



Introduction
The aging population is rapidly growing worldwide; in 2050, the population ages 60 and over is estimated to reach 2.1 billion in the world. Aging is associated with impaired organ function and increased susceptibility to various diseases, including chronic lung diseases [1–5], which are associated with life-threatening cardiopulmonary complications (e.g., pulmonary hypertension, right-sided heart failure) [6, 7]. Angiogenesis is impaired in aging animals [8–12], resulting in inhibition of organ regeneration [12]. For example, post-pneumonectomy (PNX) compensatory lung growth, which requires angiogenesis, is stimulated in young people, while it is diminished in older people [13, 14]. Inhibition of angiogenesis and attenuation of lung regeneration and repair abilities in aged people contribute to the pathogenesis of age-related lung diseases [15, 16]. Thus, we need to understand how aging disrupts angiogenesis and lung regeneration.
A transcription factor, Twist1, contributes to the age- and angiogenesis-related diseases, including pulmonary fibrosis [17, 18], diabetes [19], chronic obstructive pulmonary disease (COPD) [20], cancer [21], and atherosclerosis [22, 23]. Twist1 regulates vascular development [24] and function [25] through multiple angiogenic signaling (e.g., Tie2, platelet-derived growth factor (PDGF), VEGFR2, transforming growth factor beta receptor (TGFβR)) [18, 24–28]. Inhibition of Twist1 activity increases the expression of PGC1α that stimulates mitochondrial biogenesis [29–32] and angiogenesis [29, 33–35] in fat cells [36]. PGC1α controls age-dependent mitochondrial metabolism [31] and mediates aging-related cardiovascular diseases [29, 37–41]. The involvement of Twist1-PGC1α signaling in age-dependent inhibition of angiogenesis and how it contributes to the inhibition of lung regeneration in aged animals remains unclear.
Here we have used ECs isolated from human adipose tissue and the subcutaneous gel implantation system to examine the mechanism by which aging disrupts angiogenesis. We have then picked up lung as an organ-specific model and studied the effects of aging on lung regeneration using a PNX model. We have demonstrated that angiogenesis is impaired in aged ECs through Twist1-PGC1α signaling. Knockdown of Twist1 in aged ECs increases VEGFR2 expression and restores age-related decline in angiogenesis, while these effects are suppressed by knockdown of PGC1α. Knockdown of endothelial Twist1 also restores angiogenesis and post-PNX lung growth in aged mouse lungs. Modulation of Twist1-PGC1α signaling may be a novel intervention to rejuvenate angiogenic ability in aged adults and will lead to the development of efficient strategies for aging-associated diseases.
Results
Twist1-PGC1α mediates age-dependent decline in VEGFR2 expression
Twist1 contributes to the age- and angiogenesis-related diseases, including pulmonary fibrosis [17, 18], diabetes [19], COPD [20], cancer [21], and atherosclerosis [22, 23]. We have reported that Twist1 expression is higher in the bleomycin-induced fibrotic mouse lungs [18]. Twist1 mRNA levels were 3.4- times higher in ECs isolated from discarded de-identified aged (>50 years old) human adipose tissues compared to those in the younger tissues (<50 years old) (Figure 1A). It is reported that PGC1α activates angiogenic signaling [29, 33–35] and mediates aging-related cardiovascular diseases [29, 37–41] and that inhibition of Twist1 activity in fat cells increases the activity of PGC1α [36]. Consistently, the mRNA levels of PGC1α and angiogenic factor receptor, VEGFR2 were lower in ECs isolated from aged adipose tissues by 63% and 44%, respectively, in which Twist1 expression is higher, compared to those in the younger tissues (Figure 1A). Immunoblotting results confirmed that the protein levels of Twist1 were 2.2- times higher, while the levels of PGC1α and VEGFR2 were lower by 78% and 27%, respectively, in aged human adipose ECs compared to those in the younger tissues (Figure 1B). Twist1 siRNA transfection, which decreases Twist1 mRNA levels by 62%, increased the levels of PGC1α and VEGFR2 in aged human adipose ECs by 1.5- and 1.4-times, respectively (Figure 1C).
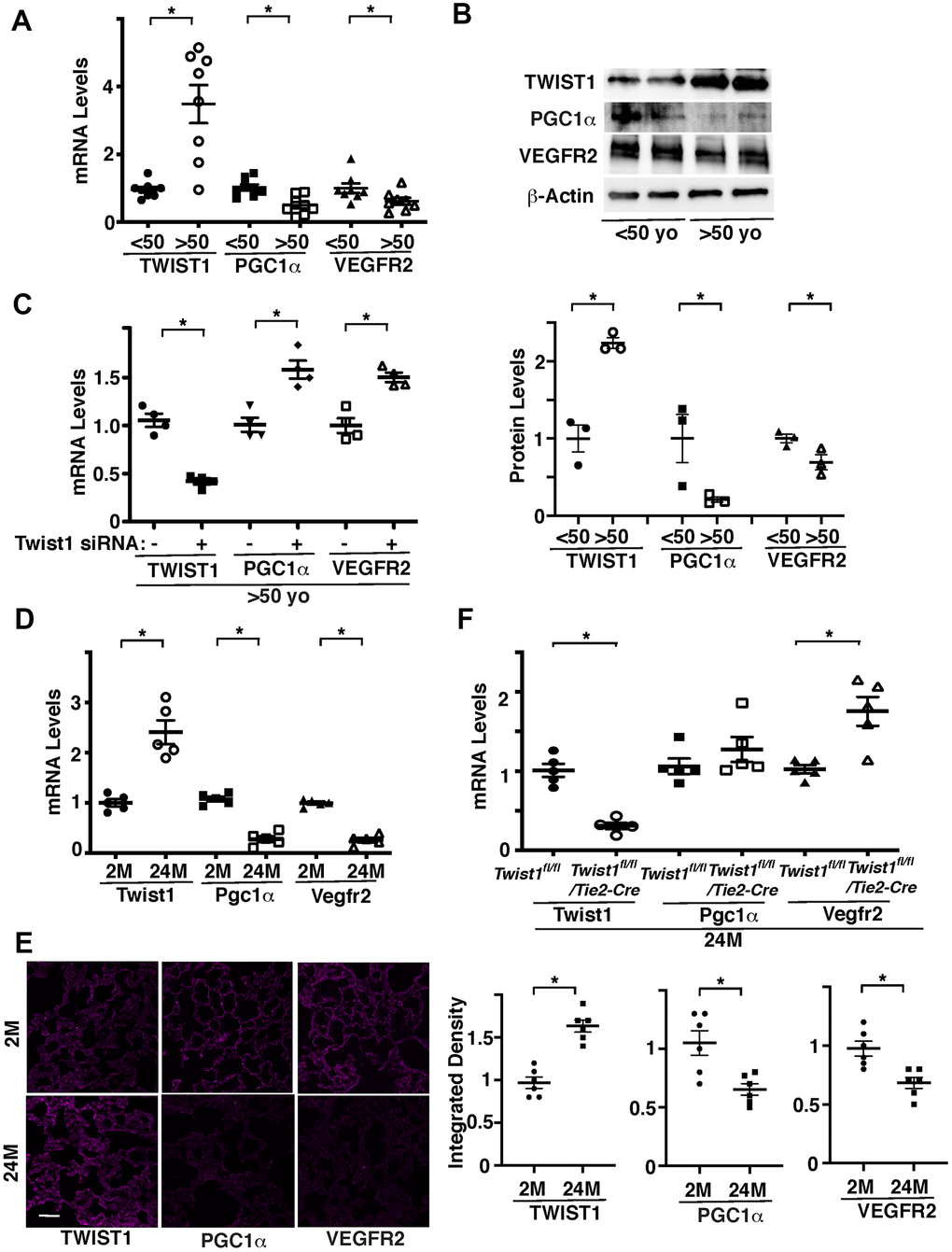
Figure 1. Twist1 mediates age-dependent decline in PGC1α and VEGFR2 expression in ECs. (A) Graph showing the mRNA levels of Twist1, PGC1α, and VEGFR2 in ECs isolated from young (<50 years old) vs. old (>50 years old) human adipose tissues (n=8, mean ± s.e.m., *, p<0.05). (B) Representative immunoblots showing Twist1, PGC1α, VEGFR2, and β-actin protein levels in young vs. aged human adipose ECs (top). Graph showing Twist1, PGC1α, and VEGFR2 protein levels normalized by β-actin protein levels in young vs. aged human adipose ECs (bottom, n=3, mean ± s.e.m., *, p<0.05). (C) Graph showing the mRNA levels of Twist1, PGC1α, and VEGFR2 in aged human adipose ECs treated with Twist1 siRNA or control siRNA with irrelevant sequences (n=4, mean ± s.e.m., *, p<0.05). (D) Graph showing the mRNA levels of Twist1, Pgc1α, and Vegfr2 in ECs isolated from 2M vs. 24M old mouse lungs (n=5, mean ± s.e.m., *, p<0.05). (E) IF micrographs showing Twist1, PGC1α, and VEGFR2 expression in 2M vs. 24M old mouse lungs. Scale bar, 50 μm. Graphs showing integrated density of Twist1, PGC1α, and VEGFR2 in the lung tissues (n=6, mean ± s.e.m., *, p<0.05). (F) Graph showing the mRNA levels of Twist1, Pgc1α, and Vegfr2 in ECs isolated from 24M old Twist1fl/fl and Twist1fl/fl/Tie2-cre mouse lungs (n=5, mean ± s.e.m., *, p<0.05).
We also confirmed the results using ECs isolated from 2 months (2M) old vs. 24M old mouse lungs. Twist1 mRNA levels were 2.4-times higher, while the mRNA levels of Pgc1α and Vegfr2 were lower by 83% and 84%, respectively, in 24M old mouse lung ECs compared to those in the 2M old mouse lung ECs (Figure 1D). Immunohistochemical analysis confirmed that the protein levels of TWIST1 were higher, while the levels of PGC1α and VEGFR2 were lower in 24M old mouse lungs compared to those in the 2M old mouse lungs (Figure 1E). The levels of Vegfr2 in ECs isolated from 24M old Tie2-specific Twist1 conditional knockout (Twist1fl/fl/Tie2-cre) mouse lungs, in which Twist1 expression is 78% decreased, were also 1.6-times higher than those in 24M old Twist1fl/fl mouse lungs (Figure 1F).
We next examined whether Twist1-PGC1α signaling controls VEGFR2 expression. Overexpression of PGC1α using lentiviral transduction, which increases PGC1α mRNA expression by 3.2-times, increased the mRNA and protein levels of VEGFR2 in ECs isolated from aged human adipose tissues (Figure 2A). PGC1α knockdown also inhibited Twist1 knockdown-induced increase in VEGFR2 in aged human adipose ECs (Figure 2B), suggesting that knockdown of Twist1 upregulates VEGFR2 expression through PGC1α in aged ECs.
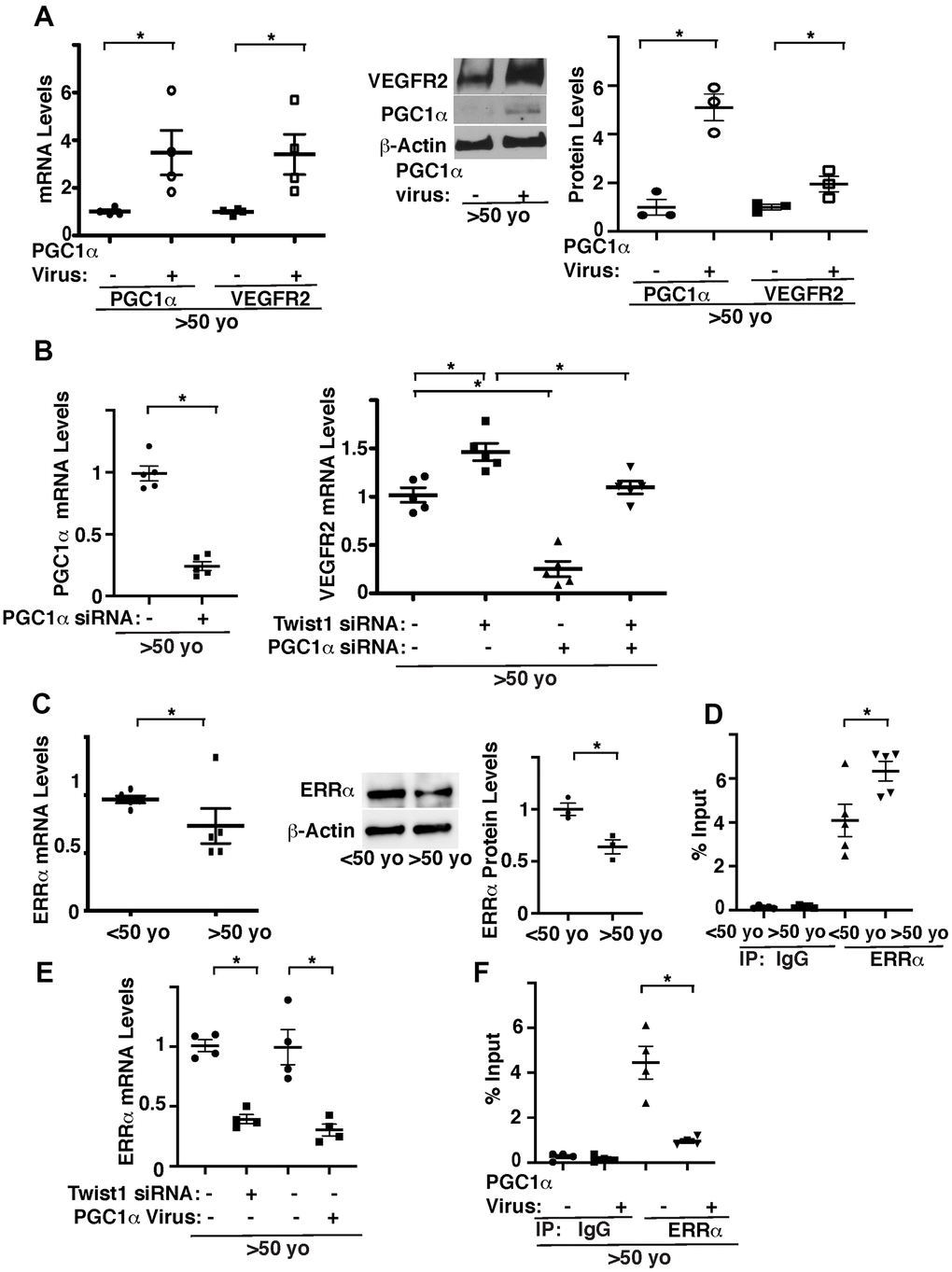
Figure 2. Twist1 controls age-dependent decline in VEGFR2 expression through PGC1α. (A) Graph showing the mRNA levels of PGC1α and VEGFR2 in aged (>50 years old) human adipose ECs treated with lentivirus overexpressing PGC1α or control virus (vector alone) (left, n=4, mean ± s.e.m., *, p<0.05). Representative immunoblots showing VEGFR2, PGC1α, and β-actin protein levels in aged human adipose ECs treated with lentivirus overexpressing PGC1α or control virus (middle). Graph showing VEGFR2 and PGC1α protein levels normalized by β-actin protein levels in aged human adipose ECs treated with lentivirus overexpressing PGC1α or control virus (right, n=3, mean ± s.e.m., *, p<0.05). (B) Graph showing the mRNA levels of PGC1α in aged human adipose ECs treated with PGC1α siRNA or control siRNA with irrelevant sequences (left, n=5, mean ± s.e.m., *, p<0.05). Graph showing the mRNA levels of VEGFR2 in aged human adipose ECs treated with Twist1 siRNA, PGC1α siRNA, or in combination (right, n=5, mean ± s.e.m., *, p<0.05). (C) Graph showing the mRNA levels of ERRα in young vs. aged human adipose ECs (left, n=5, mean ± s.e.m., *, p<0.05). Representative immunoblots showing ERRα and β-actin protein levels in young vs. aged human adipose ECs (middle). Graph showing ERRα protein levels normalized by β-actin protein levels in young vs. aged human adipose ECs (right, n=3, mean ± s.e.m., *, p<0.05). (D) ChIP analysis showing the immunoprecipitation levels of VEGFR2 promoter coimmunoprecipitating with control IgG or ERRα antibody in human adipose ECs isolated from young (<50 years old) vs. old (>50 years old) adipose tissues (n=5, mean ± s.e.m., *, p<0.05). (E) Graph showing the mRNA levels of ERRα in aged human adipose ECs treated with Twist1 siRNA or PGC1α virus (n=4, mean ± s.e.m., *, p<0.05). (F) ChIP analysis showing the immunoprecipitation levels of VEGFR2 promoter coimmunoprecipitating with control IgG or ERRα antibody in human adipose ECs isolated from old (>50 years old) adipose tissues treated with PGC1α virus or control virus (n=4, mean ± s.e.m., *, p<0.05).
It is known that PGC1α controls angiogenesis [33–35, 42] by binding to the transcription factor, estrogen-related receptor α (ERRα) [30, 43]. While ERRα stimulates angiogenic factor expression in muscle cells, ERRα represses angiogenic factor expression in ECs [44]. ERRα binds to DNA sites with the consensus sequence TCAAGGTCA (ERR response element (ERREs)) [45, 46]. Since VEGFR2 promoter sequence contains ERRE sites, we examined the effects of aging on ERRα expression and interaction of ERRα and the VEGFR2 promoter region using young vs. aged human adipose ECs. The mRNA and protein levels of ERRα in aged human ECs were 23% and 38% lower, respectively, compared to those in young ECs (Figure 2C). ERRα binds to the VEGFR2 promoter region 1.5-times higher in aged adipose ECs when analyzed using chromatin immunoprecipitation (ChIP) assay (Figure 2D). Twist1 knockdown or PGC1α overexpression decreased ERRα expression (Figure 2E) and PGC1α overexpression decreased ERRα binding ability to VEGFR2 promoter region in aged ECs (Figure 2F). These results are consistent with previous report demonstrating that ERRα acts as a transcriptional repressor in ECs [44] and suggest that overexpression of PGC1α stimulates VEGFR2 transcription by decreasing the binding ability of ERRα to the VEGFR2 promoter region in aged ECs.
Twist1 and PGC1α mediate age-related decline in angiogenic activities
We and other groups have demonstrated that angiogenesis is inhibited in aged ECs [8–12]. We next examined whether Twist1-PGC1α signaling mediates age-related decline in angiogenic activities. When we manipulated the expression of PGC1α or Twist1 in human adipose ECs of different ages using lentiviral transduction, PGC1α overexpression stimulated DNA synthesis and EC migration (Figure 3A, 3B). Twist1 knockdown inhibited DNA synthesis by 41% in young human adipose ECs, whereas it stimulated DNA synthesis and EC migration by 42% and 28%, respectively in aged ECs (Figure 3A, 3B). Twist1 knockdown-induced stimulation of DNA synthesis and migration in aged human adipose ECs was inhibited when treated with PGC1α shRNA in combination (Figure 3C, 3D). Stimulation of DNA synthesis and migration by Twist1 knockdown or PGC1α overexpression in aged human adipose ECs were also inhibited when treated with VEGFR2 inhibitor (SU5416) in combination (Figure 3E, 3F), suggesting that endothelial Twist1-PGC1α signaling mediates age-dependent inhibition of DNA synthesis and migration through VEGFR2.
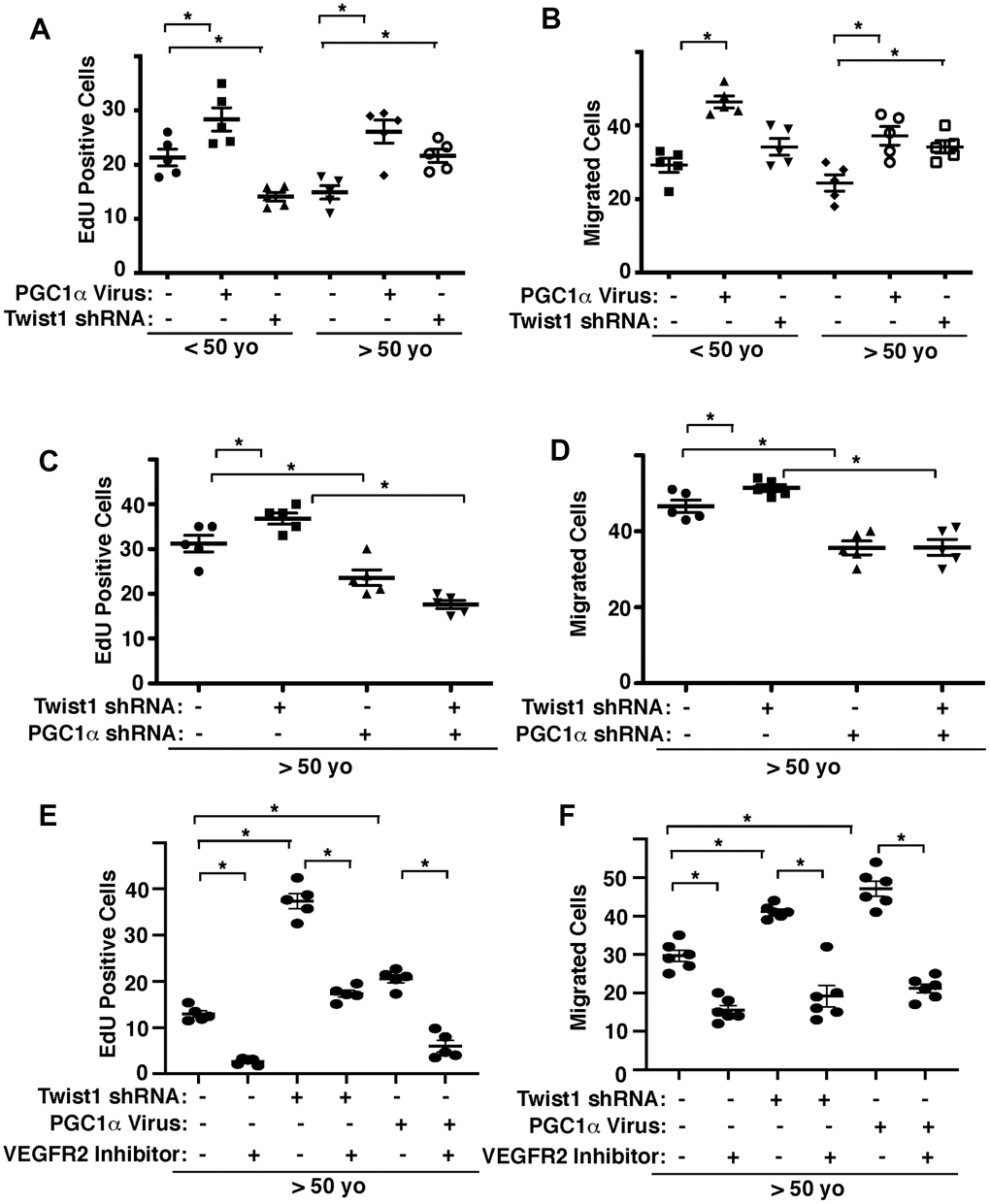
Figure 3. Twist1-PGC1α signaling controls EC DNA synthesis and migration in young vs. aged ECs. (A) Graph showing EdU-positive young (<50 years old) vs. aged (>50 years old) human adipose ECs treated with lentivirus overexpressing PGC1α or Twist1 shRNA (n=5, mean ± s.e.m., *, p<0.05). As a control, young vs. aged human adipose ECs were treated with lentivirus encoding control shRNA with irrelevant sequences or control virus (vector alone). (B) Graph showing young vs. aged human adipose ECs treated with lentivirus overexpressing PGC1α or Twist1 shRNA migrating towards 5% FBS (n=5, mean±s.e.m., *, p<0.05). (C) Graph showing EdU-positive aged human adipose ECs treated with lentivirus encoding Twist1 shRNA, PGC1α shRNA, or in combination (n=5, mean ± s.e.m., *, p<0.05). As a control, aged human adipose ECs were treated with lentivirus encoding control shRNA with irrelevant sequences. (D) Graph showing aged human adipose ECs treated with lentivirus encoding Twist1 shRNA, PGC1α shRNA, or in combination migrating towards 5% FBS (n=5, mean ± s.e.m., *, p<0.05). (E) Graph showing EdU-positive aged human adipose ECs treated with lentivirus encoding Twist1 shRNA, PGC1α, or in combination with VEGFR2 inhibitor SU5416 (n=5, mean ± s.e.m., *, p<0.05). As a control, aged human adipose ECs were treated with lentivirus encoding control shRNA with irrelevant sequences, control virus (vector alone) or control vehicle. (F) Graph showing aged human adipose ECs treated with lentivirus encoding Twist1 shRNA, PGC1α, or in combination with VEGFR2 inhibitor SU5416 migrating towards 5% FBS (n=6, mean ± s.e.m., *, p<0.05).
To further study age-dependent changes in vascular formation in vivo, we used the fibrin gel implantation system, in which young vs. aged human adipose ECs were mixed in the gel [47], and characterized vascular formation in the gel. Consistent with previous report [47], vessel formation derived from supplemented ECs in the gel was attenuated when gel mixed with GFP-labeled aged ECs was implanted under the skin of adult immunocompromised NSG mice. Vascular area was 23% lower than that in the gel supplemented with young ECs (Figure 4A). Blood vessel formation derived from supplemented ECs was stimulated when young and aged human adipose ECs overexpressing PGC1α were supplemented in the gel and subcutaneously implanted (Figure 4A). Twist1 knockdown in young human adipose ECs, which inhibits EC DNA synthesis (Figure 3), tended to inhibit supplemented EC-derived vascular formation (Figure 4A), while Twist1 knockdown in supplemented aged ECs, which increases VEGFR2 expression (Figures 1, 2), restored blood vessel formation in the implanted gel (Figure 4A). Knockdown of PGC1α or VEGFR2 inhibitor suppressed restoration of vascular formation induced by Twist1 knockdown in aged ECs in the gel (Figure 4B), suggesting that Twist1 knockdown restores vascular formation in aged ECs through PGC1α and VEGFR2.
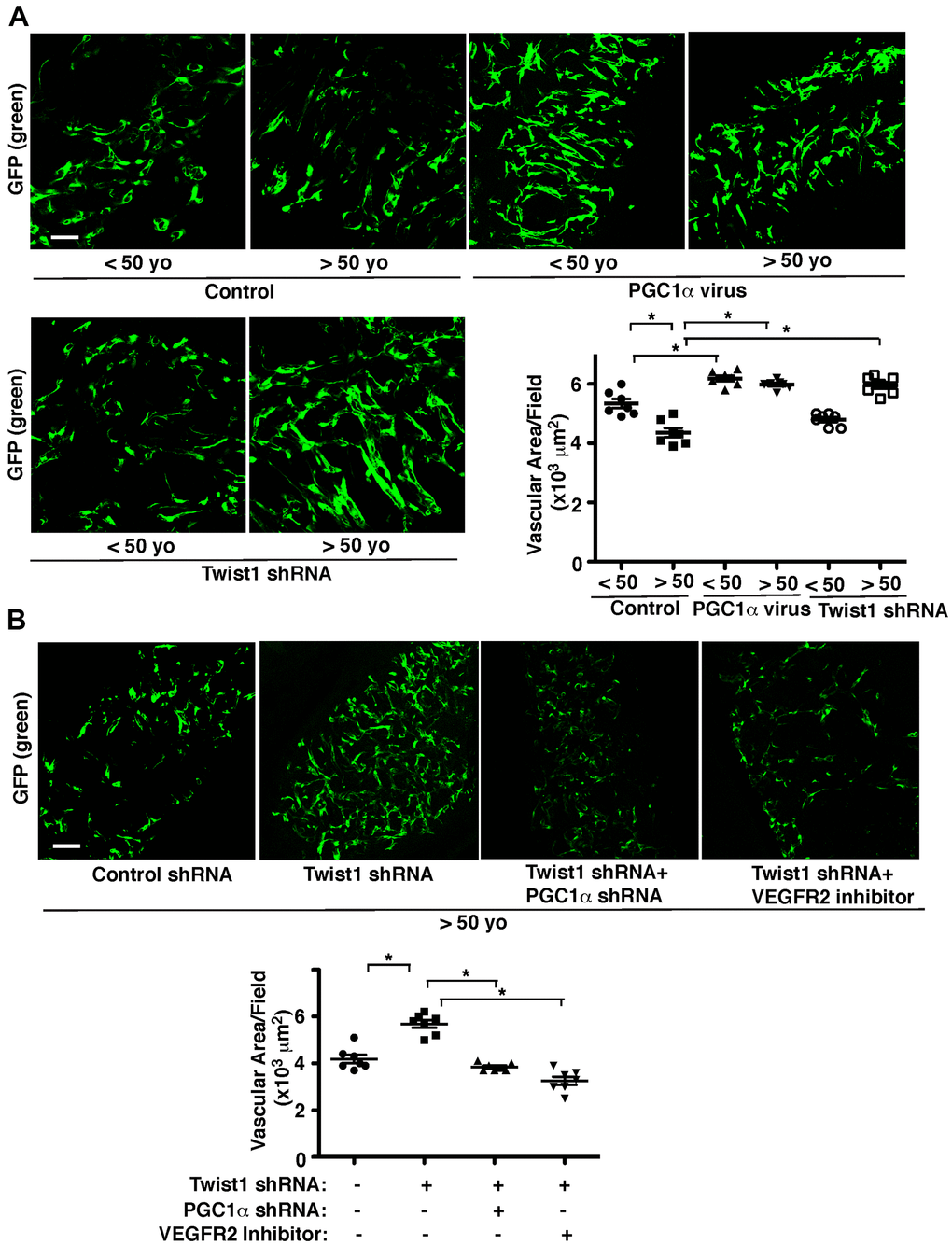
Figure 4. Twist1-PGC1α signaling mediates age-dependent decline in vascular formation in the subcutaneously implanted gel. (A) IF micrographs of fibrin gel supplemented with GFP-labeled young (<50 years old) vs. aged (>50 years old) human adipose ECs treated with lentivirus overexpressing PGC1α or Twist1 shRNA and subcutaneously implanted on NSG mice for 7 days. As a control, young vs. aged human adipose ECs were treated with lentivirus encoding control shRNA with irrelevant sequences or vector alone. Scale bar, 100 μm. Graph showing vascular area in the gel (n=7, mean ± s.e.m., *, p<0.05). (B) IF micrographs of fibrin gel supplemented with GFP-labeled aged human adipose ECs treated with lentivirus encoding Twist1 shRNA or in combination with PGC1α shRNA or VEGFR2 inhibitor (SU5416) and subcutaneously implanted on NSG mice for 7 days. As a control, aged human adipose ECs were treated with lentivirus encoding control shRNA with irrelevant sequences or control vehicle. Scale bar, 100 μm. Graph showing vascular area in the gel (n=7, mean ± s.e.m., *, p<0.05).
Endothelial Twist1 mediates age-related decline in post-PNX lung growth
Angiogenesis plays important roles in compensatory lung growth after unilateral PNX, while these effects are inhibited in aged animals [12–14, 48]. Knockdown of Twist1 in aged human adipose ECs reverses age-dependent inhibition of EC DNA synthesis and migration in cultured ECs and blood vessel formation in the gel implantation system (Figures 3, 4). Therefore, we examined the role of endothelial Twist1 in age-dependent inhibition of lung growth after PNX. The ratio of the weight of right cardiac lobe to mouse body weight (BW) increased by 1.8-fold in the 2M old Twist1fl/fl mouse lungs 7 days after PNX (Figure 5A). The post-PNX lung growth was attenuated in the 24M old Twist1fl/fl mouse lungs (Figure 5A). The alveolar size measured by mean linear intercept (MLI) decreased by 33% and the number of alveoli increased by 1.7- times in the hematoxylin and eosin (H&E)-stained histological sections of the 2M old Twist1fl/fl mouse right lung lobe after left PNX (Supplementary Figure 1). These effects were attenuated in 24M old Twist1fl/fl mouse lungs (Supplementary Figure 1). The mRNA and protein levels of VEGFR2 in the 2M old Twist1fl/fl mouse lungs 7 days after left PNX also increased by 1.3- and 2.6-times, respectively, while the effects were also attenuated in 24M old post-PNX Twist1fl/fl mouse lungs (Figure 5B–5D).
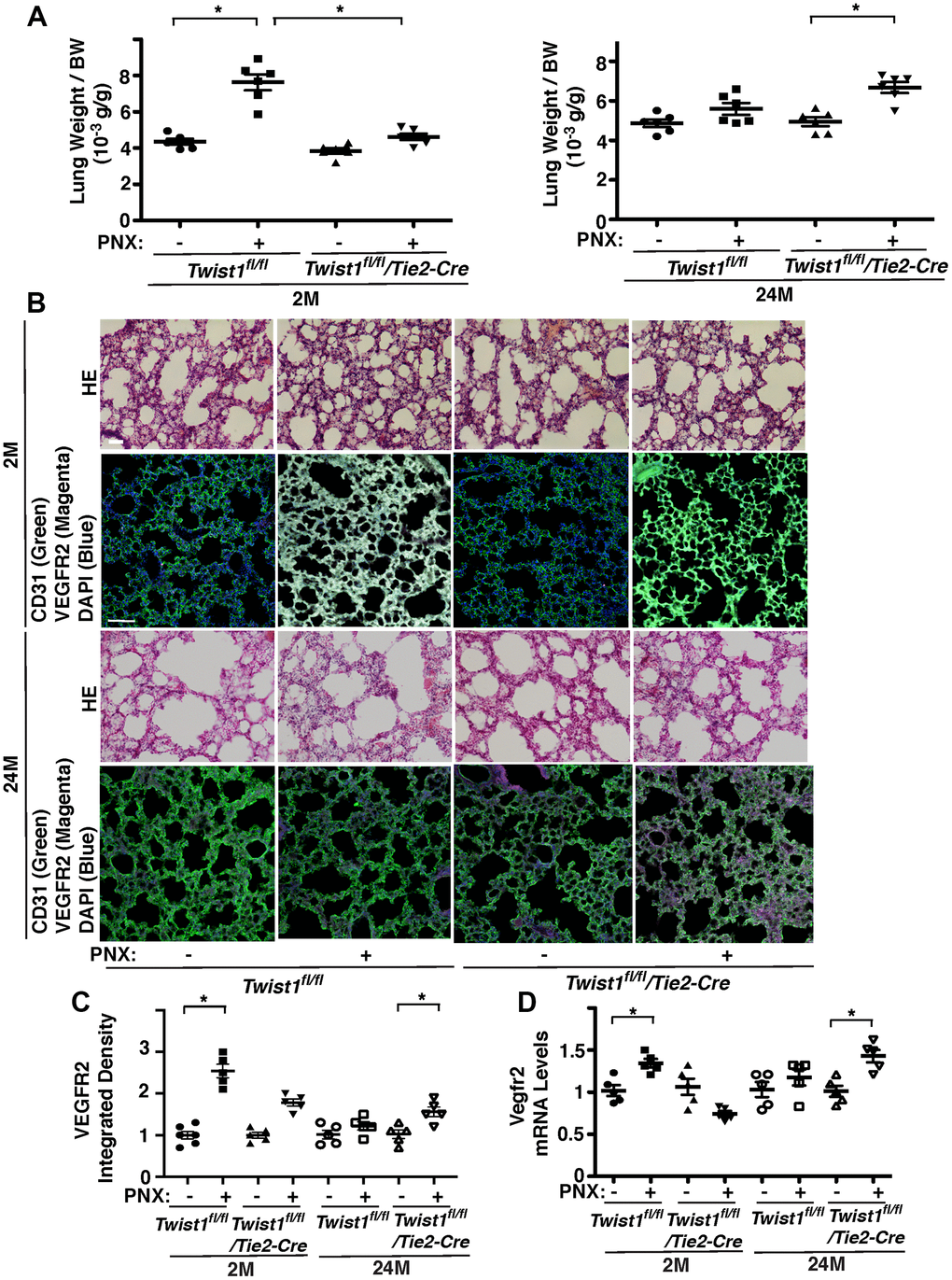
Figure 5. Endothelial Twist1 mediates age-dependent inhibition of post-PNX compensatory lung growth. (A) Graphs showing the ratio of the weight of right lung cardiac lobe to mouse BW in the 2M vs. 24M old Twist1fl/fl or Twist1fl/fl/Tie2-cre mice after PNX (n=6, mean ± s.e.m., *, p<0.05). (B) H&E-stained mouse lungs in the cardiac lobe of 2M vs. 24M old Twist1fl/fl or Twist1fl/fl/Tie2-cre mice after PNX (top, 3rd) Scale bar, 25 μm. IF micrographs showing staining of CD31, VEGFR2 and DAPI in 2M vs. 24M old Twist1fl/fl or Twist1fl/fl/Tie2-cre mice after PNX (2nd, bottom). Scale bar, 100 μm. (C) Graph showing integrated density of VEGFR2 in 2M vs. 24M old Twist1fl/fl or Twist1fl/fl/Tie2-cre mice after PNX (n=5-6, mean ± s.e.m., *, p<0.05). (D) Graph showing the mRNA levels of Vegfr2 in the ECs isolated from 2M vs. 24M old Twist1fl/fl or Twist1fl/fl/Tie2-cre mouse lungs after PNX (n=5, mean ± s.e.m., *, p<0.05).
To examine the effects of endothelial Twist1 on post-PNX lung growth in the aged lung, we compared lung growth between 2M and 24M old Twist1fl/fl/Tie2-cre mice, in which Twist1 mRNA levels in mouse lung ECs were 78% lower than those in Twist1fl/fl mice (Figure 1F). Post-PNX lung growth was inhibited in 2M old Twist1fl/fl/Tie2-cre mice compared to that in 2M old Twist1fl/fl mice, while post-PNX lung growth was restored in 24M old Twist1fl/fl/Tie2-cre mice (Figure 5A). The alveolar size and number were also restored in 24M old Twist1fl/fl/Tie2-cre mouse lungs after left unilateral PNX (Supplementary Figure 1). The mRNA levels of VEGFR2 did not increase in the 24M old Twist1fl/fl mouse lung ECs after PNX, while the levels increased by 1.4-times in the 24M old Twist1fl/fl/Tie2-cre mouse lungs after PNX (Figure 5D). The immunohistochemical analysis also confirmed that the protein levels of VEGFR2 increased by 1.4-times in the post-PNX 24M old Twist1fl/fl/Tie2-cre mouse lungs (Figure 5C), suggesting that knockdown of endothelial Twist1 increases angiogenic factor expression and restores post-PNX lung growth in aged lungs.
Discussion
Here, we have demonstrated that the levels of Twist1 are higher, while the levels of PGC1α and VEGFR2 are lower in aged human adipose ECs and mouse lung ECs compared to those from young animals. Twist1 knockdown or PGC1α overexpression upregulates VEGFR2 expression in aged ECs, which reverses age-dependent decline in EC proliferation and migration. Vascular formation was suppressed in the fibrin gel mixed with aged ECs, while PGC1α overexpression or Twist1 knockdown in aged ECs restored the effects. Post-PNX lung growth inhibited in 24M old mice was restored in Twist1fl/fl/Tie2-cre mice. These results suggest that age-dependent upregulation of Twist1 expression in ECs inhibits lung vascular and alveolar morphogenesis in aged mice by decreasing VEGFR2 expression (Figure 6). Modulation of Twist1 expression in ECs could be one of the promising strategies for age-related lung diseases and may be able to delay the aging processes in the lungs.
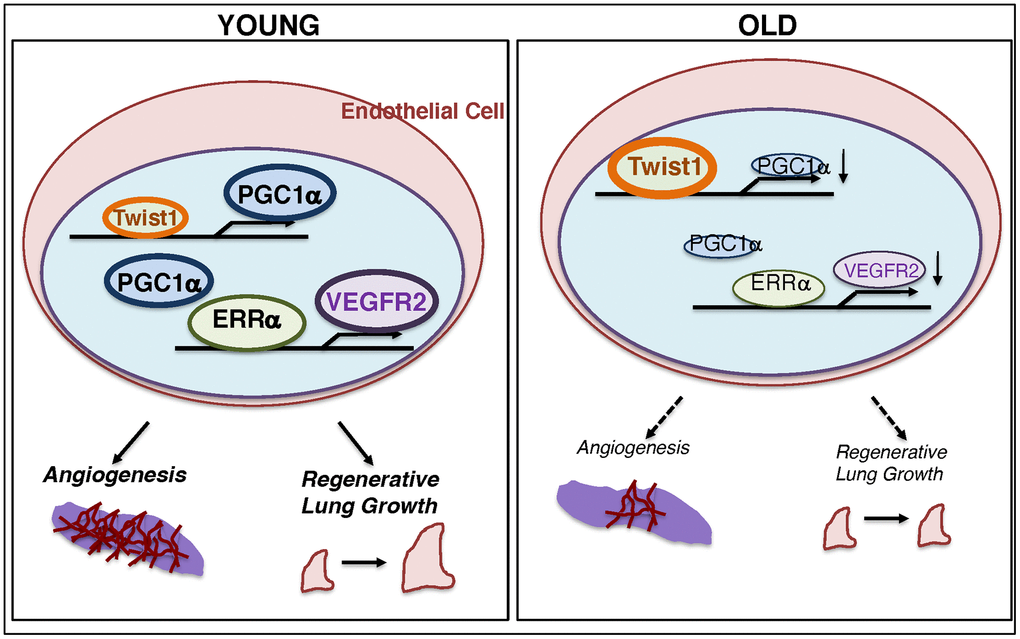
Figure 6. Schematic illustration of the angiogenic signaling pathways in young vs. aged ECs. Schematic illustration showing that Twist1 expression is higher in aged ECs, which decreases the expression of PGC1α and VEGFR2, and inhibits EC DNA synthesis and migration in vitro and blood vessel formation and alveolar regeneration in aged mice.
We examined the mechanism of aging-dependent inhibition of angiogenesis using ECs isolated from human adipose tissues. We then characterized the effects of aging on regenerative lung growth using a PNX model. We used a unilateral PNX model because (1) post-PNX regenerative lung growth takes place after the organs are damaged or partially removed, while the process is attenuated in older people [13, 14], (2) angiogenic signaling mediates post-PNX lung growth [12, 49, 50], (3) inhibition of angiogenesis and attenuation of lung regeneration and repair abilities in aged people contribute to the pathogenesis of age-related lung diseases, including COPD [15, 16]. Although lung transplantation is one of the options for end-stage lung diseases, it is not an optimal approach [51–53]. Stimulating the intrinsic ability of lung regeneration may be a promising strategy to restore structures and functions after resection of injured lungs. Thus, although the PNX model does not directly mimic certain diseases, understanding the mechanism of age-dependent inhibition of angiogenesis and regenerative lung growth using this model would improve the strategies for repair from age-related lung diseases.
We found that upregulation of Twist1 mediates age-dependent decline in angiogenesis through PGC1α and VEGFR2 expression. Twist1 controls multiple angiogenic pathways (e.g., angiopoietins (Angs)-Tie2 [18, 25], VEGF-VEGFR2 [24], PDGF [28]). For example, we have reported that Twist1 controls Tie2 expression by binding to its promoter region, E-box [18, 25]. Since the VEGFR2 promoter also contains an E-box, in addition to the pathway through PGC1α-ERRα, Twist1 may control VEGFR2 expression by binding to the E-box promoter sequence. Twist1 also interacts with other pathways controlling angiogenesis (HIF1α [54], Wnt [55], Notch [56, 57], PI3K-AKT [58, 59], and TGF-β [27]). Given that Twist1 binds to PGC1α and inhibits its co-transcriptional activity in fat cells [36], Twist1 may control angiogenesis by changing metabolic signaling. Twist1 is also involved in DNA methylation and cellular senescence, which are associated with aging processes [60–62]. Aged senescent cells secrete a number of senescence-associated secretory phenotype (SASP) factors, which may have broad effects on lung regeneration in aged lungs [63, 64]. Thus, multiple indirect signaling pathways are involved in age-dependent decline in angiogenesis through Twist1. Given that the multiple signaling pathways are required for physiological and functional blood vessel formation [65–67], manipulation of the expression of endothelial Twist1, which is involved in multiple angiogenic signaling mechanisms, could be an optimal strategy to reverse lung vascular and alveolar formation in the aged lung.
Our results have demonstrated that post-PNX lung growth is restored in 24M old Twist1fl/fl/Tie2-cre mouse lungs. Twist1 knockdown stimulates angiogenic activity in aged ECs and vascular formation in the gel implantation system. Thus, knockdown of Twist1 may stimulate lung growth in aged mice through increasing angiogenesis. In addition to ECs, Twist1 is expressed in other cell types as well (e.g., fibroblasts, epithelial cells) [17, 68], which may also influence lung vascular and alveolar morphogenesis. We used Tie2-specific Twist1 knockout mice in this study and found that Twist1 knockdown in Tie2-expressing cells restores regenerative lung growth in the aged mouse lungs. Since Tie2 is expressed in other cell types such as fibroblasts and immune cells, which also contribute to vascular and alveolar epithelial morphogenesis [69], Twist1 expression in these other cells may contribute to post-PNX lung growth in the aged lung. Twist1 knockdown in ECs in other organs may also indirectly affect post-PNX lung growth. The effects of Twist1 on angiogenesis may be different among ages and tissues. Although knockdown of Twist1 increases VEGFR2 expression in aged ECs and restores vascular and alveolar morphogenesis in aged mice, endothelial Twist1 knockdown decreases VEGFR2 expression and impairs retinal angiogenesis in neonatal mice [24] and inhibits post-PNX lung growth in young mice (Figure 5). Consistently, Twist1 overexpression did not decrease VEGFR2 expression nor inhibit EC proliferation and migration in young ECs; rather Twist1 overexpression increased VEGFR2 expression and stimulated EC DNA synthesis and migration in young ECs (Supplementary Figure 2A–2C). Thus, the effects of Twist1 seem to be age dependent; increased levels of Twist1 in aged ECs decrease PGC1α and VEGFR2 expression and inhibit EC proliferation and migration, while overexpression of Twist1 increases VEGFR2 expression and induces EC proliferation and migration in young ECs. As expected from previous reports using young adipose tissues [36] or skeletal muscles [70], overexpression of Twist1 tended to but did not significantly reduce PGC1α mRNA expression in young ECs (Supplementary Figure 2A). Twist1 is known to behave as a negative regulator of PGC1α in young fat tissues [36], and may indirectly control angiogenesis by changing its activity in young ECs. The levels of Twist1 in aged ECs seem to be high enough to inhibit cell proliferation and migration; Twist1 overexpression in aged ECs did not significantly change DNA synthesis and migration (not shown). Sustained knockdown of endothelial Twist1 in aged Twist1fl/fl/Tie2-cre mice may modulate other signaling pathways in other cell types and other organs. In fact, knockdown of endothelial Twist1 in aged Twist1fl/fl/Tie2-cre mice increased VEGFR2 expression but not PGC1α expression (Figure 1D). Further investigation using inducible endothelial-specific Twist1 knockout mice will elucidate the effects of endothelial Twist1 on age-dependent changes in angiogenesis and alveolar regeneration during specific time frames.
We found that binding of ERRα to the VEGFR2 promoter region increased in aged ECs (Figure 2D), in which VEGFR2 expression is suppressed. This is consistent with the previous report demonstrating that ERRα acts as a transcriptional repressor in ECs [44]. However, ERRα expression in aged ECs is lower than that in young ECs (Figure 2C). How aging downregulates ERRα, while increasing ERRα recruitment to the VEGFR2 promoter region remains unclear. Even at the lower expression level, the remaining ERRα recruited to the promoter region may exert its activity. Post-translational modifications of ERRα induced in aged ECs may increase its recruitment to the promoter region. ERRα may also interact with co-repressors and activators on its promoter regions and modulation of these interactions could contribute to regulation of ERRα activity.
In addition to its role in angiogenesis [29, 33–35, 38, 42, 71], PGC1α controls mitochondrial biogenesis [29–32, 42, 71]. PGC1α also regulates a number of anti-reactive oxygen species (ROS) genes [72, 73], which contribute to age-related pathologies, and protects against endothelial dysfunction [29, 74]. Thus, modulation of Twist1-PGC1α signaling may reverse age-dependent impairment of angiogenesis through multiple mechanisms.
Mechanical forces control vascular formation and function [75–78], and appropriate micromechanical environment is necessary for lung development and regeneration [69, 77, 79]. Twist1 is known to mediate the effects of mechanical forces, including shear stress [22], ECM stiffness [80, 81] and stretching forces [82]. Increases in the ratio of collagen and elastin in aged fibroblasts increase pulmonary stiffness and lower compliance [83]. The response to shear stress is also altered during aging [84]. In fact, premature aged ECs from Hutchinson-Gilford progeria syndrome patients impair normal mechanosensing, leading to accelerated fibrosis [84]. We have reported that age-related lung diseases, such as pulmonary fibrosis and accompanied pulmonary hypertension, in which changes in mechanical environment are involved in the disease progression [85–87], are prevented in Twist1fl/fl/Tie2-cre mice [18, 27]. Other transcription factors and co-factors (e.g., TFII-I, GATA2, YAP1) that sense mechanical forces interact with Twist1, control angiogenesis [47, 50, 71, 75, 88], and contribute to age-related lung diseases (e.g., pulmonary fibrosis, pulmonary hypertension) [85–87]. Thus, Twist1 senses age-dependent changes in the mechanical forces to control angiogenesis and lung regeneration.
We used ECs isolated from human adipose tissues of different conditions (e.g., body mass index (BMI), sex, pre-existing diseases, visceral vs. subcutaneous adipose tissues). The heterogeneity of the samples may impact angiogenic signaling. Investigation of the effects of aging on angiogenesis using a more specific cohort with larger sample size will further elucidate the mechanism.
In summary, endothelial Twist1 mediates age-related decline in blood vessel formation and post-PNX compensatory lung growth through PGC1α-VEGFR2 signaling. Modulation of endothelial Twist1 would potentially be a new strategy for aging-associated lung diseases.
Materials and Methods
Materials
Anti-β-actin (A5316) and –PGC1α (AB3242) monoclonal antibodies were from Sigma (St. Louis, MO). Anti-VEGFR2 (2479) and –ERRα (13826) antibodies were from Cell Signaling (Danvers, MA). Anti-CD31 (102409) and –CD45 (103113) antibodies were from BioLegend (San Diego, CA). Anti-VE-cadherin (562243) and -CD31 (553370) antibodies were from BD Pharmingen (San Diego, CA). Anti-Twist1 antibody (sc-15393) was from Santa Cruze Biotechnology (Dallas, TX). SU5416 was purchased from Millipore Sigma (Burlington, MA).
EC isolation
Mouse lung ECs were isolated from C57BL6 mice (2M and 24M old) using anti-CD31 conjugated magnetic beads and cultured as reported [27, 28, 50]. De-identified discarded human adipose tissues were collected from Medical College of Wisconsin (MCW) Tissue Bank. These de-identified human adipose tissue-derived ECs have been determined as Non-Human Subjects Research by the MCW Institutional Review Board. Sample demographic information collected using the Generic Clinical Research Database (GCRD) is summarized in Table 1. The samples from cancer patients were excluded. ECs were isolated and cultured as described before [47, 50] and used between passages 1-2.
Table 1. Sample demographics.
| Sample demographics (n=16) | Young (< 50 y.o., n=8) | Old (> 50 y.o., n=8) |
| Gender, Male/Female | 3 (37.5%)/5 (62.5%) | 5 (62.5%)/3 (37.5%) |
| Age, year (mean ± s.e.m) | 34.62±2.96 | 70.37±2.50 |
| Body mass index (mean ± s.e.m) | 28.82±2.91 | 28.38±1.33 |
| Underlying diseases | ||
| Coronary artery disease | 0 (0%) | 3 (37.5%) |
| Hypertension | 0 (0%) | 3 (37.5%) |
| Hyperlipidemia | 0 (0%) | 3 (37.5%) |
| Diabetes mellitus | 0 (0%) | 1 (12.5%) |
| Atrial fibrillation | 0 (0%) | 3 (37.5%) |
| None of the above | 1 (12.5%) | 3 (37.5%) |
Molecular biological and biochemical methods
pLenti-PGC1α (mouse) [89] and pTRIPZ-PGC1α shRNA (human) [90] were obtained as described. Lentiviral construct for human Twist1 shRNA was CCGGGCTGGACTCCAAGATGGCAAGCTCGAGCTTGCCATCTTGGAGTCCAGCTTTTT [28]. pHAGE-Twist1 was constructed as described [27]. The lentiviral pHAGE-GFP construct [75] was used for labeling human adipose ECs. Plasmid with vector only was used as a control. Lentiviral vectors were generated as reported [49, 50, 75]. The siRNA sequences for human PGC1α and Twist1 were described before [18, 25, 27, 28, 71] and siLentFect (BioRad, Hercules, CA) was used for transfection. Transfected human adipose ECs were used for the assays 3 days later. A scrambled siRNA (QIAGEN) was used as a control.
The primers for mouse Twist1, Pgc1α, Vegfr2, and cyclophilin and human Twist1, PGC1α, VEGFR2, and B2M for quantitative reverse transcription (qRT)-PCR were previously described [25, 27, 49, 71, 75]. The primers used for human ERRα were forward; AGGGTTCCTCGAGACAGAG and reverse; TCACAGGATGCCACACCATAG. The iScript reverse transcription kit and iTaq SYBR Green qPCR kit (BioRad, Hercules, CA) were used for qRT-PCR, which was performed using the BioRad real time PCR system (BioRad).
For the chromatin immunoprecipitation (ChIP) assay, DNA from human adipose young vs. aged ECs was immunoprecipitated with the ERRα antibody or control immunoglobulin (Thermo Fisher Scientific, Waltham, MA) [25, 27, 75]. The promoter region of human VEGFR2 binding to ERRα was analyzed with primers, 5’- GTGCCGGTAGGAGAGGATA-3’ and 5’- AGCGGTCAATGTGTGGTC-3’.
In vitro EC DNA synthesis and migration
Human adipose EC DNA synthesis was analyzed by an EdU incorporation assay. Human adipose ECs (EBM2 with 2% serum), in which PGC1α and/or Twist1 expression was manipulated using lentiviral transduction, were treated with EdU (10 μM, 4 h), and analyzed using a confocal Leica SP5 microscope [28]. A modified transwell migration assay was used for analysis of EC migration [28].
In vivo animal experiments
The animal study was conducted following the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Animal Care and Use Committee of MCW reviewed and approved the protocols. Nonobese diabetic/severe combined immunodeficiency gamma (NSG) mice (8 week old; Jackson Laboratory, stock # 005557), C57BL6 mice (Jackson Laboratory, stock # 000664 and NIA/NIH rodent colonies), and Twist1fl/fl/Tie2-cre and Twist1fl/fl mice [18, 25, 27, 28] were used for the study. The study used both male and female mice. For gel implantation, we implanted the fibrin gel [27, 28, 47, 50] on the back of NSG mice for 7 days and histological analysis was performed as described [47, 75]. We mixed the gel with SU5416 (final concentration; 3 μM) for VEGFR2 inhibition. Unilateral PNX was conducted as previously described [12, 49, 50]. Histological (MLI, alveolar number) and immunohistochemical analysis was performed as described [12, 49, 50].
Statistics
All phenotypic analysis was performed by masked observers. Power analysis was conducted to provide 80% power to detect an effective 20-30% difference between the experimental groups. Three or more independent experiments were conducted to determine error bars (SEM) and p values. Student’s t-test (two groups) and one-way ANOVA with a post-hoc analysis using the Bonferroni test (more than two groups) were conducted to analyze statistical significance.
Supplementary Materials
Author Contributions
Conceived and designed the experiments: AM, TM. Performed the experiments: KH, TH, KM, MT, AM, TM. Analyzed the data: KH, AM, TM. Contributed reagents/materials/analysis tools: AM, TM. Wrote the paper: AM, TM.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by funds from NIH R21AG054830 (to A.M., to T.M.), R01HL139638 (to A.M., to T.M.), R21AG062893 (to A.M., to T.M.), NIH R01HL142578 (to A.M., to T.M.), and American Heart Association (AHA) 18TPA34170129 (to A.M.).
References
- 1. Adnot S, Amsellem V, Boyer L, Marcos E, Saker M, Houssaini A, Kebe K, Dagouassat M, Lipskaia L, Boczkowski J. Telomere dysfunction and cell senescence in chronic lung diseases: therapeutic potential. Pharmacol Ther. 2015; 153:125–34. https://doi.org/10.1016/j.pharmthera.2015.06.007 [PubMed]
- 2. Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. Thorax. 2015; 70:482–89. https://doi.org/10.1136/thoraxjnl-2014-206084 [PubMed]
- 3. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012; 122:2749–55. https://doi.org/10.1172/JCI60324 [PubMed]
- 4. Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009; 135:173–80. https://doi.org/10.1378/chest.08-1419 [PubMed]
- 5. Selman M, Rojas M, Mora AL, Pardo A. Aging and interstitial lung diseases: unraveling an old forgotten player in the pathogenesis of lung fibrosis. Semin Respir Crit Care Med. 2010; 31:607–17. https://doi.org/10.1055/s-0030-1265901 [PubMed]
- 6. Falk JA, Kadiev S, Criner GJ, Scharf SM, Minai OA, Diaz P. Cardiac disease in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008; 5:543–48. https://doi.org/10.1513/pats.200708-142ET [PubMed]
- 7. Hunninghake DB. Cardiovascular disease in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005; 2:44–49. https://doi.org/10.1513/pats.200410-050SF [PubMed]
- 8. Tarnawski AS, Ahluwalia A, Jones MK. Angiogenesis in gastric mucosa: an important component of gastric erosion and ulcer healing and its impairment in aging. J Gastroenterol Hepatol. 2014 (Suppl 4); 29:112–23. https://doi.org/10.1111/jgh.12734 [PubMed]
- 9. Lähteenvuo J, Rosenzweig A. Effects of aging on angiogenesis. Circ Res. 2012; 110:1252–64. https://doi.org/10.1161/CIRCRESAHA.111.246116 [PubMed]
- 10. Rivard A, Fabre JE, Silver M, Chen D, Murohara T, Kearney M, Magner M, Asahara T, Isner JM. Age-dependent impairment of angiogenesis. Circulation. 1999; 99:111–20. https://doi.org/10.1161/01.cir.99.1.111 [PubMed]
- 11. Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, Zhou YF, McDonald KR, Na Y, et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res. 2007; 101:1310–18. https://doi.org/10.1161/CIRCRESAHA.107.153346 [PubMed]
- 12. Mammoto A, Muyleart M, Mammoto T. LRP5 in age-related changes in vascular and alveolar morphogenesis in the lung. Aging (Albany NY). 2019; 11:89–103. https://doi.org/10.18632/aging.101722 [PubMed]
- 13. Laros CD, Westermann CJ. Dilatation, compensatory growth, or both after pneumonectomy during childhood and adolescence. A thirty-year follow-up study. J Thorac Cardiovasc Surg. 1987; 93:570–76. [PubMed]
- 14. Butler JP, Loring SH, Patz S, Tsuda A, Yablonskiy DA, Mentzer SJ. Evidence for adult lung growth in humans. N Engl J Med. 2012; 367:244–47. https://doi.org/10.1056/NEJMoa1203983 [PubMed]
- 15. Voelkel NF, Douglas IS, Nicolls M. Angiogenesis in chronic lung disease. Chest. 2007; 131:874–79. https://doi.org/10.1378/chest.06-2453 [PubMed]
- 16. Alagappan VK, de Boer WI, Misra VK, Mooi WJ, Sharma HS. Angiogenesis and vascular remodeling in chronic airway diseases. Cell Biochem Biophys. 2013; 67:219–34. https://doi.org/10.1007/s12013-013-9713-6 [PubMed]
- 17. Pozharskaya V, Torres-González E, Rojas M, Gal A, Amin M, Dollard S, Roman J, Stecenko AA, Mora AL. Twist: A regulator of epithelial-mesenchymal transition in lung fibrosis. PLoS One. 2009; 4:e7559. https://doi.org/10.1371/journal.pone.0007559 [PubMed]
- 18. Mammoto T, Jiang A, Jiang E, Mammoto A. Role of Twist1 phosphorylation in angiogenesis and pulmonary fibrosis. Am J Respir Cell Mol Biol. 2016; 55:633–44. https://doi.org/10.1165/rcmb.2016-0012OC [PubMed]
- 19. Pettersson AT, Laurencikiene J, Mejhert N, Näslund E, Bouloumié A, Dahlman I, Arner P, Rydén M. A possible inflammatory role of twist1 in human white adipocytes. Diabetes. 2010; 59:564–71. https://doi.org/10.2337/db09-0997 [PubMed]
- 20. Nishioka M, Venkatesan N, Dessalle K, Mogas A, Kyoh S, Lin TY, Nair P, Baglole CJ, Eidelman DH, Ludwig MS, Hamid Q. Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir Res. 2015; 16:72. https://doi.org/10.1186/s12931-015-0232-4 [PubMed]
- 21. Wang G, Dong W, Shen H, Mu X, Li Z, Lin X, Liu Y, Du J. A comparison of twist and E-cadherin protein expression in primary non-small-cell lung carcinoma and corresponding metastases. Eur J Cardiothorac Surg. 2011; 39:1028–32. https://doi.org/10.1016/j.ejcts.2011.01.023 [PubMed]
- 22. Mahmoud MM, Kim HR, Xing R, Hsiao S, Mammoto A, Chen J, Serbanovic-Canic J, Feng S, Bowden NP, Maguire R, Ariaans M, Francis SE, Weinberg PD, et al. TWIST1 integrates endothelial responses to flow in vascular dysfunction and atherosclerosis. Circ Res. 2016; 119:450–62. https://doi.org/10.1161/CIRCRESAHA.116.308870 [PubMed]
- 23. Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, Mammoto A, Chen J, Ariaans M, Francis SE, Van der Heiden K, Ridger V, Evans PC. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor snail. Sci Rep. 2017; 7:3375. https://doi.org/10.1038/s41598-017-03532-z [PubMed]
- 24. Li J, Liu CH, Sun Y, Gong Y, Fu Z, Evans LP, Tian KT, Juan AM, Hurst CG, Mammoto A, Chen J. Endothelial TWIST1 promotes pathological ocular angiogenesis. Invest Ophthalmol Vis Sci. 2014; 55:8267–77. https://doi.org/10.1167/iovs.14-15623 [PubMed]
- 25. Mammoto T, Jiang E, Jiang A, Lu Y, Juan AM, Chen J, Mammoto A. Twist1 controls lung vascular permeability and endotoxin-induced pulmonary edema by altering Tie2 expression. PLoS One. 2013; 8:e73407. https://doi.org/10.1371/journal.pone.0073407 [PubMed]
- 26. Chen J, Yuan W, Wu L, Tang Q, Xia Q, Ji J, Liu Z, Ma Z, Zhou Z, Cheng Y, Shu X. PDGF-D promotes cell growth, aggressiveness, angiogenesis and EMT transformation of colorectal cancer by activation of Notch1/Twist1 pathway. Oncotarget. 2017; 8:9961–73. https://doi.org/10.18632/oncotarget.14283 [PubMed]
- 27. Mammoto T, Muyleart M, Konduri GG, Mammoto A. Twist1 in hypoxia-induced pulmonary hypertension through transforming growth factor-β-smad signaling. Am J Respir Cell Mol Biol. 2018; 58:194–207. https://doi.org/10.1165/rcmb.2016-0323OC [PubMed]
- 28. Mammoto A, Hendee K, Muyleart M, Mammoto T. Endothelial Twist1-PDGFB signaling mediates hypoxia-induced proliferation and migration of αSMA-positive cells. Sci Rep. 2020; 10:7563. https://doi.org/10.1038/s41598-020-64298-5 [PubMed]
- 29. Patten IS, Arany Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol Metab. 2012; 23:90–97. https://doi.org/10.1016/j.tem.2011.09.007 [PubMed]
- 30. Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol. 2015; 33:49–54. https://doi.org/10.1016/j.ceb.2014.11.002 [PubMed]
- 31. Austin S, St-Pierre J. PGC1α and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci. 2012; 125:4963–71. https://doi.org/10.1242/jcs.113662 [PubMed]
- 32. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006; 116:615–22. https://doi.org/10.1172/JCI27794 [PubMed]
- 33. Rowe GC, Raghuram S, Jang C, Nagy JA, Patten IS, Goyal A, Chan MC, Liu LX, Jiang A, Spokes KC, Beeler D, Dvorak H, Aird WC, Arany Z. PGC-1α induces SPP1 to activate macrophages and orchestrate functional angiogenesis in skeletal muscle. Circ Res. 2014; 115:504–17. https://doi.org/10.1161/CIRCRESAHA.115.303829 [PubMed]
- 34. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008; 451:1008–12. https://doi.org/10.1038/nature06613 [PubMed]
- 35. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013; 112:1171–88. https://doi.org/10.1161/CIRCRESAHA.111.300233 [PubMed]
- 36. Pan D, Fujimoto M, Lopes A, Wang YX. Twist-1 is a PPARdelta-inducible, negative-feedback regulator of PGC-1alpha in brown fat metabolism. Cell. 2009; 137:73–86. https://doi.org/10.1016/j.cell.2009.01.051 [PubMed]
- 37. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. 2017; 152:618–26. https://doi.org/10.1016/j.chest.2017.03.020 [PubMed]
- 38. Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, Tesmenitsky Y, Kang KT, Bischoff J, Kalwa H, Sartoretto JL, Kamei Y, Benjamin LE, Watada H, et al. Endothelial PGC-1α mediates vascular dysfunction in diabetes. Cell Metab. 2014; 19:246–58. https://doi.org/10.1016/j.cmet.2013.12.014 [PubMed]
- 39. Kadlec AO, Chabowski DS, Ait-Aissa K, Gutterman DD. Role of PGC-1α in vascular regulation: implications for atherosclerosis. Arterioscler Thromb Vasc Biol. 2016; 36:1467–74. https://doi.org/10.1161/ATVBAHA.116.307123 [PubMed]
- 40. Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest. 2017; 127:405–14. https://doi.org/10.1172/JCI87440 [PubMed]
- 41. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016; 118:1593–611. https://doi.org/10.1161/CIRCRESAHA.116.307505 [PubMed]
- 42. Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J, Mahmood F, Hess P, Farrell C, Koulisis N, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012; 485:333–38. https://doi.org/10.1038/nature11040 [PubMed]
- 43. Thom R, Rowe GC, Jang C, Safdar A, Arany Z. Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor γ coactivator (PGC)-1α. J Biol Chem. 2014; 289:8810–17. https://doi.org/10.1074/jbc.M114.554394 [PubMed]
- 44. Likhite N, Yadav V, Milliman EJ, Sopariwala DH, Lorca S, Narayana NP, Sheth M, Reineke EL, Giguère V, Narkar V. Loss of estrogen-related receptor alpha facilitates angiogenesis in endothelial cells. Mol Cell Biol. 2019; 39:e00411–18. https://doi.org/10.1128/MCB.00411-18 [PubMed]
- 45. Vanacker JM, Pettersson K, Gustafsson JA, Laudet V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. EMBO J. 1999; 18:4270–79. https://doi.org/10.1093/emboj/18.15.4270 [PubMed]
- 46. Sladek R, Bader JA, Giguère V. The orphan nuclear receptor estrogen-related receptor alpha is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol. 1997; 17:5400–09. https://doi.org/10.1128/mcb.17.9.5400 [PubMed]
- 47. Mammoto T, Torisawa YS, Muyleart M, Hendee K, Anugwom C, Gutterman D, Mammoto A. Effects of age-dependent changes in cell size on endothelial cell proliferation and senescence through YAP1. Aging (Albany NY). 2019; 11:7051–69. https://doi.org/10.18632/aging.102236 [PubMed]
- 48. Paxson JA, Gruntman A, Parkin CD, Mazan MR, Davis A, Ingenito EP, Hoffman AM. Age-dependent decline in mouse lung regeneration with loss of lung fibroblast clonogenicity and increased myofibroblastic differentiation. PLoS One. 2011; 6:e23232. https://doi.org/10.1371/journal.pone.0023232 [PubMed]
- 49. Mammoto T, Chen Z, Jiang A, Jiang E, Ingber DE, Mammoto A. Acceleration of lung regeneration by platelet-rich plasma extract through the low-density lipoprotein receptor-related protein 5-Tie2 pathway. Am J Respir Cell Mol Biol. 2016; 54:103–13. https://doi.org/10.1165/rcmb.2015-0045OC [PubMed]
- 50. Mammoto T, Muyleart M, Mammoto A. Endothelial YAP1 in regenerative lung growth through the angiopoietin-Tie2 pathway. Am J Respir Cell Mol Biol. 2019; 60:117–27. https://doi.org/10.1165/rcmb.2018-0105OC [PubMed]
- 51. Orens JB, Garrity ER
Jr . General overview of lung transplantation and review of organ allocation. Proc Am Thorac Soc. 2009; 6:13–19. https://doi.org/10.1513/pats.200807-072GO [PubMed] - 52. Benden C. Specific aspects of children and adolescents undergoing lung transplantation. Curr Opin Organ Transplant. 2012; 17:509–14. https://doi.org/10.1097/MOT.0b013e3283564fba [PubMed]
- 53. Lyu DM, Zamora MR. Medical complications of lung transplantation. Proc Am Thorac Soc. 2009; 6:101–07. https://doi.org/10.1513/pats.200808-077GO [PubMed]
- 54. Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008; 7:2090–96. https://doi.org/10.4161/cc.7.14.6324 [PubMed]
- 55. Guo Y, Zi X, Koontz Z, Kim A, Xie J, Gorlick R, Holcombe RF, Hoang BH. Blocking Wnt/LRP5 signaling by a soluble receptor modulates the epithelial to mesenchymal transition and suppresses met and metalloproteinases in osteosarcoma Saos-2 cells. J Orthop Res. 2007; 25:964–71. https://doi.org/10.1002/jor.20356 [PubMed]
- 56. Chen HF, Huang CH, Liu CJ, Hung JJ, Hsu CC, Teng SC, Wu KJ. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun. 2014; 5:4697. https://doi.org/10.1038/ncomms5697 [PubMed]
- 57. Wirrig EE, Yutzey KE. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler Thromb Vasc Biol. 2014; 34:737–41. https://doi.org/10.1161/ATVBAHA.113.302071 [PubMed]
- 58. Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration, and invasion by twist: AKT2 comes to interplay. Cancer Res. 2008; 68:957–60. https://doi.org/10.1158/0008-5472.CAN-07-5067 [PubMed]
- 59. Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, Rüegg C, Hemmings BA. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2012; 2:248–59. https://doi.org/10.1158/2159-8290.CD-11-0270 [PubMed]
- 60. Degerman S, Siwicki JK, Osterman P, Lafferty-Whyte K, Keith WN, Roos G. Telomerase upregulation is a postcrisis event during senescence bypass and immortalization of two nijmegen breakage syndrome T cell cultures. Aging Cell. 2010; 9:220–35. https://doi.org/10.1111/j.1474-9726.2010.00550.x [PubMed]
- 61. Cakouros D, Isenmann S, Cooper L, Zannettino A, Anderson P, Glackin C, Gronthos S. Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol Cell Biol. 2012; 32:1433–41. https://doi.org/10.1128/MCB.06315-11 [PubMed]
- 62. Johnson AA, Akman K, Calimport SR, Wuttke D, Stolzing A, de Magalhães JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012; 15:483–94. https://doi.org/10.1089/rej.2012.1324 [PubMed]
- 63. Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev. 2011; 21:107–12. https://doi.org/10.1016/j.gde.2010.10.005 [PubMed]
- 64. van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509:439–46. https://doi.org/10.1038/nature13193 [PubMed]
- 65. Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011; 12:551–64. https://doi.org/10.1038/nrm3176 [PubMed]
- 66. Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011; 27:563–84. https://doi.org/10.1146/annurev-cellbio-092910-154002 [PubMed]
- 67. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011; 473:298–307. https://doi.org/10.1038/nature10144 [PubMed]
- 68. Yeo SY, Lee KW, Shin D, An S, Cho KH, Kim SH. A positive feedback loop bi-stably activates fibroblasts. Nat Commun. 2018; 9:3016. https://doi.org/10.1038/s41467-018-05274-6 [PubMed]
- 69. Mammoto A, Mammoto T. Vascular niche in lung alveolar development, homeostasis, and regeneration. Front Bioeng Biotechnol. 2019; 7:318. https://doi.org/10.3389/fbioe.2019.00318 [PubMed]
- 70. Mudry JM, Massart J, Szekeres FL, Krook A. TWIST1 and TWIST2 regulate glycogen storage and inflammatory genes in skeletal muscle. J Endocrinol. 2015; 224:303–13. https://doi.org/10.1530/JOE-14-0474 [PubMed]
- 71. Mammoto A, Muyleart M, Kadlec A, Gutterman D, Mammoto T. YAP1-TEAD1 signaling controls angiogenesis and mitochondrial biogenesis through PGC1α. Microvasc Res. 2018; 119:73–83. https://doi.org/10.1016/j.mvr.2018.04.003 [PubMed]
- 72. Won JC, Park JY, Kim YM, Koh EH, Seol S, Jeon BH, Han J, Kim JR, Park TS, Choi CS, Lee WJ, Kim MS, Lee IK, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha overexpression prevents endothelial apoptosis by increasing ATP/ADP translocase activity. Arterioscler Thromb Vasc Biol. 2010; 30:290–97. https://doi.org/10.1161/ATVBAHA.109.198721 [PubMed]
- 73. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005; 66:562–73. https://doi.org/10.1016/j.cardiores.2005.01.026 [PubMed]
- 74. Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, Wenzel P, Münzel T, Keaney JF
Jr . Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation. 2008; 118:1347–57. https://doi.org/10.1161/CIRCULATIONAHA.108.784298 [PubMed] - 75. Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009; 457:1103–08. https://doi.org/10.1038/nature07765 [PubMed]
- 76. Mammoto A, Mammoto T, Kanapathipillai M, Wing Yung C, Jiang E, Jiang A, Lofgren K, Gee EP, Ingber DE. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat Commun. 2013; 4:1759. https://doi.org/10.1038/ncomms2774 [PubMed]
- 77. Mammoto T, Jiang E, Jiang A, Mammoto A. Extracellular matrix structure and tissue stiffness control postnatal lung development through the lipoprotein receptor-related protein 5/Tie2 signaling system. Am J Respir Cell Mol Biol. 2013; 49:1009–18. https://doi.org/10.1165/rcmb.2013-0147OC [PubMed]
- 78. Mammoto T, Jiang A, Jiang E, Panigrahy D, Kieran MW, Mammoto A. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am J Pathol. 2013; 183:1293–305. https://doi.org/10.1016/j.ajpath.2013.06.026 [PubMed]
- 79. Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, Li J, Huang H, Cai T, Ji H, Yang C, Tang N. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 2016; 16:1810–19. https://doi.org/10.1016/j.celrep.2016.07.020 [PubMed]
- 80. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, Yang J. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015; 17:678–88. https://doi.org/10.1038/ncb3157 [PubMed]
- 81. Ondeck MG, Kumar A, Placone JK, Plunkett CM, Matte BF, Wong KC, Fattet L, Yang J, Engler AJ. Dynamically stiffened matrix promotes Malignant transformation of mammary epithelial cells via collective mechanical signaling. Proc Natl Acad Sci USA. 2019; 116:3502–07. https://doi.org/10.1073/pnas.1814204116 [PubMed]
- 82. Farge E. Mechanical induction of twist in the drosophila foregut/stomodeal primordium. Curr Biol. 2003; 13:1365–77. https://doi.org/10.1016/s0960-9822(03)00576-1 [PubMed]
- 83. Brandenberger C, Mühlfeld C. Mechanisms of lung aging. Cell Tissue Res. 2017; 367:469–80. https://doi.org/10.1007/s00441-016-2511-x [PubMed]
- 84. Lowenstein CJ, Bennett JA. New vascular insights into premature aging. J Clin Invest. 2019; 129:492–93. https://doi.org/10.1172/JCI125616 [PubMed]
- 85. Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB, Kumar R, Saggar R, et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep. 2015; 13:1016–32. https://doi.org/10.1016/j.celrep.2015.09.049 [PubMed]
- 86. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang YY, Rehman S, Sugahara M, Qi Z, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. 2016; 126:3313–35. https://doi.org/10.1172/JCI86387 [PubMed]
- 87. Alsamman S, Christenson SA, Yu A, Ayad NM, Mooring MS, Segal JM, Hu JK, Schaub JR, Ho SS, Rao V, Marlow MM, Turner SM, Sedki M, et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci Transl Med. 2020; 12:eaay8798. https://doi.org/10.1126/scitranslmed.aay8798 [PubMed]
- 88. Zhang H, von Gise A, Liu Q, Hu T, Tian X, He L, Pu W, Huang X, He L, Cai CL, Camargo FD, Pu WT, Zhou B. Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J Biol Chem. 2014; 289:18681–92. https://doi.org/10.1074/jbc.M114.554584 [PubMed]
- 89. Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006; 127:59–69. https://doi.org/10.1016/j.cell.2006.09.015 [PubMed]
- 90. Kadlec AO, Chabowski DS, Ait-Aissa K, Hockenberry JC, Otterson MF, Durand MJ, Freed JK, Beyer AM, Gutterman DD. PGC-1α (peroxisome proliferator-activated receptor γ coactivator 1-α) overexpression in coronary artery disease recruits NO and hydrogen peroxide during flow-mediated dilation and protects against increased intraluminal pressure. Hypertension. 2017; 70:166–73. https://doi.org/10.1161/HYPERTENSIONAHA.117.09289 [PubMed]