



Introduction
Gastric cancer (GC) is one of the most common gastrointestinal cancers, with a high incidence in East Asian countries. The pathogenesis of GC is a multifactorial and multi-step process [1]. GC is diagnosed using endoscopy, biopsy, and pathology. Its five-year survival rate is related to the stage of the disease at diagnosis [2]. Early diagnosis has increased due to the application of advanced detection methods, but the prognosis of patients with advanced GC remains very poor [3, 4]. The occurrence of GC reflects the abnormal regulation of tumor-related genes [1, 5, 6]. Understanding the molecular mechanisms underlying GC will facilitate the diagnosis and enhance the treatment of GC, and the development of biomarkers for early detection will improve the prognosis of GC patients.
Dysregulation of the metabolic environment in the body plays a key role in cancer. The Warburg effect is a form of glycolysis that occurs in tumors in an aerobic environment [7]. One study found that a long non-coding RNA, lncRNA-MACC1, can enhance the Warburg effect in GC cells and up-regulate expression of glycolytic enzymes [8]. There are also changes in amino acid metabolism in patients with GC; the levels of cysteine, serine, isoleucine, tyrosine, and valine are increased [9, 10]. Exploring these metabolic changes in cancer may yield new treatments. In recent years, various metabolic enzymes and their products have become important as potential drug targets [11–13]. Drugs developed for the treatment of metabolic disorders may also be effective in the treatment of some cancers [14]. To study the prognostic utility of metabolism-related genes (MRGs) in GC, we established a GC prognostic risk model and studied its clinical application.
Results
Identification of PDEMRGs in GC
Four hundred and seven mRNA samples (375 GC tissues and 32 adjacent nontumor tissues) were analyzed in The Cancer Genome Atlas (TCGA). Through the Wilcoxon signed-rank test, 194 differentially expressed MRGs (DEMRGs) were obtained, including 122 up-regulated genes and 72 down-regulated genes of GC tissues compared with adjacent nontumor tissues (Figure 1A, 1B).
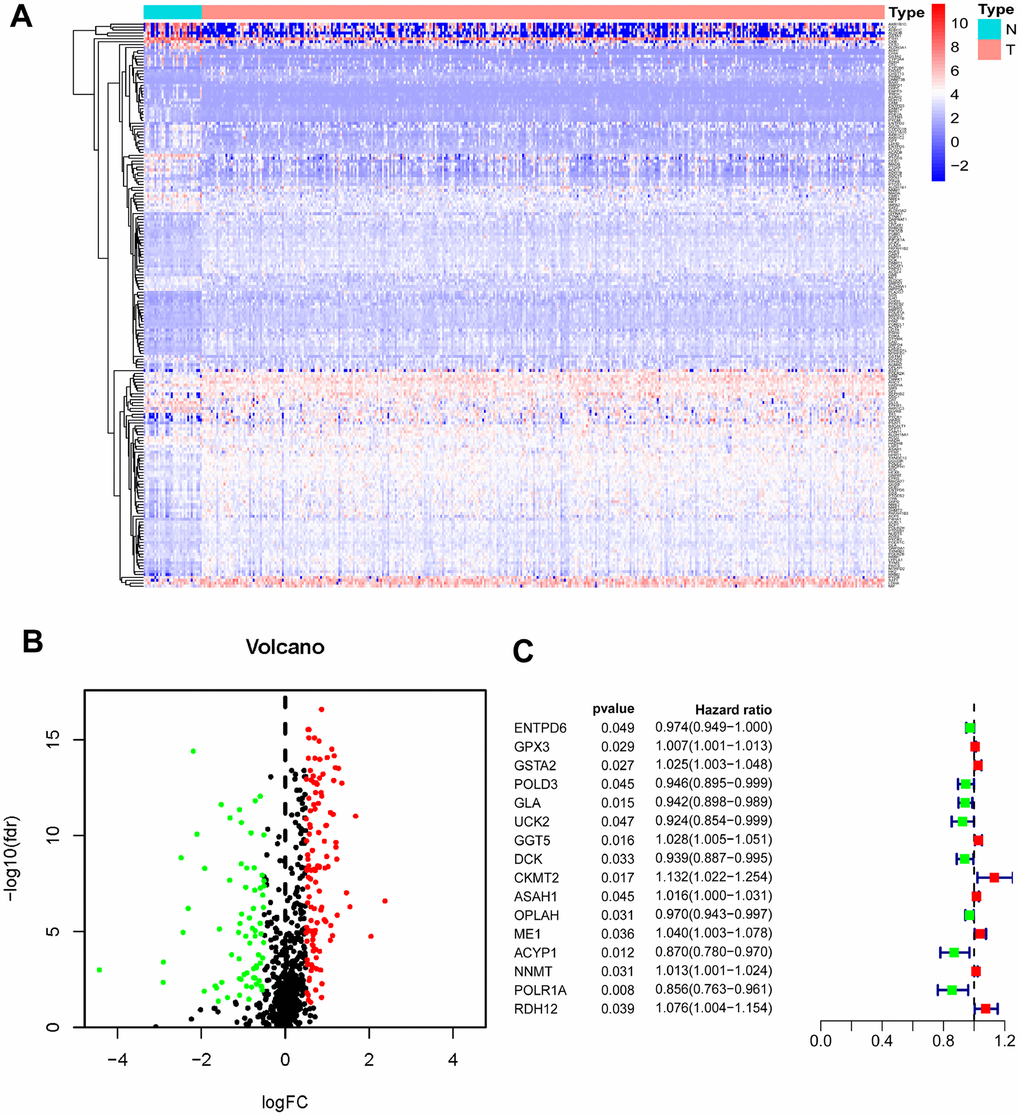
Figure 1. Identification of PDEMRGs. (A) Heatmap of DEMRGs: the red to blue spectrum signifies high to low gene expression. (B) Volcano plot of DEMRGs: red indicates upregulated DEMRGs, green indicates downregulated DEMRG, and black indicates DEMRGs that were not significantly differentially expressed. (C) Forrest plot of PDEMRGs: The red represents high-risk genes (hazard ratios, HR > 1); the green represents low-risk genes (HR < 1).
To determine the prognostic DEMRGs (PDEMRGs), univariate Cox regression analysis was used to screen the expression of DEMRGs in a training cohort. Sixteen DEMRGs (8 high-risk genes and 8 low-risk genes) were identified to be related to the overall survival (OS) of GC patients (Figure 1C).
Establishment and validation of the prognostic risk model
Lasso regression was performed to remove PDEMRGs that are related to each other to prevent the model from overfitting (Figure 2A, 2B). We obtained 13 candidate PDEMRGs (risk genes) to construct the prognostic risk model (Table 1). CKMT2, ME1, GSTA2, ASAH1, GGT5, RDH12, and NNMT were identified as high-risk genes, while POLR1A, ACYP1, GLA, OPLAH, DCK, and POLD3 were identified as low-risk genes.
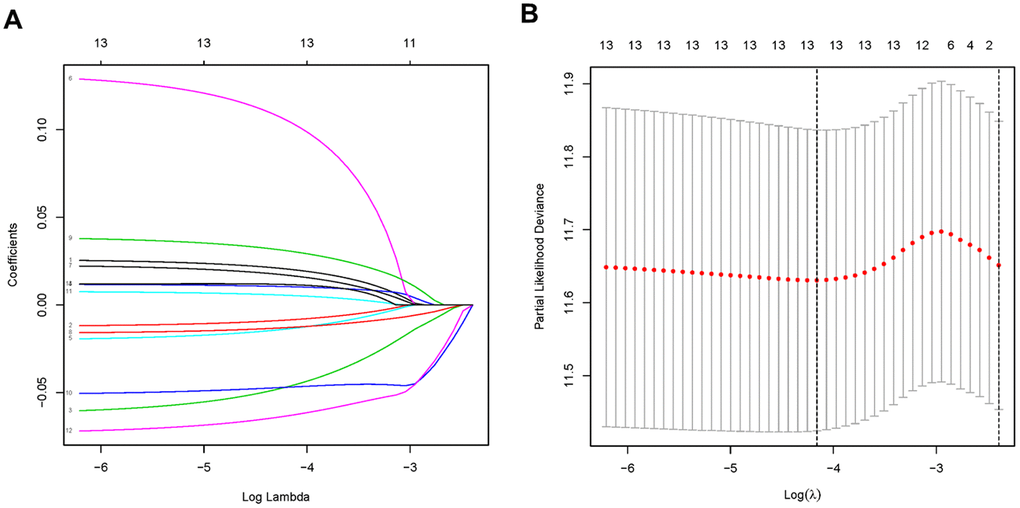
Figure 2. PDEMRGs selected by lasso regression. (A) Constructing the lasso coefficient prediction model. (B) Selecting variables in lasso regression with minimum criteria by 1000 times cross-validation.
Table 1. Lasso Cox analysis.
| Gene | Full name of gene | Coefficient | Metabolism-related KEGG pathways |
| CKMT2 | Creatine kinase, mitochondrial 2 | 0.07837 | Amino acid metabolism; Arginine and proline metabolism |
| ME1 | Malic enzyme 1 | 0.02443 | Lipid metabolism; Pyruvate metabolism; |
| GSTA2 | Glutathione S-transferase alpha 2 | 0.01516 | Other amino acids metabolism; Glutathione metabolism Xenobiotics biodegradation and metabolism; Metabolism of xenobiotics by cytochrome P450; Drug metabolism - cytochrome P450; Drug metabolism - other enzymes |
| ASAH1 | N-acylsphingosine amidohydrolase 1 | 0.01175 | Lipid metabolism; Sphingolipid metabolism |
| GGT5 | Gamma-glutamyl transferase 5 | 0.00916 | Lipid metabolism; Arachidonic acid metabolism; other amino acids metabolism; Taurine and hypotaurine metabolism; Glutathione metabolism |
| RDH12 | Retinol dehydrogenase 12 | 0.00904 | Cofactors and vitamins metabolism; Retinol metabolism |
| NNMT | Nicotinamide N-methyltransferase | 0.00346 | Cofactors and vitamins metabolism; Nicotinate and nicotinamide metabolism |
| POLR1A | RNA polymerase I subunit A | -0.05663 | - |
| ACYP1 | Acylphosphatase 1 | -0.04524 | Carbohydrate metabolism; Pyruvate metabolism |
| GLA | Galactosidase alpha | -0.03499 | Galactose metabolism; Glycosphingolipid metabolism; Sphingolipid metabolism |
| OPLAH | 5-oxoprolinase (ATP-hydrolysing) | -0.0105 | Other amino acids metabolism; Glutathione metabolism |
| DCK | Deoxycytidine kinase | -0.00875 | Nucleotide metabolism; Pyrimidine metabolism; Purine metabolism |
| POLD3 | DNA polymerase delta 3 | -0.00575 | Pyrimidine metabolism |
To study the role of the risk model in predicting the overall survival (OS) of GC patients, we used the expression levels of genes and regression coefficients to calculate the risk score for each patient. The risk score = (0.0152×expression of GSTA2) – (0.0058×expression of POLD3) – (0.0350×expression of GLA) + (0.0092×expression of GGT5) – (0.0088× expression of DCK) + (0.0784×expression of CKMT2) + (0.0117×expression of ASAH1) – (0.0105×expression of OPLAH) + (0.0244×expression of ME1) – (0.0452×expression of ACYP1) + (0.0035×expression of NNMT) – (0.0566×expression of POLR1A) + (0.0090×expression of RDH12).
Patients in the training cohort were divided into a high-risk (n=167) and a low-risk group (n=167) by the median risk score. To identify the prognostic difference between them we made a Kaplan-Meier curve. The OS was poorer in the high-risk group than the low-risk group (p < 0.001) (Figure 3A). We ranked the risk score of patients in the TCGA dataset (training cohort). The dot chart showed the survival state of patients and a heat map described the expression pattern of the high-risk and low-risk genes in the two groups (Figure 3C). Seven high-risk genes (CKMT2, ME1, GSTA2, ASAH1, GGT5, RDH12, and NNMT) were up-regulated, while six low-risk genes (POLR1A, ACYP1, GLA, OPLAH, DCK, and POLD3) were down-regulated. The risk genes showed opposite expression patterns for patients with low-risk scores. The prognostic model was validated in the Gene Expression Omnibus dataset (GEO, verification cohort), and the results were consistent with the training group (Figure 3B, 3D).
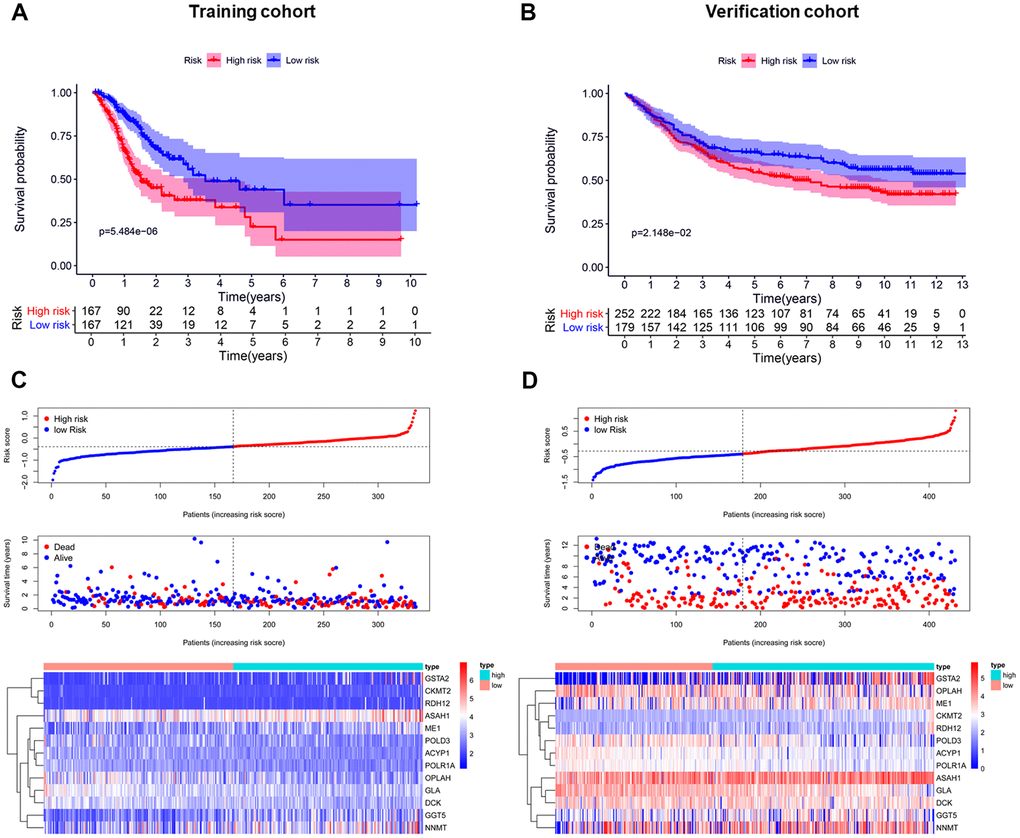
Figure 3. Establishment and validation of the prognostic risk model. (A, B) Kaplan-Meier curve analysis of the high-risk and low-risk groups. (C, D) From top to bottom=Risk score distribution of patients. Survival status scatter plots of patients. Expression patterns of risk genes.
These results indicated that this risk model can accurately predict the prognosis for GC patients.
Independent prognostic value of the risk model
We performed univariate and multivariate Cox regression analysis to determine if the risk score generated by the prognostic model was independent of other clinical indices.
In the training cohort, univariate Cox regression analysis representing age, stage, tumor (T), node (N), and risk score were significantly correlated with the OS (p < 0.05) (Figure 4A). Multivariate Cox regression analysis indicated the variables of age, gender, and risk score were independently correlated with the OS (p < 0.05) (Figure 4B). In the verification cohort, univariate and multivariate Cox regression analysis showed the variables of age, T, N, and risk score were significantly associated with the OS (p < 0.05) (Figure 4C, 4D). The results indicated that the risk model could serve as an independent prognostic factor independent of other clinical indices.
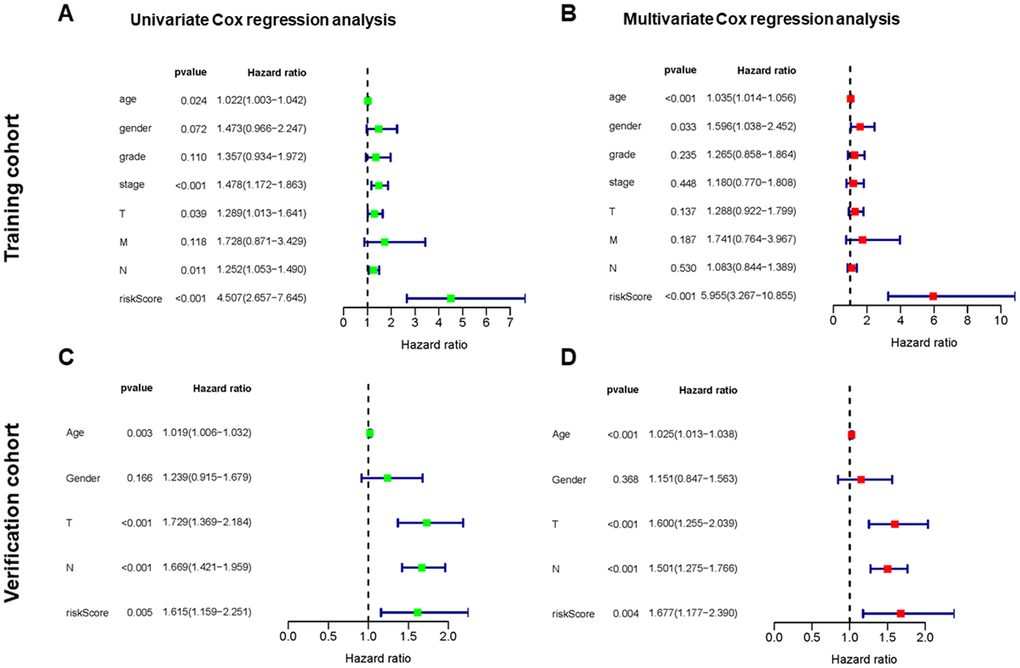
Figure 4. Independent value of the prognostic risk model. (A, B) Forrest plots of the univariate and multivariate Cox regression analysis in training cohort. (C, D) Forrest plot of the univariate and multivariate Cox regression analysis in verification cohort.
The risk score was more precise than other clinical indices. The AUCs (area under the curve) of the training cohort at risk score, age, gender, T, and N were 0.695, 0.572, 0.536, 0.558 and 0.574, respectively (Figure 5A). However, the risk score in the verification cohort is not the largest, which is not consistent with the result of the training cohort. This may be related to the relatively small sample size of the verification cohort (Figure 5C). To better predict the prognosis of GC patients, we established a nomogram model that accurately predicted the OS at 1, 3, and 5 years based on the variables associated with OS (age, gender, grade, stage, T, N, metastasis (M) and risk score) (Figure 5B, 5D).
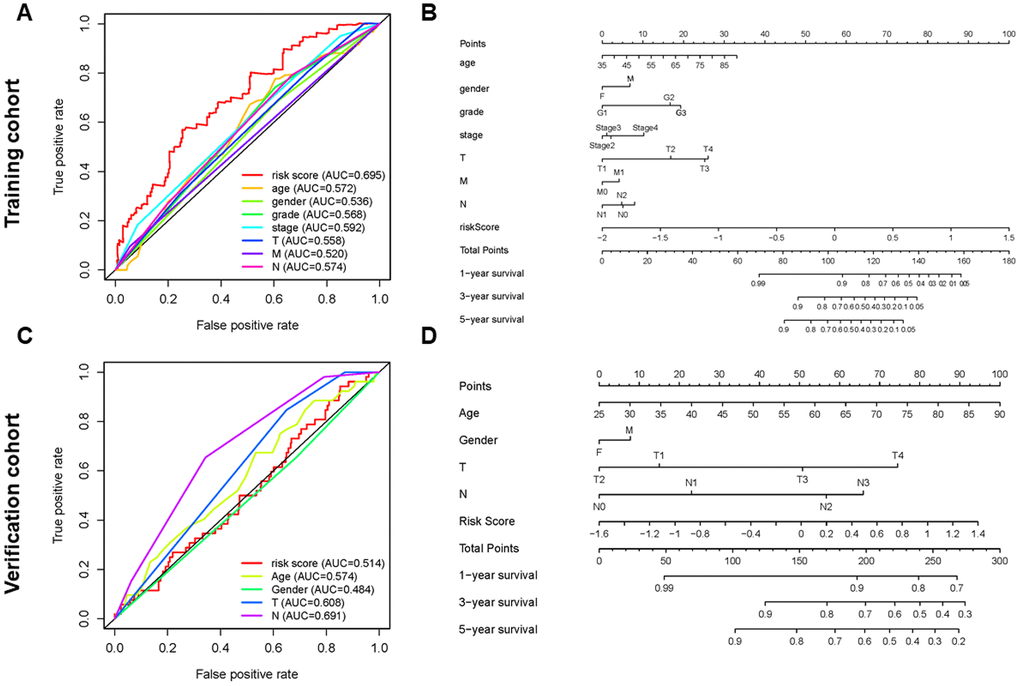
Figure 5. Establishment of ROC curves and nomograms. (A, C) ROC curves (receiver operating characteristics) of the risk score and other clinical indices. (B, D) The nomogram was established based on the independent prognosis model.
Gene set enrichment analyses
Gene Set Enrichment Analysis software (GSEA) was used and identified 60 significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the training cohort or verification cohort (Nominal p-value < 0.05). The majority of enrichment pathways were associated with metabolism, including arachidonic acid metabolism, drug metabolism by cytochrome p450, xenobiotics metabolism by cytochrome p450, pyrimidine and purine metabolism, glyoxylate and dicarboxylate metabolism, alanine aspartate and glutamate metabolism, cysteine and methionine metabolism, and fructose and mannose metabolism. Physiological processes such as glycan degradation and ubiquitin-mediated proteolysis significantly inhibit the occurrence and development of cancer, and there are common signaling pathways in cancer, MAPK signaling pathway, and the p53 signaling pathway (Figure 6).
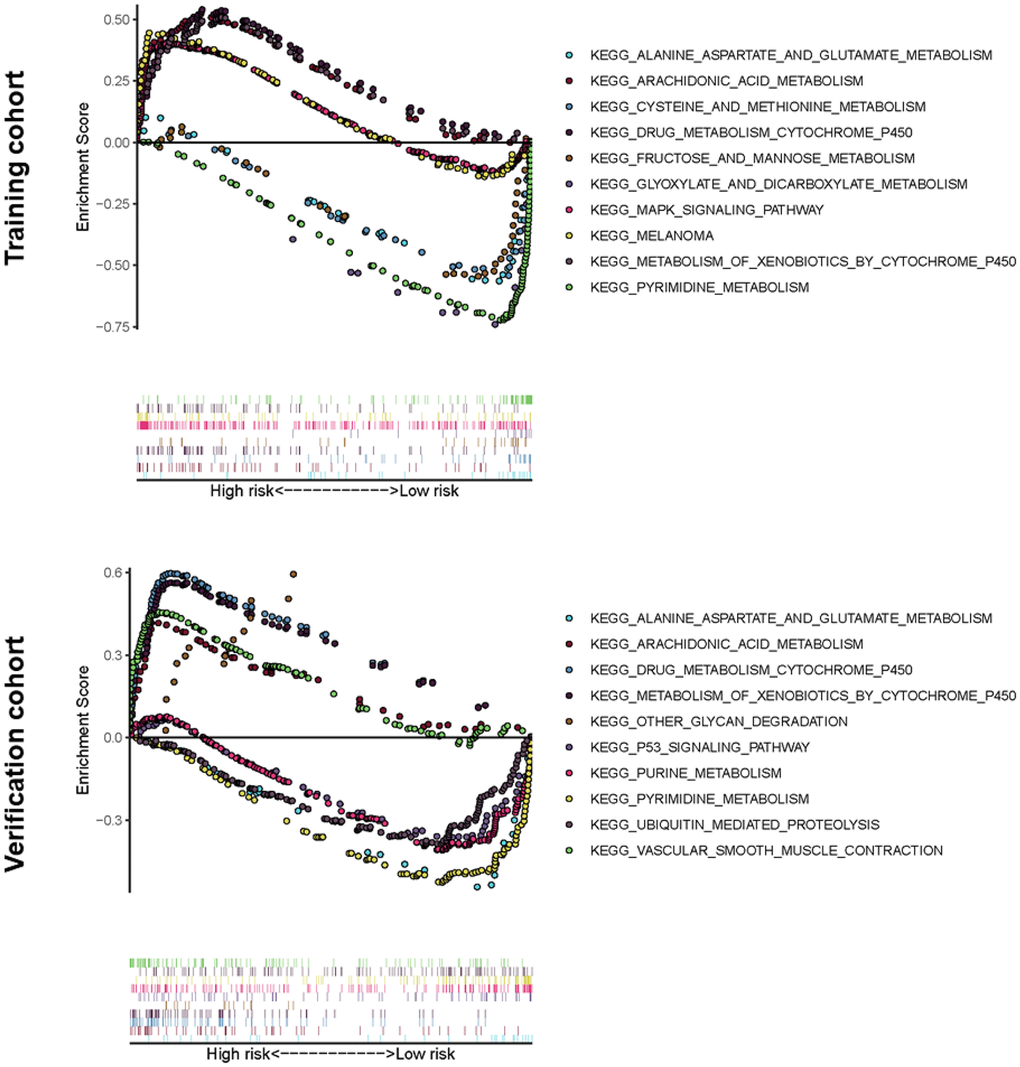
Figure 6. Significantly enriched KEGG pathways in training or verification cohort by GSEA. Above the horizontal axis indicated the pathways are in the high-risk group, and below the horizontal axis indicated that the pathways are in the low-risk group.
Clinical utility of the PDEMRGs
Exploration of the PDEMRGs during GC clinical progression indicated that the levels of GGT5 and NNMT were increased with clinical stage. This correlation between the expression levels of these two genes and GC progression may be useful in GC diagnosis (Figure 7A).
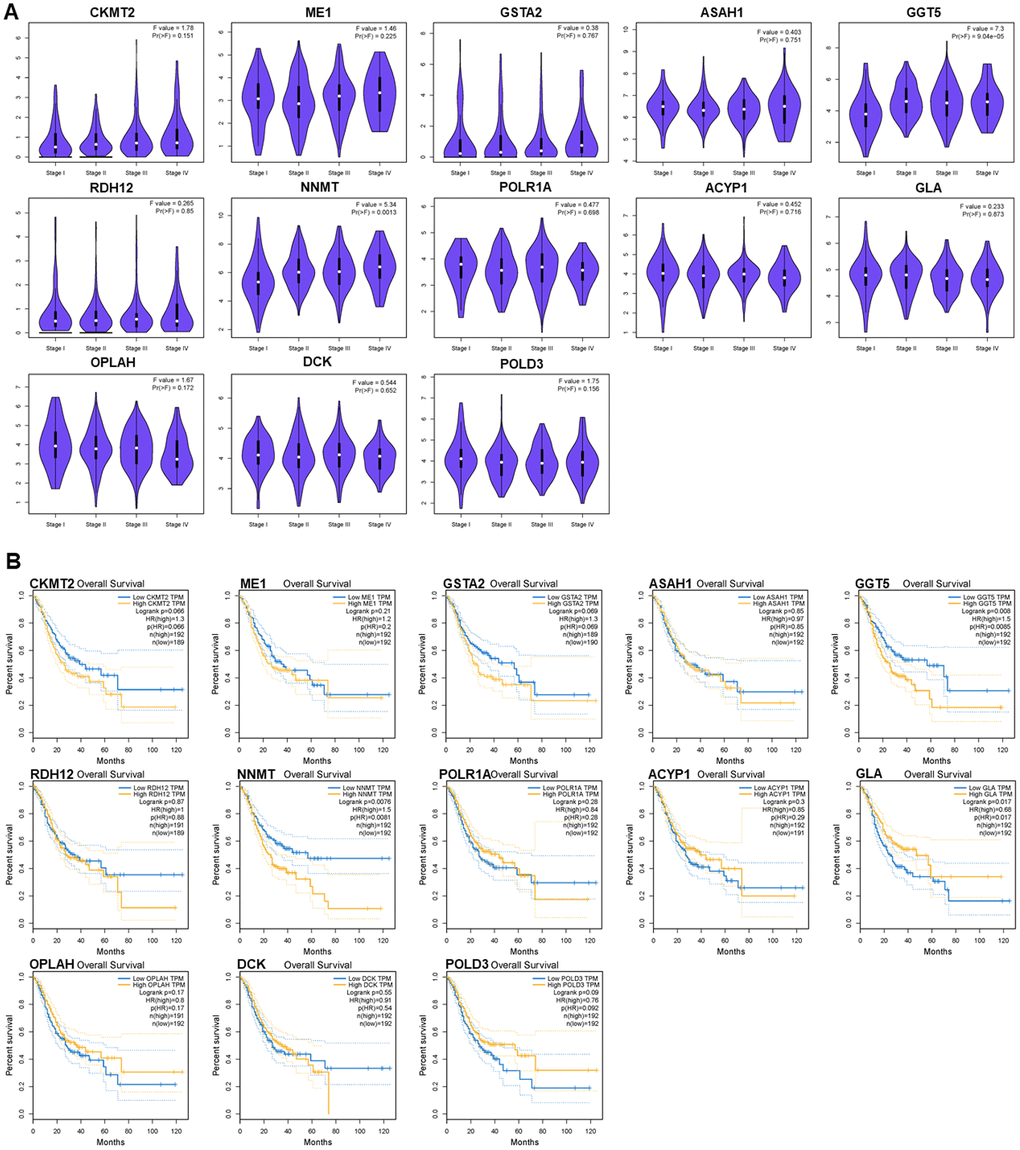
Figure 7. Relationships of the PDEMRGs with pathological stage and survival time. (A) The violin pot shows the expression levels of GGT5 and NNMT is significantly correlated with the pathological stage. (B) Survival plot indicates that the expression levels of GGT5, NNMT, and GLA are significantly correlated with OS.
Survival analysis indicated that the expression of GGT5, NNMT, and GLA had a significant association with patient survival (P < 0.05). Higher expression (yellow line) of GGT5 and NNMT indicated poorer prognosis. Lower expression (blue line) of GLA indicated lower patient survival (P < 0.05). The correlation between PDEMRGs and GC prognosis showed that PDEMRGs contribute to the progression of GC (Figure 7B).
External validation of the PDEMRGs using the online database
The Gene Expression Profiling Interactive Analysis database (GEPIA) corroborated the differences in gene expression between GC and normal gastric tissues. Boxplot showed most genes in the model had differences in GC mRNA expression compared with normal gastric tissues (P < 0.05) (Figure 8A). Genes such as CKMT2, GSTA2, and RDH12 were down-regulated, while POLR1A, ASAH1, GLA, DCK, and POLD3 were up-regulated. Representative protein expression was determined in the Human Protein Atlas (Figure 8B). The immunohistochemistry of GC genes was positive compared with normal gastric tissues, suggesting that the protein expression was increased. These results are consistent with the results of mRNA expression. However, RDH12 was not found in the database.
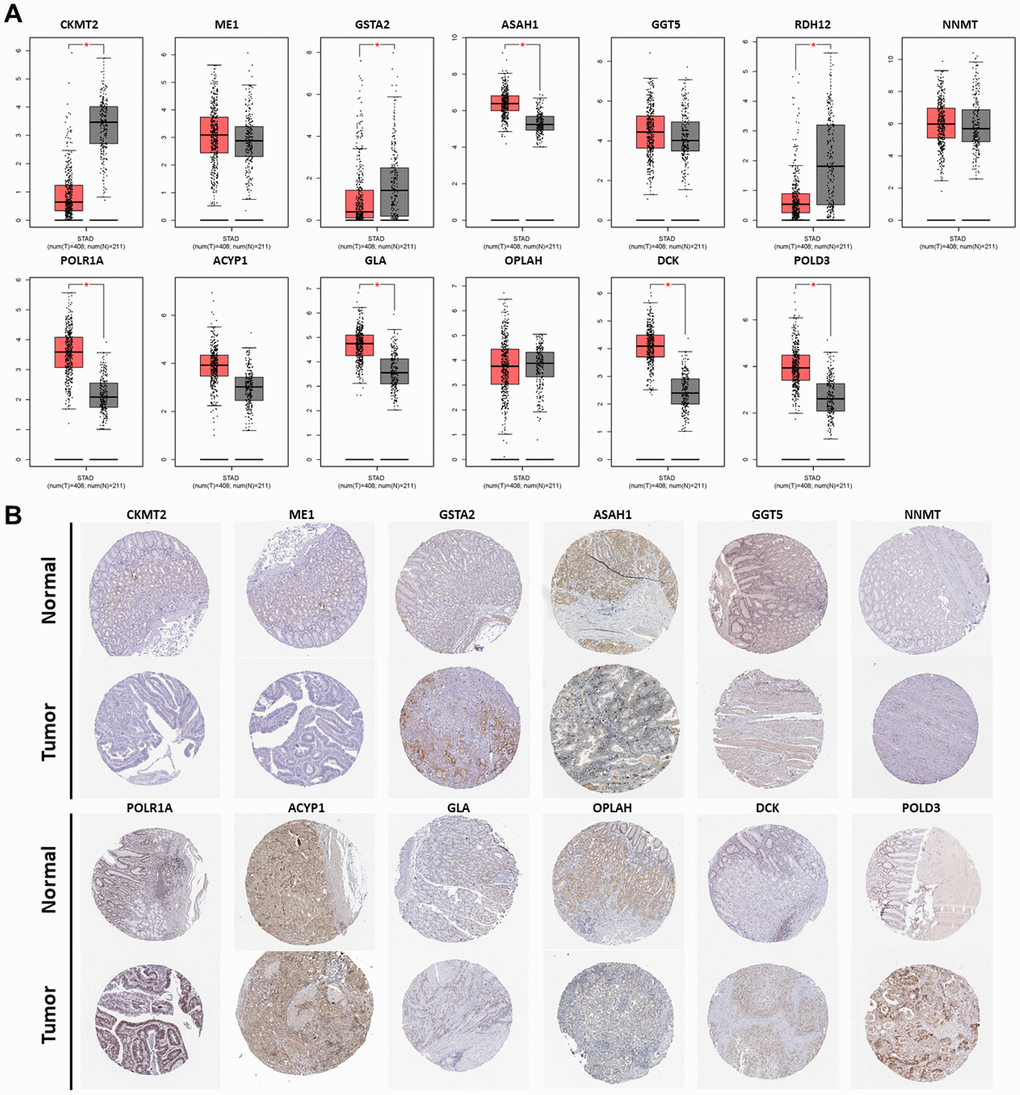
Figure 8. Expression of the PDEMRGs. (A) The mRNA expression levels of the PDEMRGs in GC and normal gastric tissue (*P < 0.05). Red represents GC and gray represents normal gastric tissue. (B) The representative protein expression of the PDEMRGs in GC and normal gastric tissue.
NNMT, ACYP1, and GLA were significantly over-expressed in GC, while CKMT2, ME1, GSTA2, and RDH12 were significantly under-expressed in the Oncomine database (Figure 9A). There is no mRNA expression of ASAH1, GGT5, POLR1A, OPLAH, DCK, and POLD3 in GC in the Oncomine database, but these genes have been confirmed to be over-expressed in GC both in the GEPIA and The Human Protein Atlas. Mutations in the form of amplification and deletion was expressed in the high-risk group, while amplification was expressed in the low-risk group. OPLAH had the most common genetic alterations (12%), and amplification was the most frequent genetic alteration (Figure 9B).
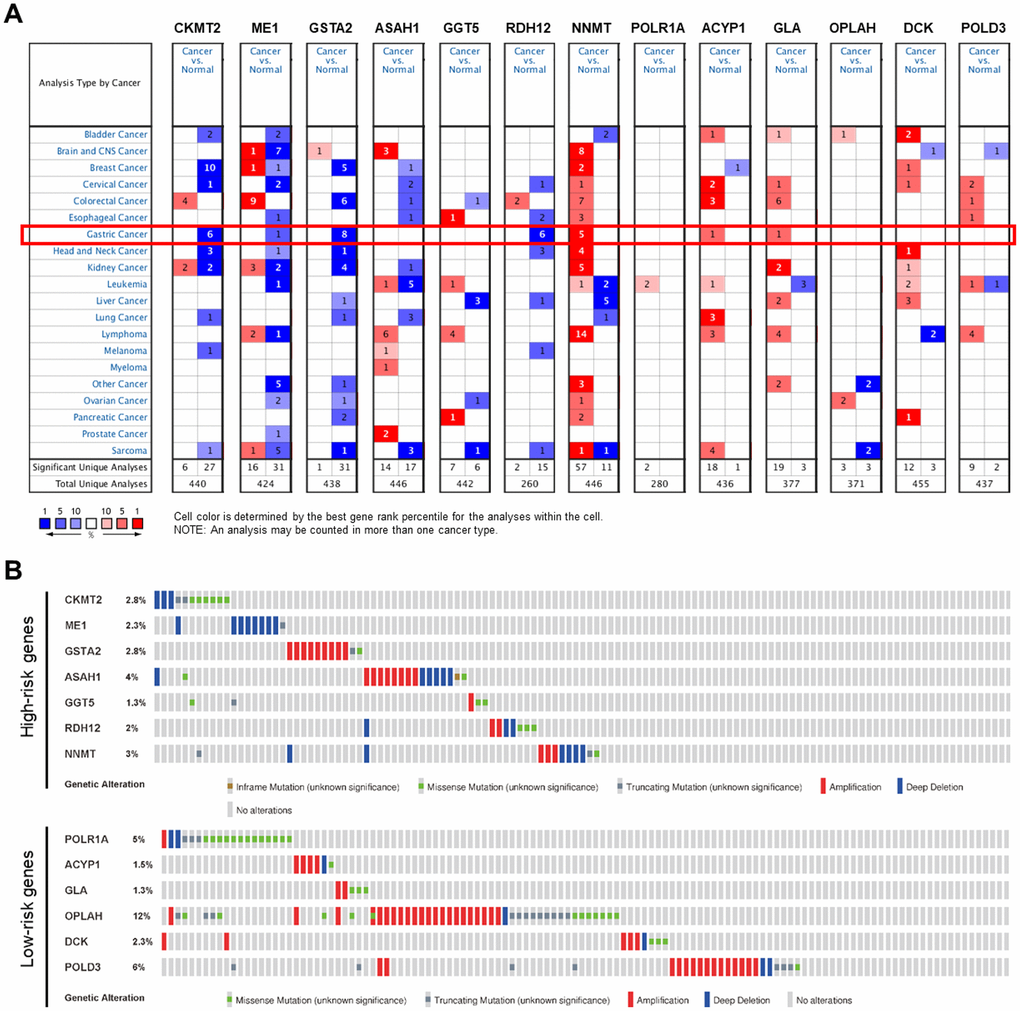
Figure 9. Genetic alterations of the PDEMRGs. (A) The expression profiles of the PDEMRGs in the Oncomine database. Red represents over-expressed; blue represents under-expressed. (B) Genetic alterations of the PDEMRGs from cBioportal for Cancer Genomics.
We evaluated the tumor mutation burden (TMB) of the GC dataset and found the mutation count in the high TMB group was higher compared with the low-risk group (p < 0.05) (Figure 10). This result illustrates that our model can stratify patients for personalized treatment.
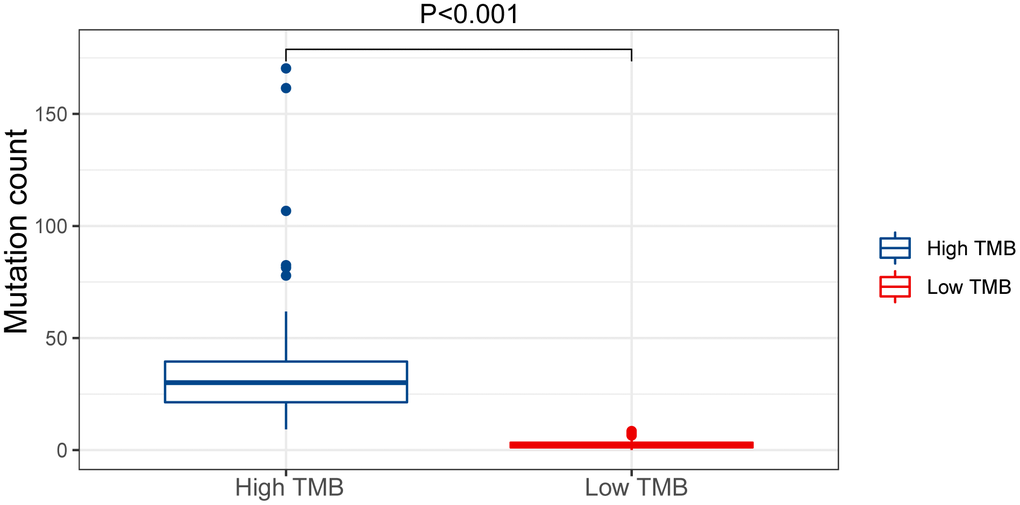
Figure 10. Mutation count of GC patients in the low and high TMB groups of the TCGA cohort.
Discussion
The development of genome sequencing and combining prognosis-related genes with traditional parameters has advantages in predicting cancer [15, 16]. Metabolism-related research is a new research focus [7]. Recent studies have analyzed the association between metabolism-related genes, the risk of GC, and diagnostic value [17]. Wei Zheng found that single nucleotide polymorphisms (SNPs) in trace element-related metabolic genes were related to GC risk [18]. Therefore, the expression of PDEMRGs may predict the progression of GC and the prognosis of GC patients. We identified the PDEMRGs based on a training cohort and utilized the PDEMRGs to build a reliable model to predict the OS in GC patients, which we verified with a verification cohort.
Univariate and multivariate Cox regression analysis showed that this model was an independent prognostic factor independent of other clinical indices. The risk score derived with the model was more accurate than other clinical parameters in predicting OS. Nomogram analysis showed that this model combined with clinical indices (age, gender, grade, stage, and TNM) could be used to accurately predict the OS of GC patients at 1, 3, and 5 years. This may help plan short-term follow-up of individualized treatment, so that early intervention can improve prognosis.
GSEA analysis showed that abundant metabolic pathways were related to tumors, which confirmed the close relationship between the model and the metabolic system. Patients in the high-risk group were associated with arachidonic acid and cytochrome P450 metabolic pathways, while patients in the low-risk group were associated with pyrimidine, glyoxylic acid and dicarboxylates, aspartic acid and glutamate, alanine, cysteine, and methionine, fructose and mannose, and purine metabolic pathways. Interestingly, the low-risk group involved more diverse metabolic pathways, and they may benefit from metabolic-related treatments.
We also analyzed the clinical application of the model. The expression of several genes, including GGT5, NNMT and ME1, was increased in stage I and II GC, and there was a negative association with patient survival. This indicates that this model has high prognostic value, especially for the short-term survival of GC patients. The PDEMRGs in the prognostic risk model are associated with the occurrence and development of cancer. For instance, ME1, a well-known oncogene, promotes GC growth, lung metastasis, and peritoneal dissemination, and over- expression of ME1 correlates with shorter and disease-free GC survival [19]. In addition, cancer cells expressing NNMT can alter the epigenetic state and increase expression of pro-tumorigenic gene products [20].
DNA replication stress induced by oncogenes is considered a driving factor of tumorigenesis. Research has shown that POLD3 plays a unique role in the process of RS-induced DNA break repair, so targeting POLD3 could provide a priority opportunity to target cancer cells [21–23]. DCK negatively regulates the transcriptional activity of NRF2, resulting in a decrease in the expression of antioxidant genes, and negatively regulates intracellular redox homeostasis and ROS production. DCK has a negative regulatory effect on the proliferation and metastasis of pancreatic cancer cells. The low expression of DCK promoted NRF2-mediated antioxidant transcription, which enhanced drug resistance to gemcitabine [24]. OPLAH encodes the 5-oxoproline enzyme, which controls the synthesis and degradation of glutathione. Hypermethylation of OPLAH3 is a common feature of some tumors. Naumov et al. sequenced the genomes of 22 pairs of colorectal cancer (CRC) and adjacent tissues and found that OPLAH is the initiation gene of DNA methylation in CRC [25]. Roy et al. found that GLA promotes mitochondrial death and apoptosis and reduces hypoxia. It combines limitation of de novo fatty acid synthesis and the cholinergic anti-inflammatory pathway that confirms anticancer function [26]. Cao et al. reported that ACYP1 was lower in imatinib-resistant gastrointestinal stromal tumor T1 cells [27]. Silencing POLR1A can hinder G1-S cell cycle progression in p53-inactivated human cancer cell lines [28]. Guo et al. observed that in cervical squamous cell carcinoma (CSCC) tissues, RDH12 expression was reduced by 74.5%. The expression of RDH12 was negatively associated with tumor size and infiltration depth in cervical cancer [29]. Studies have confirmed that GGT5 gene amplification contributes to non-small cell lung cancer (NSCLC). Cells produce high levels of glutamate and promote glutamine metabolism [30]. Immunohistochemical staining was used to detect the expression of ASAH1 in 120 cases of non-special invasive ductal carcinoma (IDC-NOS). The expression of ASAH1 correlated with lymph node metastasis, suggesting that ASAH1 is a biomarker predictive of lymph node status [31]. Low expression of glutathione S-transferases (GSTs) in the liver reduces the detoxification of chemical carcinogens. GSTA2 was found to be in linkage disequilibrium in Caucasians [32]. It was concluded that CKMT2 was a key regulatory factor in the development of osteosarcoma, and significantly correlated with patient OS [33].
Mutations in the genome can alter gene expression [34]. Research by DeBerardinis and Chandel confirmed that glycolysis is correlated with activated oncogenes and mutated tumor suppressors [11]. When Laskowski and her colleagues studied aging complement factor H-deficient mice, they observed spontaneous hepatic tumor formation in more than 50% [35]. The results illustrate the interaction between aging, genetic mutation, and cancer. We believe that aging is closely related to gastric cancer. Additionally, Hamada et al. found that tumor mutation burden (TMB) is related to the emergence of new antigens [36]. Therefore, we checked whether this model reflected the TMB of GC patients. The results showed the high TMB group was significantly higher than the low TMB group. We found the overall probability of genetic alterations was higher in the low-risk group than in the high-risk group. These findings indicate that this model can be used in patients with different metabolic abnormalities, making individualized therapy strategies possible.
In summary, we used 13 PDEMRGs to build a risk model that accurately predicts the prognosis in patients with GC. In addition, this model reflects the dysregulated metabolic microenvironment in tumor patients, and provides biomarkers for metabolic treatment of these patients. However, further in vitro and in vivo experiments are needed to validate the results of this research.
Materials and Methods
Data collection
Transcriptome data and the relevant clinical data were downloaded from TCGA (https://portal.gdc.cancer.gov/) and GEO (https://www.ncbi.nlm.nih.gov/geo/). The somatic mutation data were obtained from TCGA. The candidate metabolic gene sets were searched from “c2.cp.kegg. v7.0. symbols” of Gene Set Enrichment Analysis (GSEA).
Establishment of experimental model
We used univariate Cox analysis to initially identify potential PDEMRGs. Lasso penalty Cox regression analysis [37] was used for confirmation. The penalized maximum likelihood estimator with 1000-fold cross validation was used to construct the prognostic risk model. The expression values of PDEMRGs were weighted by the regression coefficient of the Cox regression model to calculate the risk score of every patient. Risk score = (CoefficientmRNA1×mRNA1 expression) + (CoefficientmRNA2×mRNA2 expression) + ⋯ + (CoefficientmRNAn× mRNAn expression). Taking the median risk score of the training cohort as the cut-off value, all patients with GC were divided into a high-risk group and a low-risk group. R packages “survival” [38] and “survminer” were performed to compare the survival differences between the high-and low-risk group, and a significant p-value was obtained. The verification cohort was used for verification.
Independence of the PDEMRGs
Univariate and multivariate Cox regression analysis were performed to analyze the independent prognosis of GC patients with forwarding stepwise procedure. P < 0.05 indicated statistical significance. The “SurvivalROC” [39] of R package was used to determine the prognostic value of the risk score. The nomogram [40] was constructed by including all independent prognostic factors to predict the survival of GC patients at 1, 3, and 5 years.
Gene set enrichment analyses
GSEA v4.0.1 software (https://www.gsea-msigdb.org/gsea/login.jsp) was run to reveal potential biological pathways and mechanisms in the KEGG. P < 0.05 was considered statistically significant.
External verification of PDEMRGs
To verify the expression of PDEMRGs in this model, the mRNA level was validated by Gene Expression Profiling Interactive Analysis database (GEPIA, http://gepia.cancer-pku.cn/) and the Oncomine database (https://www.oncomine.org/resource/main.html). The Human Protein Atlas database (http://www.proteinatlas.org) was used for the protein level. GEPIA was also used to determine the pathological stage and survival of PMRGs in this model. The genetic alterations of PMRGs in this model were evaluated by cBioportal for Cancer Genomics (http://www.cbioportal.org/).
Statistical analysis
All statistical analyses were run through R software version 3.6.1 (https://www.r-project.org/). Differences between variables were evaluated using independent t-tests. Log-rank test was used to compare the high-risk with the low-risk group in the Kaplan-Meier curve. Qualitative variables were compared by Pearson χ2 test or Fisher's exact test. A two-sided P<0.05 was considered statistically significant.
Author Contributions
Fang Wen conceived, designed and write the manuscript. Jiani Huang analyzed the data and generated the figures and tables. Xiaona Lu, Wenjie Huang and Yulan Wang performed the literature search and collected data for the manuscript. Yingfeng Bai and Shuai Ruan revised the images. SuPing Gu and Xiaoxue Chen revised the tables and checked the manuscript. Peng Shu directed the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors have declared that no conflicts of interest exists.
Funding
This study was supported by National Natural Science Foundation of China [No.81673918], Pilot gastric cancer project of clinical cooperation of traditional Chinese and western medicine for major and difficult diseases, The Open Projects of the Discipline of Chinese Medicine of Nanjing University of Chinese Medicine Supported by the Subject of Academic priority discipline of Jiangsu Higher Education Institutions [No.ZYX03KF020], and National Administration of Traditional Chinese Medicine : 2019 Project of building evidence based practice capacity for TCM[No.2019XZZX-ZL003].
References
- 1. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992; 52:6735–40. [PubMed]
- 2. Tan YK, Fielding JW. Early diagnosis of early gastric cancer. Eur J Gastroenterol Hepatol. 2006; 18:821–29. https://doi.org/10.1097/00042737-200608000-00004 [PubMed]
- 3. Chen S, Li T, Zhao Q, Xiao B, Guo J. Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin Chim Acta. 2017; 466:167–71. https://doi.org/10.1016/j.cca.2017.01.025 [PubMed]
- 4. Wang J, Qu J, Li Z, Che X, Liu J, Teng Y, Jin B, Zhao M, Liu Y, Qu X. Pretreatment platelet-to-lymphocyte ratio is associated with the response to first-line chemotherapy and survival in patients with metastatic gastric cancer. J Clin Lab Anal. 2018; 32:e22185. https://doi.org/10.1002/jcla.22185 [PubMed]
- 5. Li P, Chen H, Chen S, Mo X, Li T, Xiao B, Yu R, Guo J. Circular RNA 0000096 affects cell growth and migration in gastric cancer. Br J Cancer. 2017; 116:626–33. https://doi.org/10.1038/bjc.2016.451 [PubMed]
- 6. Xie Y, Shao Y, Sun W, Ye G, Zhang X, Xiao B, Guo J. Downregulated expression of hsa_circ_0074362 in gastric cancer and its potential diagnostic values. Biomark Med. 2018; 12:11–20. https://doi.org/10.2217/bmm-2017-0114 [PubMed]
- 7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013 [PubMed]
- 8. Lin L, Huang H, Liao W, Ma H, Liu J, Wang L, Huang N, Liao Y, Liao W. MACC1 supports human gastric cancer growth under metabolic stress by enhancing the warburg effect. Oncogene. 2015; 34:2700–10. https://doi.org/10.1038/onc.2014.204 [PubMed]
- 9. Wu H, Xue R, Tang Z, Deng C, Liu T, Zeng H, Sun Y, Shen X. Metabolomic investigation of gastric cancer tissue using gas chromatography/mass spectrometry. Anal Bioanal Chem. 2010; 396:1385–95. https://doi.org/10.1007/s00216-009-3317-4 [PubMed]
- 10. Song H, Peng JS, Dong-Sheng Y, Yang ZL, Liu HL, Zeng YK, Shi XP, Lu BY. Serum metabolic profiling of human gastric cancer based on gas chromatography/mass spectrometry. Braz J Med Biol Res. 2012; 45:78–85. https://doi.org/10.1590/s0100-879x2011007500158 [PubMed]
- 11. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016; 2:e1600200. https://doi.org/10.1126/sciadv.1600200 [PubMed]
- 12. Sciacovelli M, Frezza C. Oncometabolites: unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med. 2016; 100:175–81. https://doi.org/10.1016/j.freeradbiomed.2016.04.025 [PubMed]
- 13. Beger RD, Dunn W, Schmidt MA, Gross SS, Kirwan JA, Cascante M, Brennan L, Wishart DS, Oresic M, Hankemeier T, Broadhurst DI, Lane AN, Suhre K, et al, and for “Precision Medicine and Pharmacometabolomics Task Group”-Metabolomics Society Initiative. Metabolomics enables precision medicine: “a white paper, community perspective”. Metabolomics. 2016; 12:149. https://doi.org/10.1007/s11306-016-1094-6 [PubMed]
- 14. Piccinin E, Villani G, Moschetta A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol. 2019; 16:160–74. https://doi.org/10.1038/s41575-018-0089-3 [PubMed]
- 15. Wan B, Liu B, Huang Y, Yu G, Lv C. Prognostic value of immune-related genes in clear cell renal cell carcinoma. Aging (Albany NY). 2019; 11:11474–89. https://doi.org/10.18632/aging.102548 [PubMed]
- 16. Liu GM, Xie WX, Zhang CY, Xu JW. Identification of a four-gene metabolic signature predicting overall survival for hepatocellular carcinoma. J Cell Physiol. 2020; 235:1624–36. https://doi.org/10.1002/jcp.29081 [PubMed]
- 17. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492 [PubMed]
- 18. Zheng W, Li H, Liu B, Wu C. Association between the SNPs in trace element-related metabolic genes and the risk of gastric cancer: a case-control study in xianyou of China. J Genet. 2019; 98:67. [PubMed]
- 19. Lu YX, Ju HQ, Liu ZX, Chen DL, Wang Y, Zhao Q, Wu QN, Zeng ZL, Qiu HB, Hu PS, Wang ZQ, Zhang DS, Wang F, et al. ME1 Regulates NADPH Homeostasis to Promote Gastric Cancer Growth and Metastasis. Cancer Res. 2018; 78:1972–1985. https://doi.org/10.1158/0008-5472.CAN-17-3155 [PubMed]
- 20. Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. 2013; 9:300–06. https://doi.org/10.1038/nchembio.1204 [PubMed]
- 21. Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015; 528:286–90. https://doi.org/10.1038/nature16139 [PubMed]
- 22. Murga M, Lecona E, Kamileri I, Díaz M, Lugli N, Sotiriou SK, Anton ME, Méndez J, Halazonetis TD, Fernandez-Capetillo O. POLD3 is haploinsufficient for DNA replication in mice. Mol Cell. 2016; 63:877–83. https://doi.org/10.1016/j.molcel.2016.07.007 [PubMed]
- 23. Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015; 349:742–47. https://doi.org/10.1126/science.aaa8391 [PubMed]
- 24. Hu Q, Qin Y, Xiang J, Liu W, Xu W, Sun Q, Ji S, Liu J, Zhang Z, Ni Q, Xu J, Yu X, Zhang B. dCK negatively regulates the NRF2/ARE axis and ROS production in pancreatic cancer. Cell Prolif. 2018; 51:e12456. https://doi.org/10.1111/cpr.12456 [PubMed]
- 25. Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, Alekseev MV, Shelygin YA, Govorun VM. Genome-scale analysis of DNA methylation in colorectal cancer using infinium HumanMethylation450 BeadChips. Epigenetics. 2013; 8:921–34. https://doi.org/10.4161/epi.25577 [PubMed]
- 26. Roy S, Singh M, Rawat A, Devi U, Gautam S, Yadav RK, Rawat JK, Ansari MN, Saeedan AS, Kumar D, Kaithwas G. GLA supplementation regulates PHD2 mediated hypoxia and mitochondrial apoptosis in DMBA induced mammary gland carcinoma. Int J Biochem Cell Biol. 2018; 96:51–62. https://doi.org/10.1016/j.biocel.2018.01.011 [PubMed]
- 27. Cao J, Wei J, Yang P, Zhang T, Chen Z, He F, Wei F, Chen H, Hu H, Zhong J, Yang Z, Cai W, Li W, Wang Q. Genome-scale CRISPR-Cas9 knockout screening in gastrointestinal stromal tumor with imatinib resistance. Mol Cancer. 2018; 17:121. https://doi.org/10.1186/s12943-018-0865-2 [PubMed]
- 28. Donati G, Brighenti E, Vici M, Mazzini G, Treré D, Montanaro L, Derenzini M. Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J Cell Sci. 2011; 124:3017–28. https://doi.org/10.1242/jcs.086074 [PubMed]
- 29. Peng G, Dan W, Jun W, Junjun Y, Tong R, Baoli Z, Yang X. Transcriptome profiling of the cancer and adjacent nontumor tissues from cervical squamous cell carcinoma patients by RNA sequencing. Tumour Biol. 2015; 36:3309–17. https://doi.org/10.1007/s13277-014-2963-0 [PubMed]
- 30. Serizawa M, Kusuhara M, Zangiacomi V, Urakami K, Watanabe M, Takahashi T, Yamaguchi K, Yamamoto N, Koh Y. Identification of metabolic signatures associated with erlotinib resistance of non-small cell lung cancer cells. Anticancer Res. 2014; 34:2779–87. [PubMed]
- 31. Li YH, Liu HT, Xu J, Xing AY, Zhang J, Wang YW, Yin G, Gao P. The value of detection of S100A8 and ASAH1 in predicting the chemotherapy response for breast cancer patients. Hum Pathol. 2018; 74:156–63. https://doi.org/10.1016/j.humpath.2018.01.004 [PubMed]
- 32. Ning B, Wang C, Morel F, Nowell S, Ratnasinghe DL, Carter W, Kadlubar FF, Coles B. Human glutathione s-transferase A2 polymorphisms: variant expression, distribution in prostate cancer cases/controls and a novel form. Pharmacogenetics. 2004; 14:35–44. https://doi.org/10.1097/00008571-200401000-00004 [PubMed]
- 33. Wang H, Tang M, Ou L, Hou M, Feng T, Huang YE, Jin Y, Zhang H, Zuo G. Biological analysis of cancer specific microRNAs on function modeling in osteosarcoma. Sci Rep. 2017; 7:5382. https://doi.org/10.1038/s41598-017-05819-7 [PubMed]
- 34. Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002; 99:12963–68. https://doi.org/10.1073/pnas.162471999 [PubMed]
- 35. Laskowski J, Renner B, Pickering MC, Serkova NJ, Smith-Jones PM, Clambey ET, Nemenoff RA, Thurman JM. Complement factor H-deficient mice develop spontaneous hepatic tumors. J Clin Invest. 2020; 135105. https://doi.org/10.1172/JCI135105 [PubMed]
- 36. Hamada T, Soong TR, Masugi Y, Kosumi K, Nowak JA, da Silva A, Mu XJ, Twombly TS, Koh H, Yang J, Song M, Liu L, Gu M, et al. TIME (tumor immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology. 2018; 7:e1442999. https://doi.org/10.1080/2162402X.2018.1442999 [PubMed]
- 37. Tibshirani R. The lasso method for variable selection in the cox model. Stat Med. 1997; 16:385–95. https://doi.org/10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3 [PubMed]
- 38. Chan AW, Zhong J, Berhane S, Toyoda H, Cucchetti A, Shi K, Tada T, Chong CC, Xiang BD, Li LQ, Lai PB, Mazzaferro V, García-Fiñana M, et al. Development of pre and post-operative models to predict early recurrence of hepatocellular carcinoma after surgical resection. J Hepatol. 2018; 69:1284–93. https://doi.org/10.1016/j.jhep.2018.08.027 [PubMed]
- 39. Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000; 56:337–44. https://doi.org/10.1111/j.0006-341x.2000.00337.x [PubMed]
- 40. Iasonos A, Schrag D, Raj GV, Panageas KS. How to build and interpret a nomogram for cancer prognosis. J Clin Oncol. 2008; 26:1364–70. https://doi.org/10.1200/JCO.2007.12.9791 [PubMed]