Gross T, Abramovitch H, La Ferla M, Franceschi S, Lessi F, Naccarato AG, Mazzanti CM, Bevilacqua G, Hochman J. A new immunization and treatment strategy for Mouse Mammary Tumor Virus (MMTV) associated cancers. Oncotarget. 2016; 7:21168–80. https://doi.org/10.18632/oncotarget.7762 [PubMed]
- 42. Melis MG. Monte d’Accoddi and the end of the Neolithic in Sardinia (Italy). Documenta Praehistorica. 2011; 38:207–20. https://doi.org/10.4312/dp.38.16
- 43. Marcus JH, Posth C, Ringbauer H, Lai L, Skeates R, Sidore C, Beckett J, Furtwängler A, Olivieri A, Chiang CW, Al-Asadi H, Dey K, Joseph TA, et al. Genetic history from the middle neolithic to present on the mediterranean island of sardinia. Nat Commun. 2020; 11:939. https://doi.org/10.1038/s41467-020-14523-6 [PubMed]
- 44. Ferembach D, Schwindezky I, Stoukal M. Recommendation for Age and Sex Diagnoses of Skeletons. J Hum Evol. 1980; 9:517–49. https://doi.org/10.1016/0047-2484(80)90061-5
- 45. Lovejoy CO. Dental wear in the libben population: its functional pattern and role in the determination of adult skeletal age at death. Am J Phys Anthropol. 1985; 68:47–56. https://doi.org/10.1002/ajpa.1330680105 [PubMed]
- 46. Ling Z, Xiang C. Molecular Microecological Techniques. In: Li L. (eds) Infectious Microecology. Advanced Topics in Science and Technology in China. . Springer, Berlin, Heidelberg.. 2014. p. 153–88. https://doi.org/10.1007/978-3-662-43883-1_7
- 47. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012; 28:1647–49. https://doi.org/10.1093/bioinformatics/bts199 [PubMed]
- 48. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013; 30:772–80. https://doi.org/10.1093/molbev/mst010 [PubMed]
- 49. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013; 30:2725–29. https://doi.org/10.1093/molbev/mst197 [PubMed]